The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment


Abstract
:1. The Physiology of Cyclic Nucleotide Signaling
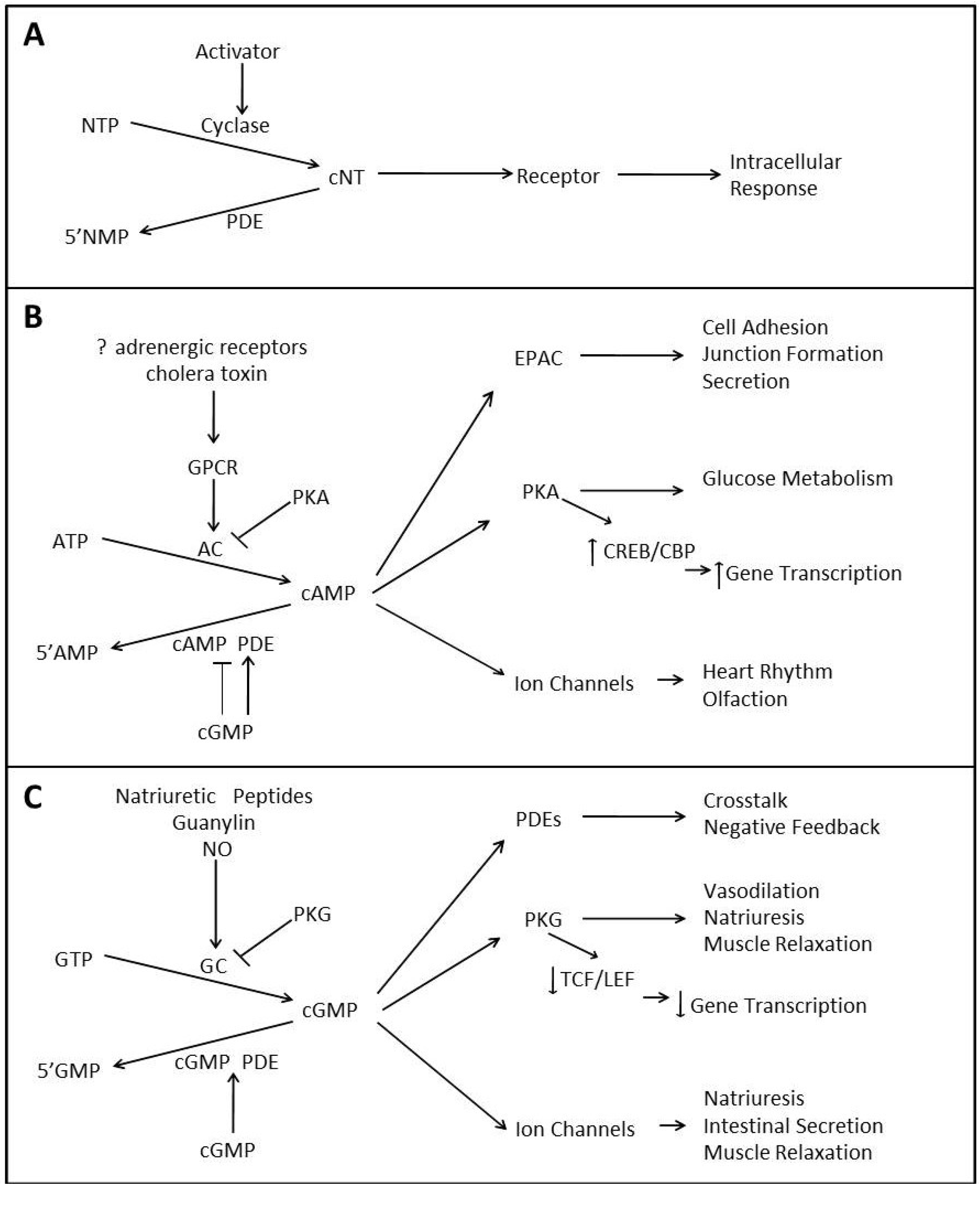
1.1. cAMP Signaling

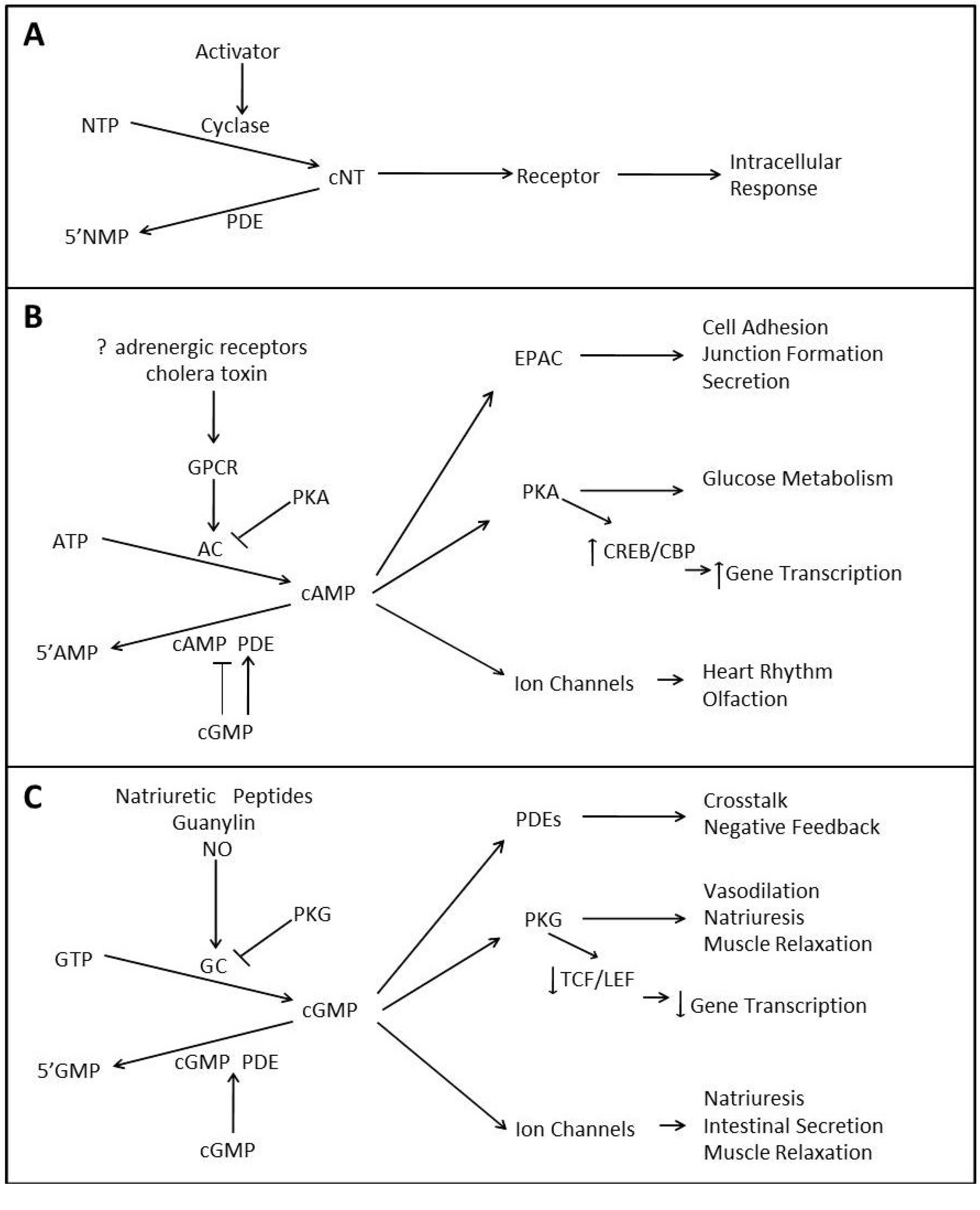{kind=link}
| Isozyme Family | Number of Genes | Putative Number of Isozymes * | Substrate Specificity | Regulators | Inhibitors |
|---|---|---|---|---|---|
| 1 | 3 | 21 | dual | Ca2+-CaM: ↑ | IC224, SH51866, |
| 2 | 1 | 3 | dual | cGMP: ↑ | EHNA, BAY 60-7550, PDP, IC933 |
| 3 | 2 | 4 | dual | cGMP:↓ | Milrinone, Tolafentrine, Cilostazol, Cilostamide, OPC-33540 |
| 4 | 4 | 31 | cAMP | PKA: ↓ | Rolipram, Cilomilast, Roflumilast, Ro20-1724, Denbufylline, AWD12281 |
| 5 | 1 | 3 | cGMP | cGMP: ↑ | Sildenafil, Zaprinast, |
| 6 | 3 | 3 | cGMP | Transducin: ↑ | Sildenafil, |
| 7 | 2 | 7 | cAMP | unknown | BRL 50481, IC242, Dipyridamole, Thiadiazoles |
| 8 | 2 | 9 | cAMP | unknown | Dipyridamole |
| 9 | 1 | 2 | cGMP | unknown | BAY73-669, SCH 51886, Zaprinast |
| 10 | 1 | 10 | dual | PKA: ↑ | Papaverine, PF-2545920, PQ-10, Dipyridamole |
| 11 | 1 | 4 | dual | unknown | BC 11-38, Dipyridamole |
1.2. cGMP Signaling
1.3. Crosstalk between Cyclic Nucleotide Signaling Pathways
2. Cyclic Nucleotide Signaling in Cancer
| Cancer Type | Observed Alteration | Reference |
|---|---|---|
| Bladder | ↑ PDE5 expression | [40,61] |
| ↑ MRP5 expression | ||
| Breast | ↑ PDE expression and activity | [39,52,62,63,64] |
| Altered PDE localization | ||
| ↑ MRP5 expression | ||
| Colon | ↑ GC-C expression; ↓ ligand expression | [36,51,65] |
| ↓ PKG expression | ||
| ↓ PKA expression | ||
| Hepatoma | ↑ basal levels of cAMP and cGMP | [53] |
| Leukemia | Altered PDE isozyme expression | [46,66] |
| ↑ PDE activity | ||
| Lung | ↑ PDE expression and activity | [61,67] |
| ↑ MRP5 expression | ||
| Lymphoma | ↑ PDE activity | [49,50] |
| ↓ basal levels of cAMP and cGMP | ||
| Ovarian | ↓ basal levels of cAMP | [54,61] |
| ↑ MRP5 expression | ||
| Pituitary | ↓ AC activity | [55] |
| ↑ PDE expression and activity | ||
| Prostate | ↑ MRP5 expression | [61] |
| Skin | ↑ PDE activity | [67] |
2.1. Cyclic Nucleotide Signaling in Hematological Malignancies
2.2. Cyclic Nucleotide Signaling in Epithelial Tumors
3. Targeting Cyclic Nucleotide Signaling for the Prevention and/or Treatment of Cancer
3.1. Targeting Cyclases
3.2. Targeting Phosphodiesterases
3.3. Targeting Kinases
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Gerits, N.; Kostenko, S.; Shiryaev, A.; Johannessen, M.; Moens, U. Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: Comradeship and hostility. Cell. Signal. 2008, 20, 1592–1607. [Google Scholar] [CrossRef]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Rehmann, H.; Wittinghofer, A.; Bos, J.L. Capturing cyclic nucleotides in action: Snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 2007, 8, 63–73. [Google Scholar] [CrossRef]
- Taussig, R.; Gilman, A.G. Mammalian membrane-bound adenylyl cyclases. J. Biol. Chem. 1995, 270, 1–4. [Google Scholar] [CrossRef]
- Hanoune, J.; Defer, N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef]
- Lee, L.C.; Maurice, D.H.; Baillie, G.S. Targeting protein-protein interactions within the cyclic AMP signaling system as a therapeutic strategy for cardiovascular disease. Future Med. Chem. 2013, 5, 451–464. [Google Scholar] [CrossRef]
- Buxton, I.L.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 1983, 258, 10233–10239. [Google Scholar]
- Sodani, K.; Patel, A.; Kathawala, R.J.; Chen, Z.-S. Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 2012, 31, 58–72. [Google Scholar] [CrossRef]
- Cheepala, S.; Hulot, J.S.; Morgan, J.A.; Sassi, Y.; Zhang, W.; Naren, A.P.; Schuetz, J.D. Cyclic nucleotide compartmentalization: Contributions of phosphodiesterases and ATP-binding cassette transporters. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 231–253. [Google Scholar] [CrossRef]
