The Interactions of microRNA and Epigenetic Modifications in Prostate Cancer

Abstract
:1. Epigenetic Contributions to Cancer
1.1. An Epigenetic Basis for Prostate Cancer
1.2. An Emergent Understanding on the Role of miRNA in Prostate Cancer
2. Interplay between Histone Modifications, DNA Methylation and miRNA
2.1. Regulation of DNA and Histone Modifications under the Control of miRNAs in Prostate Cancer
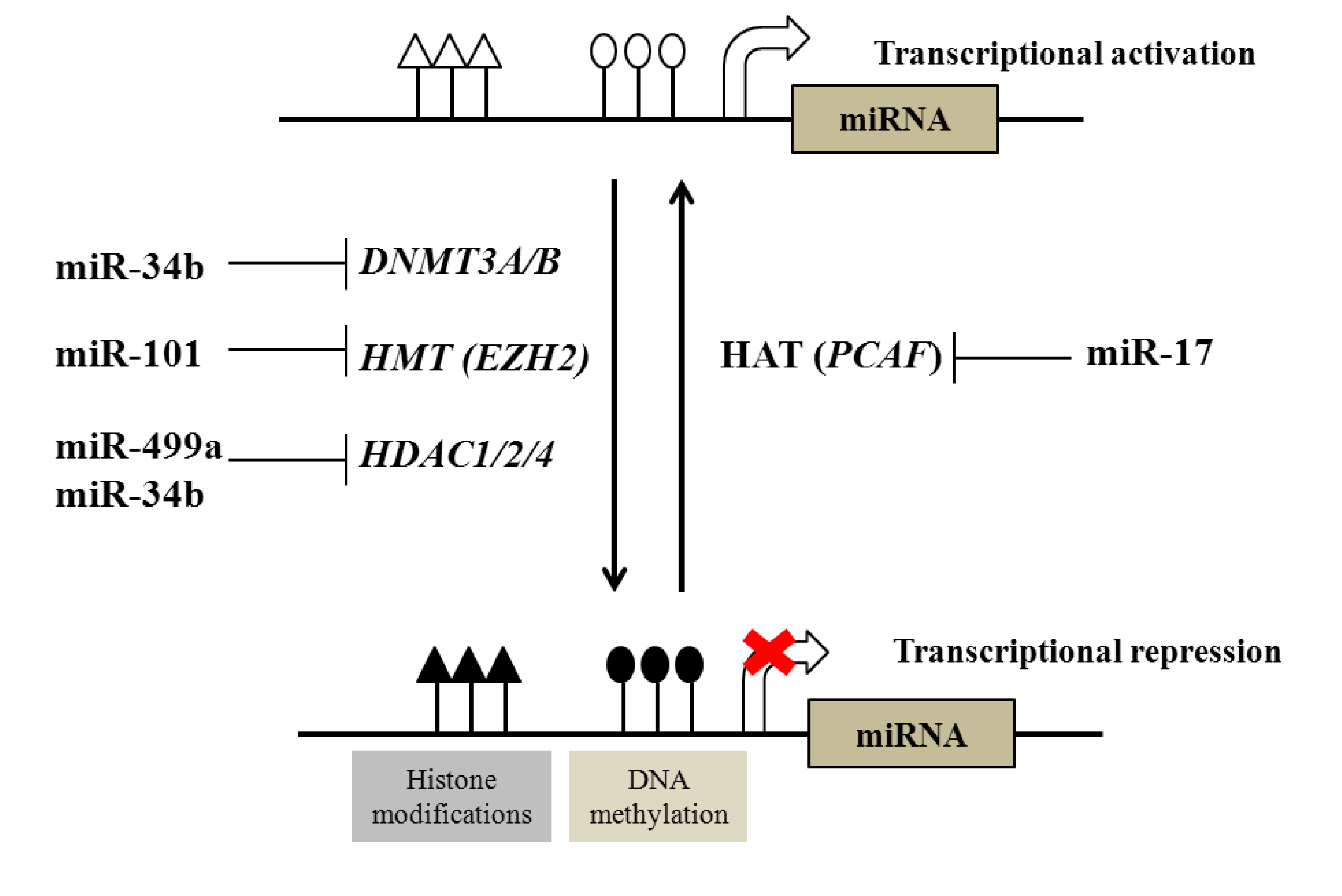

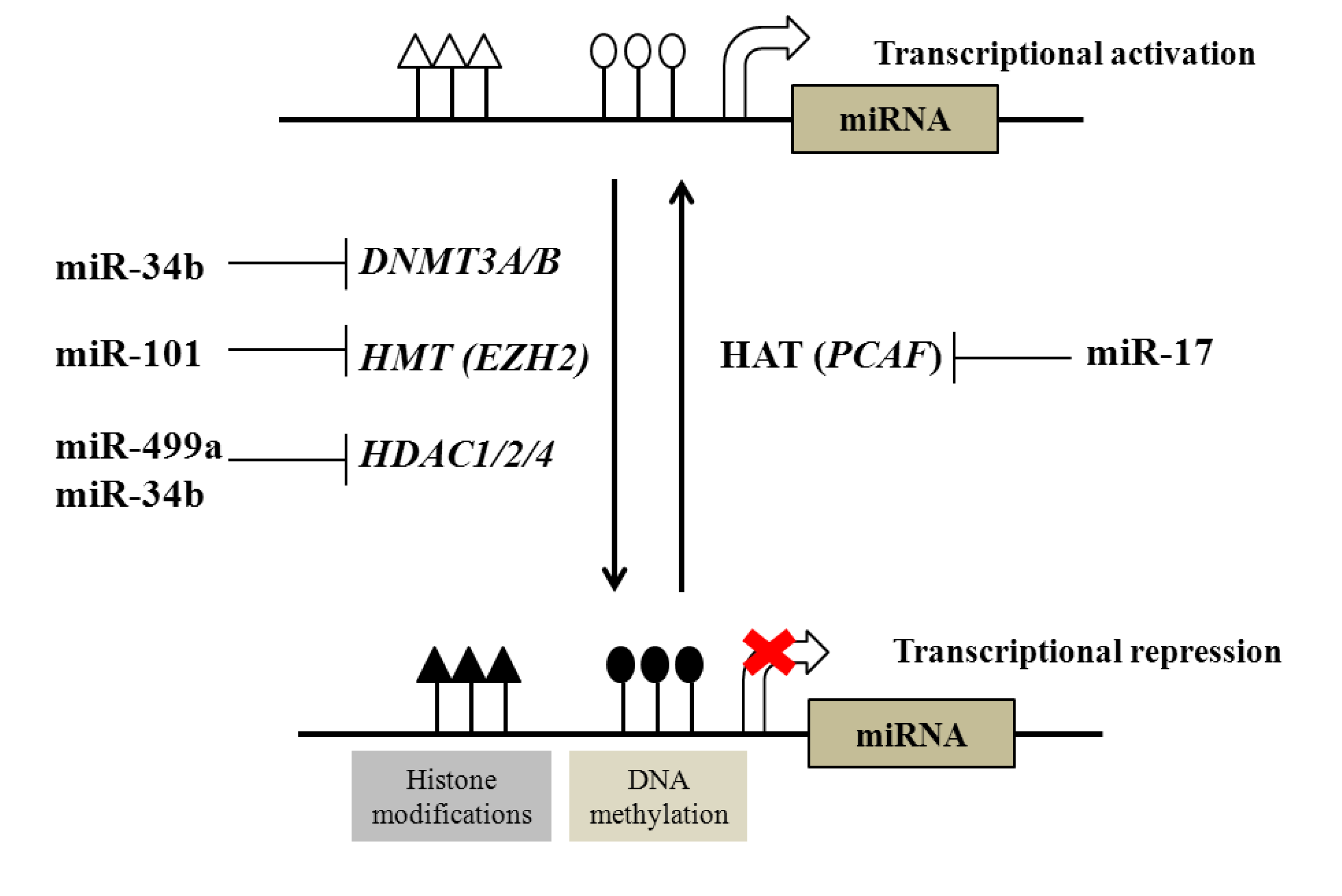{kind=link}
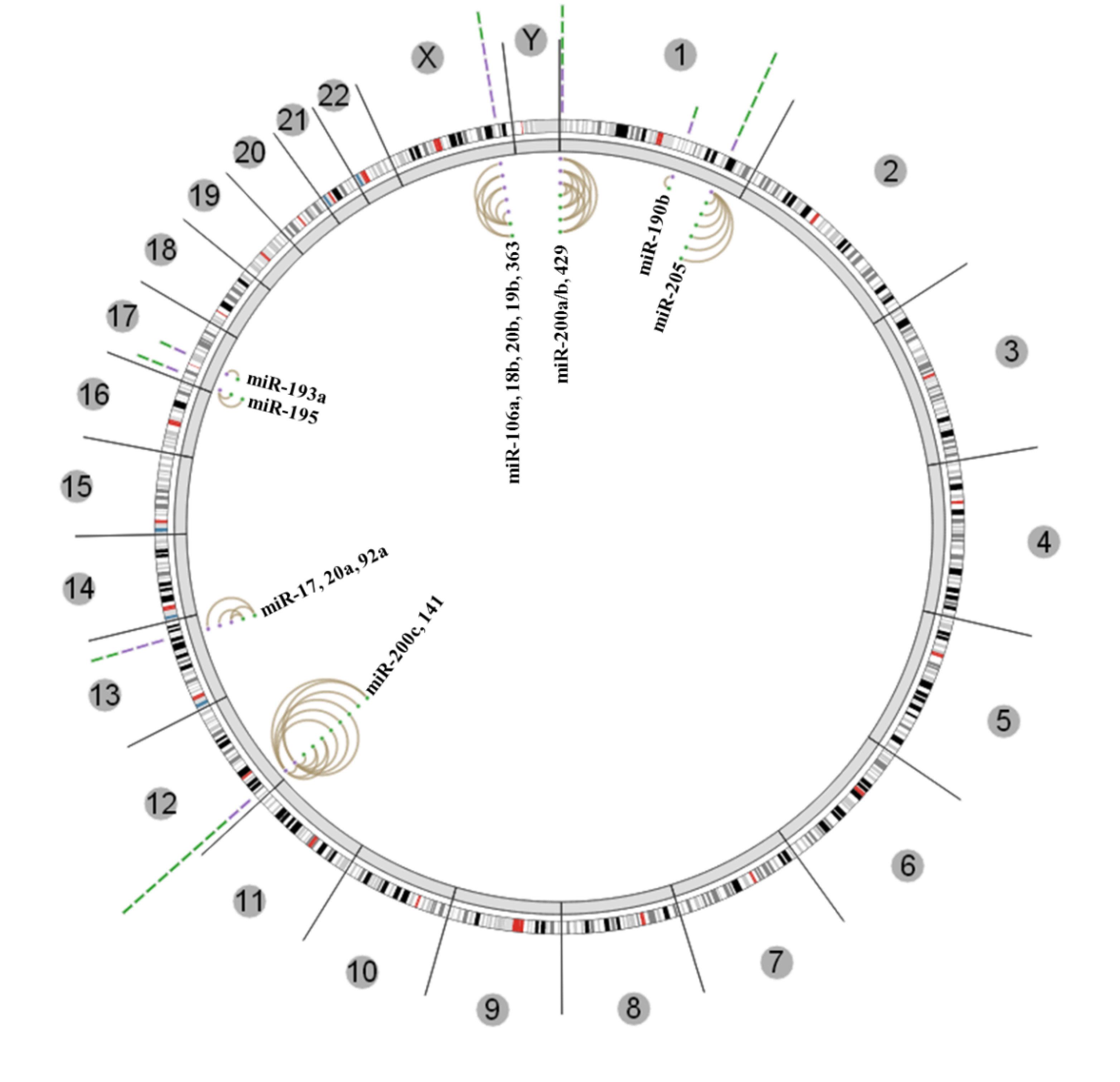
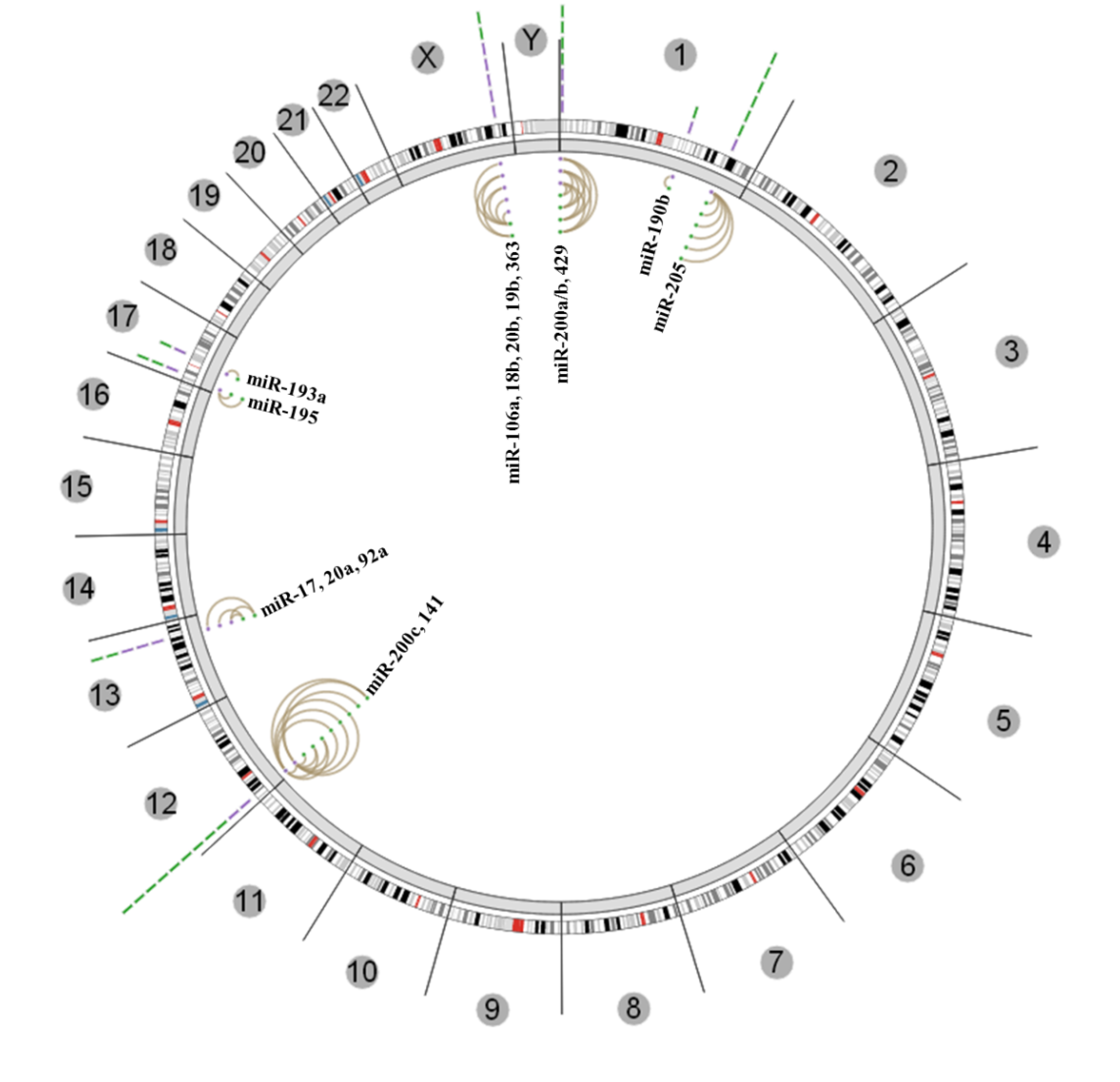{kind=link}
{kind=link}
| Examples of miRNAs regulated by DNA methylation | |||
|---|---|---|---|
| DNA methylation | miR-145, miR-193b, miR-34a, miR-34b, miR-200c, miR-141, miR-205, miR-196b, miR-21, miR-615 | ||
| Examples of epigenetic modifiers regulated by miRNAs | |||
| Epigenetic modifiers | Targeting miRNA | ||
| DNA methyltransferases (DNTMTs) | DNMT3A and DNMT3B | miR-29 family | |
| DNMT1 | miR-34b | ||
| Histone methyltransferases (HMTs) | EZH2 | miR-101 | |
| Histone deacetylases (HDACs) | HDAC1, HDAC2 and HDAC4 | miR-34b, miR-499a | |
| Histone acetyltransferases (HATs) | PCAF | miR-17-5p | |
2.2. Regulation of microRNAs by DNA and Histone Modification in Prostate Cancer
3. Clinical Exploitation of Epigenetic States in Prostate Cancer
3.1. Epigenetic Modifications as Biomarkers in Prostate Cancer
3.2. Diagnostic and Prognostic miRNA Expression Patterns
3.3. Insights from the Cancer Genome Atlas (TCGA) and ENCODE

4. Conclusions