- Wielinga, P.R.; van der Heijden, I.; Reid, G.; Beijnen, J.H.; Wijnholds, J.; Borst, P. Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J. Biol. Chem. 2003, 278, 17664–17671. [Google Scholar]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Savai, R.; Pullamsetti, S.S.; Banat, G.A.; Weissmann, N.; Ghofrani, H.A.; Grimminger, F.; Schermuly, R.T. Targeting cancer with phosphodiesterase inhibitors. Expert Opin. Investig. Drugs 2010, 19, 117–131. [Google Scholar] [CrossRef]
- Verhoest, P.R.; Chapin, D.S.; Corman, M.; Fonseca, K.; Harms, J.F.; Hou, X.; Marr, E.S.; Menniti, F.S.; Nelson, F.; O’Connor, R.; et al. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]-quinoline (PF-2545920) for the treatment of schizophrenia. J. Med. Chem. 2009, 52, 5188–5196. [Google Scholar] [CrossRef]
- Dedeurwaerdere, S.; Wintmolders, C.; Vanhoof, G.; Langlois, X. Patterns of brain glucose metabolism induced by phosphodiesterase 10A inhibitors in the mouse: A potential translational biomarker. J. Pharmacol. Exp. Ther. 2011, 339, 210–217. [Google Scholar] [CrossRef]
- Ceyhan, O.; Birsoy, K.; Hoffman, C.S. Identification of biologically active PDE11-selective inhibitors using a yeast-based high-throughput screen. Chem. Biol. 2012, 19, 155–163. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Francis, S.H.; Corbin, J.D. Cyclic nucleotide-dependent protein kinases: Intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab. Sci. 1999, 36, 275–328. [Google Scholar] [CrossRef]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef]
- Beard, M.B.; Olsen, A.E.; Jones, R.E.; Erdogan, S.; Houslay, M.D.; Bolger, G.B. UCR1 and UCR2 domains unique to the cAMP-specific phosphodiesterase family form a discrete module via electrostatic interactions. J. Biol. Chem. 2000, 275, 10349–10358. [Google Scholar]
- Goncalves, R.L.; Lugnier, C.; Keravis, T.; Lopes, M.J.; Fantini, F.A.; Schmitt, M.; Cortes, S.F.; Lemos, V.S. The flavonoid dioclein is a selective inhibitor of cyclic nucleotide phosphodiesterase type 1 (PDE1) and a cGMP-dependent protein kinase (PKG) vasorelaxant in human vascular tissue. Eur. J. Pharmacol. 2009, 620, 78–83. [Google Scholar] [CrossRef]
- Insel, P.A.; Zhang, L.; Murray, F.; Yokouchi, H.; Zambon, A.C. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol. (Oxf.) 2012, 204, 277–287. [Google Scholar] [CrossRef]
- Caretta, A.; Mucignat-Caretta, C. Protein kinase A in cancer. Cancers 2011, 3, 913–926. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Frank, D.A. CREB in the pathophysiology of cancer: Implications for targeting transcription factors for cancer therapy. Clin. Cancer Res. 2009, 15, 2583–2587. [Google Scholar] [CrossRef]
- Feil, R.; Kemp-Harper, B. cGMP signalling: From bench to bedside. Conference on cGMP generators, effectors and therapeutic implications. EMBO Rep. 2006, 7, 149–153. [Google Scholar] [CrossRef]
- Beavo, J.A. Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol. Rev. 1995, 75, 725–748. [Google Scholar]
- Lincoln, T.M.; Cornwell, T.L. Intracellular cyclic GMP receptor proteins. FASEB J. 1993, 7, 328–338. [Google Scholar]
- Browning, D.D. Protein kinase G as a therapeutic target for the treatment of metastatic colorectal cancer. Expert Opin. Ther. Targets 2008, 12, 367–376. [Google Scholar] [CrossRef]
- Das, A.; Xi, L.; Kukreja, R.C. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J. Biol. Chem. 2008, 283, 29572–29585. [Google Scholar] [CrossRef]
- Ruth, P. Cyclic GMP-dependent protein kinases: Understanding in vivo functions by gene targeting. Pharmacol. Ther. 1999, 82, 355–372. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Kwon, I.K.; Wang, R.; Thangaraju, M.; Shuang, H.; Liu, K.; Dashwood, R.; Dulin, N.; Ganapathy, V.; Browning, D.D. PKG inhibits TCF signaling in colon cancer cells by blocking beta-catenin expression and activating FOXO4. Oncogene 2010, 29, 3423–3434. [Google Scholar] [CrossRef]