Conflict of Interest
References
- Schroder, F.H.; Hugosson, J.; Roobol, M.J.; Tammela, T.L.; Ciatto, S.; Nelen, V.; Kwiatkowski, M.; Lujan, M.; Lilja, H.; Zappa, M.; et al. Screening and prostate-cancer mortality in a randomized european study. N. Engl. J. Med. 2009, 360, 1320–1328. [Google Scholar] [CrossRef]
- Andriole, G.L.; Crawford, E.D.; Grubb, R.L., 3rd; Buys, S.S.; Chia, D.; Church, T.R.; Fouad, M.N.; Gelmann, E.P.; Kvale, P.A.; Reding, D.J.; et al. Mortality results from a randomized prostate-cancer screening trial. N. Engl. J. Med. 2009, 360, 1310–1319. [Google Scholar] [CrossRef]
- He, W.W.; Sciavolino, P.J.; Wing, J.; Augustus, M.; Hudson, P.; Meissner, P.S.; Curtis, R.T.; Shell, B.K.; Bostwick, D.G.; Tindall, D.J.; et al. A novel human prostate-specific, androgen-regulated homeobox gene (nkx3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics 1997, 43, 69–77. [Google Scholar] [CrossRef]
- Cairns, P.; Okami, K.; Halachmi, S.; Halachmi, N.; Esteller, M.; Herman, J.G.; Jen, J.; Isaacs, W.B.; Bova, G.S.; Sidransky, D. Frequent inactivation of pten/mmac1 in primary prostate cancer. Cancer Res. 1997, 57, 4997–5000. [Google Scholar]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. Pten, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Perner, S.; Demichelis, F.; Beroukhim, R.; Schmidt, F.H.; Mosquera, J.M.; Setlur, S.; Tchinda, J.; Tomlins, S.A.; Hofer, M.D.; Pienta, K.G.; et al. Tmprss2:Erg fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006, 66, 8337–8341. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ets gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent spop, foxa1 and med12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef]