- Thompson, W.J.; Piazza, G.A.; Li, H.; Liu, L.; Fetter, J.; Zhu, B.; Sperl, G.; Ahnen, D.; Pamukcu, R. Exisulind induction of apoptosis involves guanosine 3',5'-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. 2000, 60, 3338–3342. [Google Scholar]
- Deguchi, A.; Thompson, W.J.; Weinstein, I.B. Activation of protein kinase G is sufficient to induce apoptosis and inhibit cell migration in colon cancer cells. Cancer Res. 2004, 64, 3966–3973. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Underwood, T.; Lloyd, M.; David, M.; Sperl, G.; Pamukcu, R.; Thompson, W.J. Cyclic GMP-dependent protein kinase activation and induction by exisulind and CP461 in colon tumor cells. J. Pharmacol. Exp. Ther. 2001, 299, 583–592. [Google Scholar]
- Deguchi, A.; Soh, J.W.; Li, H.; Pamukcu, R.; Thompson, W.J.; Weinstein, I.B. Vasodilator-stimulated phosphoprotein (VASP) phosphorylation provides a biomarker for the action of exisulind and related agents that activate protein kinase G. Mol. Cancer Ther. 2002, 1, 803–809. [Google Scholar]
- Drees, M.; Zimmermann, R.; Eisenbrand, G. 3',5'-Cyclic nucleotide phosphodiesterase in tumor cells as potential target for tumor growth inhibition. Cancer Res. 1993, 53, 3058–3061. [Google Scholar]
- Piazza, G.A.; Thompson, W.J.; Pamukcu, R.; Alila, H.W.; Whitehead, C.M.; Liu, L.; Fetter, J.R.; Gresh, W.E., Jr.; Klein-Szanto, A.J.; Farnell, D.R.; et al. Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer Res. 2001, 61, 3961–3968. [Google Scholar]
- Whitehead, C.M.; Earle, K.A.; Fetter, J.; Xu, S.; Hartman, T.; Chan, D.C.; Zhao, T.L.; Piazza, G.; Klein-Szanto, A.J.; Pamukcu, R.; et al. Exisulind-induced apoptosis in a non-small cell lung cancer orthotopic lung tumor model augments docetaxel treatment and contributes to increased survival. Mol. Cancer Ther. 2003, 2, 479–488. [Google Scholar]
- Zhu, B.; Strada, S.J. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr. Top. Med. Chem. 2007, 7, 437–454. [Google Scholar] [CrossRef]
- Zhu, B.; Strada, S.; Stevens, T. Cyclic GMP-specific phosphodiesterase 5 regulates growth and apoptosis in pulmonary endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L196–L206. [Google Scholar] [CrossRef]
- Jiang, X.; Li, J.; Paskind, M.; Epstein, P.M. Inhibition of calmodulin-dependent phosphodiesterase induces apoptosis in human leukemic cells. Proc. Natl. Acad. Sci. USA 1996, 93, 11236–11241. [Google Scholar] [CrossRef]
- Kloster, M.M.; Hafte, T.T.; Moltzau, L.R.; Naderi, E.H.; Dahle, M.K.; Skålhegg, B.S.; Gaudernack, G.; Levy, F.O.; Naderi, S.; Blomhoff, H.K. EBV infection renders B cells resistant to growth inhibition via adenylyl cyclase. Cell. Signal. 2008, 20, 1169–1178. [Google Scholar] [CrossRef]
- Lerner, A.; Epstein, P.M. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem. J. 2006, 393, 21–41. [Google Scholar] [CrossRef]
- Chawla, R.K.; Shlaer, S.M.; Lawson, D.H.; Murray, T.G.; Schmidt, F.; Shoji, M.; Nixon, D.W.; Richmond, A.; Rudman, D. Elevated plasma and urinary guanosine 3':5'-monophosphate and increased production rate in patients with neoplastic diseases. Cancer Res. 1980, 40, 3915–3920. [Google Scholar]
- Chawla, R.K.; Nixon, D.W.; Shoji, M.; Rudman, D. Plasma and urine cyclic guanosine 3':5'-monophosphate in disseminated cancer. Ann. Intern. Med. 1979, 91, 862–864. [Google Scholar] [CrossRef]
- Aleksijevic, A.; Lang, J.M.; Giron, C.; Stoclet, J.C.; Mayer, S.; Oberling, F. Alterations of peripheral blood lymphocyte cyclic AMP and cyclic GMP in untreated patients with hodgkin’s disease. Clin. Immunol. Immunopathol. 1983, 26, 398–405. [Google Scholar] [CrossRef]
- Aleksijevic, A.; Lugnier, C.; Giron, C.; Mayer, S.; Stoclet, J.C.; Lang, J.M. Cyclic AMP and cyclic GMP phosphodiesterase activities in Hodgkin’s disease lymphocytes. Int. J. Immunopharmacol. 1987, 9, 525–531. [Google Scholar] [CrossRef]
- Carlson, C.C.; Smithers, S.L.; Yeh, K.A.; Burnham, L.L.; Dransfield, D.T. Protein kinase A regulatory subunits in colon cancer. Neoplasia 1999, 1, 373–378. [Google Scholar]
- Ciardiello, F.; Pepe, S.; Bianco, C.; Baldassarre, G.; Ruggiero, A.; Bianco, C.; Selvam, M.P.; Bianco, A.R.; Tortora, G. Down-regulation of RI alpha subunit of cAMP-dependent protein kinase induces growth inhibition of human mammary epithelial cells transformed by c-Ha-ras and c-erbB-2 proto-oncogenes. Int. J. Cancer 1993, 53, 438–443. [Google Scholar] [CrossRef]
- DeRubertis, F.R.; Craven, P.A. Sequential alterations in the hepatic content and metabolism of cyclic AMP and cyclic GMP induced by DL-ethionine: Evidence for malignant transformation of liver with a sustained increase in cyclic AMP. Metabolism 1976, 25, 1611–1625. [Google Scholar] [CrossRef]
- Heinonen, P.K.; Metsa-Ketela, T. Prostanoids and cyclic nucleotides in malignant and benign ovarian tumors. Med. Oncol. Tumor Pharmacother. 1988, 5, 11–15. [Google Scholar]
- Pertuit, M.; Barlier, A.; Enjalbert, A.; Gérard, C. Signalling pathway alterations in pituitary adenomas: Involvement of Gsalpha, cAMP and mitogen-activated protein kinases. J. Neuroendocrinol. 2009, 21, 869–877. [Google Scholar] [CrossRef]
- Ahn, Y.H.; Jung, J.M.; Hong, S.H. 8-Chloro-cyclic AMP-induced growth inhibition and apoptosis is mediated by p38 mitogen-activated protein kinase activation in HL60 cells. Cancer Res. 2005, 65, 4896–4901. [Google Scholar] [CrossRef]
- Michalides, R.; Griekspoor, A.; Balkenende, A.; Verwoerd, D.; Janssen, L.; Jalink, K.; Floore, A.; Velds, A.; van’t Veer, L.; Neefjes, J. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cell 2004, 5, 597–605. [Google Scholar] [CrossRef]
- Schuller, H.M. Effects of tobacco constituents and psychological stress on the beta-adrenergic regulation of non-small cell lung cancer and pancreatic cancer: Implications for intervention. Cancer Biomark. 2013, 13, 133–144. [Google Scholar]
- Wharton, J.; Strange, J.W.; Møller, G.M.; Growcott, E.J.; Ren, X.; Franklyn, A.P.; Phillips, S.C.; Wilkins, M.R. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am. J. Respir. Crit. Care Med. 2005, 172, 105–113. [Google Scholar] [CrossRef]
- Zhu, B.; Vemavarapu, L.; Thompson, W.J.; Strada, S.J. Suppression of cyclic GMP-specific phosphodiesterase 5 promotes apoptosis and inhibits growth in HT29 cells. J. Cell. Biochem. 2005, 94, 336–350. [Google Scholar] [CrossRef]
- Kool, M.; de Haas, M.; Scheffer, G.L.; Scheper, R.J.; van Eijk, M.J.; Juijn, J.A.; Baas, F.; Borst, P. Analysis of expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the multidrug resistance-associated protein gene (MRP1), in human cancer cell lines. Cancer Res. 1997, 57, 3537–3547. [Google Scholar]
- Singer, A.L.; Sherwin, R.P.; Dunn, A.S.; Appleman, M.M. Cyclic nucleotide phosphodiesterases in neoplastic and nonneoplastic human mammary tissues. Cancer Res. 1976, 36, 60–66. [Google Scholar]
- Cohen, L.A.; Straka, D.; Chan, P.C. Cyclic nucleotide phosphodiesterase activity in normal and neoplastic rat mammary cells grown in monolayer culture. Cancer Res. 1976, 36, 2007–2012. [Google Scholar]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Zhang, W.; Abadi, A.H.; Reynolds, R.C.; Piazza, G.A. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol. Cancer Ther. 2009, 8, 3331–3340. [Google Scholar] [CrossRef]
- Li, P.; Schulz, S.; Bombonati, A.; Palazzo, J.P.; Hyslop, T.M.; Xu, Y.; Baran, A.A.; Siracusa, L.D.; Pitari, G.M.; Waldman, S.A. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology 2007, 133, 599–607. [Google Scholar] [CrossRef]
- Zhang, L.; Murray, F.; Zahno, A.; Kanter, J.R.; Chou, D.; Suda, R.; Fenlon, M.; Rassenti, L.; Cottam, H.; Kipps, T.J.; et al. Cyclic nucleotide phosphodiesterase profiling reveals increased expression of phosphodiesterase 7B in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 19532–19537. [Google Scholar] [CrossRef]
- Marko, D.; Pahlke, G.; Merz, K.H.; Eisenbrand, G. Cyclic 3',5'-nucleotide phosphodiesterases: Potential targets for anticancer therapy. Chem. Res. Toxicol. 2000, 13, 944–948. [Google Scholar] [CrossRef]