- Holliday, R. The inheritance of epigenetic defects. Science 1987, 238, 163–170. [Google Scholar]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Campbell, M.J.; Turner, B.M. Altered histone modifications in cancer. Adv. Exp. Med. Biol. 2013, 754, 81–107. [Google Scholar] [CrossRef]
- Chiam, K.; Ricciardelli, C.; Bianco-Miotto, T. Epigenetic biomarkers in prostate cancer: Current and future uses. Cancer Lett. 2012. [Google Scholar] [CrossRef]
- Jeronimo, C.; Bastian, P.J.; Bjartell, A.; Carbone, G.M.; Catto, J.W.; Clark, S.J.; Henrique, R.; Nelson, W.G.; Shariat, S.F. Epigenetics in prostate cancer: Biologic and clinical relevance. Eur. Urol. 2011, 60, 753–766. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef]
- Denis, H.; Ndlovu, M.N.; Fuks, F. Regulation of mammalian DNA methyltransferases: A route to new mechanisms. EMBO Rep. 2011, 12, 647–656. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Albert, M.; Helin, K. Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 2010, 21, 209–220. [Google Scholar] [CrossRef]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Nakayama, M.; Bennett, C.J.; Hicks, J.L.; Epstein, J.I.; Platz, E.A.; Nelson, W.G.; de Marzo, A.M. Hypermethylation of the human glutathione s-transferase-pi gene (gstp1) cpg island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: A detailed study using laser-capture microdissection. Am. J. Pathol. 