- Kobsar, A.; Heeg, S.; Krohne, K.; Opitz, A.; Walter, U.; Böck, M.; Gambaryan, S.; Eigenthaler, M. Cyclic nucleotide-regulated proliferation and differentiation vary in human hematopoietic progenitor cells derived from healthy persons, tumor patients, and chronic myelocytic leukemia patients. Stem Cells Dev. 2008, 17, 81–91. [Google Scholar] [CrossRef]
- Kiefer, J.; Okret, S.; Jondal, M.; McConkey, D.J. Functional glucocorticoid receptor expression is required for cAMP-mediated apoptosis in a human leukemic T cell line. J. Immunol. 1995, 155, 4525–4528. [Google Scholar]
- Tortorella, C.; Piazzolla, G.; Spaccavento, F.; Antonaci, S. Effects of granulocyte-macrophage colony-stimulating factor and cyclic AMP interaction on human neutrophil apoptosis. Mediat. Inflamm. 1998, 7, 391–396. [Google Scholar] [CrossRef]
- Martin, M.C.; Dransfield, I.; Haslett, C.; Rossi, A.G. Cyclic AMP regulation of neutrophil apoptosis occurs via a novel protein kinase A-independent signaling pathway. J. Biol. Chem. 2001, 276, 45041–45050. [Google Scholar]
- Garcia-Bermejo, L.; Pérez, C.; Vilaboa, N.E.; de Blas, E.; Aller, P. cAMP increasing agents attenuate the generation of apoptosis by etoposide in promonocytic leukemia cells. J. Cell Sci. 1998, 111, 637–644. [Google Scholar]
- Monahan, T.M.; Marchand, N.W.; Fritz, R.R.; Abell, C.W. Cyclic adenosine 3':5'-monophosphate levels and activities of related enzymes in normal and leukemic lymphocytes. Cancer Res. 1975, 35, 2540–2547. [Google Scholar]
- Carpentieri, U.; Monahan, T.M.; Gustavson, L.P. Observations on the level of cyclic nucleotides in three population of human lymphocytes in culture. J. Cycl. Nucleotide Res. 1980, 6, 253–259. [Google Scholar]
- Lee, R.; Wolda, S.; Moon, E.; Esselstyn, J.; Hertel, C.; Lerner, A. PDE7A is expressed in human B-lymphocytes and is up-regulated by elevation of intracellular cAMP. Cell. Signal. 2002, 14, 277–284. [Google Scholar] [CrossRef]
- Flamigni, F.; Facchini, A.; Stanic, I.; Tantini, B.; Bonavita, F.; Stefanelli, C. Control of survival of proliferating L1210 cells by soluble guanylate cyclase and p44/42 mitogen-activated protein kinase modulators. Biochem. Pharmacol. 2001, 62, 319–328. [Google Scholar] [CrossRef]
- Cho, E.C.; Mitton, B.; Sakamoto, K.M. CREB and leukemogenesis. Crit. Rev. Oncog. 2011, 16, 37–46. [Google Scholar] [CrossRef]
- Camici, M. Guanylin peptides and colorectal cancer (CRC). Biomed. Pharmacother. 2008, 62, 70–76. [Google Scholar] [CrossRef]
- Birbe, R.; Palazzo, J.P.; Walters, R.; Weinberg, D.; Schulz, S.; Waldman, S.A. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum. Pathol. 2005, 36, 170–179. [Google Scholar] [CrossRef]
- Steinbrecher, K.A.; Wowk, S.A.; Rudolph, J.A.; Witte, D.P.; Cohen, M.B. Targeted inactivation of the mouse guanylin gene results in altered dynamics of colonic epithelial proliferation. Am. J. Pathol. 2002, 161, 2169–2178. [Google Scholar] [CrossRef]
- Waldman, S.A.; Barber, M.; Pearlman, J.; Park, J.; George, R.; Parkinson, S.J. Heterogeneity of guanylyl cyclase C expressed by human colorectal cancer cell lines in vitro. Cancer Epidemiol. Biomark. Prev. 1998, 7, 505–514. [Google Scholar]
- Pitari, G.M.; Zingman, L.V.; Waldman, S.A. Bacterial enterotoxins are associated with resistance to colon cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 2695–2699. [Google Scholar] [CrossRef]
- Johansson, C.C.; Yndestad, A.; Enserink, J.M.; Ree, A.H.; Aukrust, P.; Taskén, K. The epidermal growth factor-like growth factor amphiregulin is strongly induced by the adenosine 3',5'-monophosphate pathway in various cell types. Endocrinology 2004, 145, 5177–5184. [Google Scholar] [CrossRef]
- Kung, W.; Bechtel, E.; Geyer, E.; Salokangas, A.; Preisz, J.; Huber, P.; Torhorst, J.; Jungmann, R.A.; Talmadge, K.; Eppenberger, U. Altered levels of cyclic nucleotides, cyclic AMP phosphodiesterase and adenylyl cyclase activities in normal, dysplastic and neoplastic human mammary tissue. FEBS Lett. 1977, 82, 102–106. [Google Scholar] [CrossRef]
- Whitfield, J.F.; Boynton, A.L.; MacManus, J.P.; Rixon, R.H.; Sikorska, M.; Tsang, B.; Walker, P.R.; Swierenga, S.H. The roles of calcium and cyclic AMP in cell proliferation. Ann. NY Acad. Sci. 1980, 339, 216–240. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Burchell, B.; Keppler, D. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J. Biol. Chem. 2000, 275, 30069–30074. [Google Scholar] [CrossRef]
- Borland, G.; Smith, B.O.; Yarwood, S.J. EPAC proteins transduce diverse cellular actions of cAMP. Br. J. Pharmacol. 2009, 158, 70–86. [Google Scholar] [CrossRef]
- Li, P.; Lin, J.E.; Chervoneva, I.; Schulz, S.; Waldman, S.A.; Pitari, G.M. Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase C restricts the proliferating compartment in intestine. Am. J. Pathol. 2007, 171, 1847–1858. [Google Scholar] [CrossRef]
- Pitari, G.M.; di Guglielmo, M.D.; Park, J.; Schulz, S.; Waldman, S.A. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7846–7851. [Google Scholar] [CrossRef]
- Mann, E.A.; Steinbrecher, K.A.; Stroup, C.; Witte, D.P.; Cohen, M.B.; Giannella, R.A. Lack of guanylyl cyclase C, the receptor for Escherichia coli heat-stable enterotoxin, results in reduced polyp formation and increased apoptosis in the multiple intestinal neoplasia (Min) mouse model. Int. J. Cancer 2005, 116, 500–505. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Clementi, E.; Sciorati, C.; Nistico, G. Growth factor-induced Ca2+ responses are differentially modulated by nitric oxide via activation of a cyclic GMP-dependent pathway. Mol. Pharmacol. 1995, 48, 1068–1077. [Google Scholar]
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 2000, 52, 375–414. [Google Scholar]
- Chen, H.; Levine, Y.C.; Golan, D.E.; Michel, T.; Lin, A.J. Atrial natriuretic peptide-initiated cGMP pathways regulate vasodilator-stimulated phosphoprotein phosphorylation and angiogenesis in vascular endothelium. J. Biol. Chem. 2008, 283, 4439–4447. [Google Scholar] [CrossRef]
- Idriss, S.D.; Gudi, T.; Casteel, D.E.; Kharitonov, V.G.; Pilz, R.B.; Boss, G.R. Nitric oxide regulation of gene transcription via soluble guanylate cyclase and type I cGMP-dependent protein kinase. J. Biol. Chem. 1999, 274, 9489–9493. [Google Scholar]
- Pilz, R.B.; Casteel, D.E. Regulation of gene expression by cyclic GMP. Circ. Res. 2003, 93, 1034–1046. [Google Scholar] [CrossRef]
- Wong, J.C.; Bathina, M.; Fiscus, R.R. Cyclic GMP/protein kinase G type-Ialpha (PKG-Ialpha) signaling pathway promotes CREB phosphorylation and maintains higher c-IAP1, livin, survivin, and Mcl-1 expression and the inhibition of PKG-Ialpha kinase activity synergizes with cisplatin in non-small cell lung cancer cells. J. Cell. Biochem. 2012, 113, 3587–3598. [Google Scholar] [CrossRef]
- Zhou, L.; Hosohata, K.; Gao, S.; Gu, Z.; Wang, Z. cGMP-dependent protein kinase Ibeta interacts with p44/WDR77 to regulate androgen receptor-driven gene expression. PLoS One 2013, 8, e63119. [Google Scholar]
- Lan, T.; Chen, Y.; Sang, J.; Wu, Y.; Wang, Y.; Jiang, L.; Tao, Y. Type II cGMP-dependent protein kinase inhibits EGF-induced MAPK/JNK signal transduction in breast cancer cells. Oncol. Rep. 2012, 27, 2039–2044. [Google Scholar]
- Lubbe, W.J.; Zhou, Z.Y.; Fu, W.; Zuzga, D.; Schulz, S.; Fridman, R.; Muschel, R.J.; Waldman, S.A.; Pitari, G.M. Tumor epithelial cell matrix metalloproteinase 9 is a target for antimetastatic therapy in colorectal cancer. Clin. Cancer Res. 2006, 12, 1876–1882. [Google Scholar] [CrossRef]
- Seamon, K.B.; Padgett, W.; Daly, J.W. Forskolin: Unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. USA 1981, 78, 3363–3367. [Google Scholar] [CrossRef]
- Agarwal, K.C.; Parks, R.E., Jr. Forskolin: A potential antimetastatic agent. Int. J. Cancer 1983, 32, 801–804. [Google Scholar] [CrossRef]