2003, 163, 923–933. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein ezh2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Saramaki, O.R.; Tammela, T.L.; Martikainen, P.M.; Vessella, R.L.; Visakorpi, T. The gene for polycomb group protein enhancer of zeste homolog 2 (ezh2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer 2006, 45, 639–645. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Graur, D.; Zheng, Y.; Price, N.; Azevedo, R.B.; Zufall, R.A.; Elhaik, E. On the immortality of television sets: “Function” in the human genome according to the evolution-free gospel of encode. Genome Biol. Evol. 2013, 5, 578–590. [Google Scholar] [CrossRef]
- Doolittle, W.F. Is junk DNA bunk? A critique of encode. Proc. Natl. Acad. Sci. USA 2013, 110, 5294–5300. [Google Scholar] [CrossRef]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. Microrna signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Iorio, M.V.; Croce, C.M. Microrna dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef]
- Manikandan, J.; Aarthi, J.J.; Kumar, S.D.; Pushparaj, P.N. Oncomirs: The potential role of non-coding micrornas in understanding cancer. Bioinformation 2008, 2, 330–334. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—Micrornas with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microrna expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. Microrna expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microrna targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Lussier, Y.A.; Stadler, W.M.; Chen, J.L. Advantages of genomic complexity: Bioinformatics opportunities in microrna cancer signatures. J. Am. Med. Inform. Assoc. 2012, 19, 156–160. [Google Scholar] [CrossRef]