- Chen, T.C.; Hinton, D.R.; Zidovetzki, R.; Hofman, F.M. Up-regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Lab. Investig. 1998, 78, 165–174. [Google Scholar]
- Li, Z.; Wang, J. A forskolin derivative, FSK88, induces apoptosis in human gastric cancer BGC823 cells through caspase activation involving regulation of Bcl-2 family gene expression, dissipation of mitochondrial membrane potential and cytochrome c release. Cell Biol. Int. 2006, 30, 940–946. [Google Scholar] [CrossRef]
- Shayo, C.; Legnazzi, B.L.; Monczor, F.; Fernández, N.; Riveiro, M.E.; Baldi, A.; Davio, C. The time-course of cyclic AMP signaling is critical for leukemia U-937 cell differentiation. Biochem. Biophys. Res. Commun. 2004, 314, 798–804. [Google Scholar] [CrossRef]
- Baumann, G.; Felix, S.; Sattelberger, U.; Klein, G. Cardiovascular effects of forskolin (HL 362) in patients with idiopathic congestive cardiomyopathy—A comparative study with dobutamine and sodium nitroprusside. J. Cardiovasc. Pharmacol. 1990, 16, 93–100. [Google Scholar] [CrossRef]
- Christenson, J.T.; Thulesius, O.; Nazzal, M.M. The effect of forskolin on blood flow, platelet metabolism, aggregation and ATP release. Vasa 1995, 24, 56–61. [Google Scholar]
- Ding, X.; Staudinger, J.L. Induction of drug metabolism by forskolin: The role of the pregnane X receptor and the protein kinase a signal transduction pathway. J. Pharmacol. Exp. Ther. 2005, 312, 849–856. [Google Scholar] [CrossRef]
- Barron, T.I.; Sharp, L.; Visvanathan, K. Beta-adrenergic blocking drugs in breast cancer: A perspective review. Ther. Adv. Med. Oncol. 2012, 4, 113–125. [Google Scholar] [CrossRef]
- Ganz, P.A.; Cole, S.W. Expanding our therapeutic options: Beta blockers for breast cancer? J. Clin. Oncol. 2011, 29, 2612–2616. [Google Scholar] [CrossRef]
- Fitzgerald, P.J. Beta blockers, norepinephrine, and cancer: An epidemiological viewpoint. Clin. Epidemiol. 2012, 4, 151–156. [Google Scholar] [CrossRef]
- Wang, H.M.; Liao, Z.X.; Komaki, R.; Welsh, J.W.; O’Reilly, M.S.; Chang, J.Y.; Zhuang, Y.; Levy, L.B.; Lu, C.; Gomez, D.R. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann. Oncol. 2013, 24, 1312–1319. [Google Scholar] [CrossRef]
- Garg, U.C.; Hassid, A. Inhibition of rat mesangial cell mitogenesis by nitric oxide-generating vasodilators. Am. J. Physiol. 1989, 257, F60–F66. [Google Scholar]
- Garg, U.C.; Hassid, A. Nitric oxide-generating vasodilators inhibit mitogenesis and proliferation of BALB/C 3T3 fibroblasts by a cyclic GMP-independent mechanism. Biochem. Biophys. Res. Commun. 1990, 171, 474–479. [Google Scholar] [CrossRef]
- Gu, M.; Lynch, J.; Brecher, P. Nitric oxide increases p21(Waf1/Cip1) expression by a cGMP-dependent pathway that includes activation of extracellular signal-regulated kinase and p70(S6k). J. Biol. Chem. 2000, 275, 11389–11396. [Google Scholar] [CrossRef]
- Hagos, G.K.; Abdul-Hay, S.O.; Sohn, J.; Edirisinghe, P.D.; Chandrasena, R.E.; Wang, Z.; Li, Q.; Thatcher, G.R. Anti-inflammatory, antiproliferative, and cytoprotective activity of NO chimera nitrates of use in cancer chemoprevention. Mol. Pharmacol. 2008, 74, 1381–1391. [Google Scholar] [CrossRef]
- Rigas, B.; Williams, J.L. NO-donating NSAIDs and cancer: An overview with a note on whether NO is required for their action. Nitric Oxide 2008, 19, 199–204. [Google Scholar] [CrossRef]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar]
- Shami, P.J.; Saavedra, J.E.; Bonifant, C.L.; Chu, J.; Udupi, V.; Malaviya, S.; Carr, B.I.; Kar, S.; Wang, M.; Jia, L.; et al. Antitumor activity of JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] and related O2-aryl diazeniumdiolates in vitro and in vivo. J. Med. Chem. 2006, 49, 4356–4366. [Google Scholar] [CrossRef]
- Bischoff, E. Potency, selectivity, and consequences of nonselectivity of PDE inhibition. Int. J. Impot. Res. 2004, 16, S11–S14. [Google Scholar] [CrossRef]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Lu, W.Y.; Li, Y.H.; Piazza, G.A. Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/beta-catenin-mediated transcription in human breast tumor cells. Cancer Prev. Res. (Phila.) 2011, 4, 1275–1284. [Google Scholar] [CrossRef]