- Sun, T.; Wang, Q.; Balk, S.; Brown, M.; Lee, G.S.; Kantoff, P. The role of microrna-221 and microrna-222 in androgen-independent prostate cancer cell lines. Cancer Res. 2009, 69, 3356–3363. [Google Scholar] [CrossRef]
- Medina, R.; Zaidi, S.K.; Liu, C.G.; Stein, J.L.; van Wijnen, A.J.; Croce, C.M.; Stein, G.S. Micrornas 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008, 68, 2773–2780. [Google Scholar]
- Mercatelli, N.; Coppola, V.; Bonci, D.; Miele, F.; Costantini, A.; Guadagnoli, M.; Bonanno, E.; Muto, G.; Frajese, G.V.; de Maria, R.; et al. The inhibition of the highly expressed mir-221 and mir-222 impairs the growth of prostate carcinoma xenografts in mice. PLoS One 2008, 3, e4029. [Google Scholar] [CrossRef]
- Wang, L.; Tang, H.; Thayanithy, V.; Subramanian, S.; Oberg, A.L.; Cunningham, J.M.; Cerhan, J.R.; Steer, C.J.; Thibodeau, S.N. Gene networks and micrornas implicated in aggressive prostate cancer. Cancer Res. 2009, 69, 9490–9497. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, C.; Zhang, A.; Wang, K.; Jia, Z.; Wang, G.; Han, L.; Kang, C.; Pu, P. Mir-221/222 is the regulator of cx43 expression in human glioblastoma cells. Oncol. Rep. 2012, 27, 1504–1510. [Google Scholar]
- Tucci, P.; Agostini, M.; Grespi, F.; Markert, E.K.; Terrinoni, A.; Vousden, K.H.; Muller, P.A.; Dotsch, V.; Kehrloesser, S.; Sayan, B.S.; et al. Loss of p63 and its microrna-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 15312–15317. [Google Scholar] [CrossRef]
- Zhao, J.J.; Lin, J.; Yang, H.; Kong, W.; He, L.; Ma, X.; Coppola, D.; Cheng, J.Q. Microrna-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J. Biol. Chem. 2008, 283, 31079–31086. [Google Scholar]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. Microrna-221/222 confers tamoxifen resistance in breast cancer by targeting p27kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar]
- Lin, D.; Cui, F.; Bu, Q.; Yan, C. The expression and clinical significance of gtp-binding ras-like 3 (arhi) and microrna 221 and 222 in prostate cancer. J. Int. Med. Res. 2011, 39, 1870–1875. [Google Scholar] [CrossRef]
- Tong, A.W.; Fulgham, P.; Jay, C.; Chen, P.; Khalil, I.; Liu, S.; Senzer, N.; Eklund, A.C.; Han, J.; Nemunaitis, J. Microrna profile analysis of human prostate cancers. Cancer Gene Ther. 2009, 16, 206–216. [Google Scholar]
- Spahn, M.; Kneitz, S.; Scholz, C.J.; Stenger, N.; Rudiger, T.; Strobel, P.; Riedmiller, H.; Kneitz, B. Expression of microrna-221 is progressively reduced in aggressive prostate cancer and metastasis and predicts clinical recurrence. Int. J. Cancer 2010, 127, 394–403. [Google Scholar]
- Martens-Uzunova, E.S.; Jalava, S.E.; Dits, N.F.; van Leenders, G.J.; Moller, S.; Trapman, J.; Bangma, C.H.; Litman, T.; Visakorpi, T.; Jenster, G. Diagnostic and prognostic signatures from the small non-coding rna transcriptome in prostate cancer. Oncogene 2012, 31, 978–991. [Google Scholar] [CrossRef]