- Tinsley, H.N.; Gary, B.D.; Thaiparambil, J.; Li, N.; Lu, W.; Li, Y.; Maxuitenko, Y.Y.; Keeton, A.B.; Piazza, G.A. Colon tumor cell growth-inhibitory activity of sulindac sulfide and other nonsteroidal anti-inflammatory drugs is associated with phosphodiesterase 5 inhibition. Cancer Prev. Res. (Phila.) 2010, 3, 1303–1313. [Google Scholar] [CrossRef]
- Hirsh, L.; Dantes, A.; Suh, B.S.; Yoshida, Y.; Hosokawa, K.; Tajima, K.; Kotsuji, F.; Merimsky, O.; Amsterdam, A. Phosphodiesterase inhibitors as anti-cancer drugs. Biochem. Pharmacol. 2004, 68, 981–988. [Google Scholar] [CrossRef]
- Erikstein, B.S.; McCormack, E.; Tronstad, K.J.; Schwede, F.; Berge, R.; Gjertsen, B.T. Protein kinase A activators and the pan-PPAR agonist tetradecylthioacetic acid elicit synergistic anti-leukaemic effects in AML through CREB. Leuk. Res. 2010, 34, 77–84. [Google Scholar] [CrossRef]
- Mantovani, G.; Bondioni, S.; Lania, A.G.; Rodolfo, M.; Peverelli, E.; Polentarutti, N.; Veliz Rodriguez, T.; Ferrero, S.; Bosari, S.; Beck-Peccoz, P.; et al. High expression of PKA regulatory subunit 1A protein is related to proliferation of human melanoma cells. Oncogene 2008, 27, 1834–1843. [Google Scholar] [CrossRef]
- Bouizar, Z.; Ragazzon, B.; Viou, L.; Hortane, M.; Bertherat, J.; Rizk-Rabin, M. 8Cl-cAMP modifies the balance between PKAR1 and PKAR2 and modulates the cell cycle, growth and apoptosis in human adrenocortical H295R cells. J. Mol. Endocrinol. 2010, 44, 331–347. [Google Scholar] [CrossRef]
- Cen, B.; Deguchi, A.; Weinstein, I.B. Activation of protein kinase G Increases the expression of p21CIP1, p27KIP1, and histidine triad protein 1 through Sp1. Cancer Res. 2008, 68, 5355–5362. [Google Scholar]
- Kwon, I.K.; Schoenlein, P.V.; Delk, J.; Liu, K.; Thangaraju, M.; Dulin, N.O.; Ganapathy, V.; Berger, F.G.; Browning, D.D. Expression of cyclic guanosine monophosphate-dependent protein kinase in metastatic colon carcinoma cells blocks tumor angiogenesis. Cancer 2008, 112, 1462–1470. [Google Scholar] [CrossRef]
- Hou, Y.; Wong, E.; Martin, J.; Schoenlein, P.V.; Dostmann, W.R.; Browning, D.D. A role for cyclic-GMP dependent protein kinase in anoikis. Cell. Signal. 2006, 18, 882–888. [Google Scholar] [CrossRef]
- Soh, J.W.; Kazi, J.U.; Li, H.; Thompson, W.J.; Weinstein, I.B. Celecoxib-induced growth inhibition in SW480 colon cancer cells is associated with activation of protein kinase G. Mol. Carcinog. 2008, 47, 519–525. [Google Scholar] [CrossRef]
- Fallahian, F.; Karami-Tehrani, F.; Salami, S.; Aghaei, M. Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J. 2011, 278, 3360–3369. [Google Scholar]
- Ausserlechner, M.J.; Hagenbuchner, J.; Fuchs, S.; Geiger, K.; Obexer, P. FOXO Transcription Factors as Potential Therapeutic Targets in Neuroblastoma, in Neuroblastoma—Present and Future; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Edwards, B.K.; Ward, E.; Kohler, B.A.; Eheman, C.; Zauber, A.G.; Anderson, R.N.; Jemal, A.; Schymura, M.J.; Lansdorp-Vogelaar, I.; Seeff, L.C.; et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010, 116, 544–573. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment. Cancers 2014, 6, 436-458. https://doi.org/10.3390/cancers6010436
Fajardo AM, Piazza GA, Tinsley HN. The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment. Cancers. 2014; 6(1):436-458. https://doi.org/10.3390/cancers6010436
Chicago/Turabian StyleFajardo, Alexandra M., Gary A. Piazza, and Heather N. Tinsley. 2014. "The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment" Cancers 6, no. 1: 436-458. https://doi.org/10.3390/cancers6010436
APA StyleFajardo, A. M., Piazza, G. A., & Tinsley, H. N. (2014). The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment. Cancers, 6(1), 436-458. https://doi.org/10.3390/cancers6010436