- Wach, S.; Nolte, E.; Szczyrba, J.; Stohr, R.; Hartmann, A.; Orntoft, T.; Dyrskjot, L.; Eltze, E.; Wieland, W.; Keck, B.; et al. Microrna profiles of prostate carcinoma detected by multiplatform microrna screening. Int. J. Cancer 2012, 130, 611–621. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating micrornas as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Ozen, M.; Creighton, C.J.; Ozdemir, M.; Ittmann, M. Widespread deregulation of microrna expression in human prostate cancer. Oncogene 2008, 27, 1788–1793. [Google Scholar] [CrossRef]
- Porkka, K.P.; Pfeiffer, M.J.; Waltering, K.K.; Vessella, R.L.; Tammela, T.L.; Visakorpi, T. Microrna expression profiling in prostate cancer. Cancer Res. 2007, 67, 6130–6135. [Google Scholar]
- Lee, Y.S.; Kim, H.K.; Chung, S.; Kim, K.S.; Dutta, A. Depletion of human micro-rna mir-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J. Biol. Chem. 2005, 280, 16635–16641. [Google Scholar]
- Shi, X.B.; Xue, L.; Yang, J.; Ma, A.H.; Zhao, J.; Xu, M.; Tepper, C.G.; Evans, C.P.; Kung, H.J.; deVere White, R.W. An androgen-regulated mirna suppresses bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 19983–19988. [Google Scholar] [CrossRef]
- Ribas, J.; Ni, X.; Haffner, M.; Wentzel, E.A.; Salmasi, A.H.; Chowdhury, W.H.; Kudrolli, T.A.; Yegnasubramanian, S.; Luo, J.; Rodriguez, R.; et al. Mir-21: An androgen receptor-regulated microrna that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res. 2009, 69, 7165–7169. [Google Scholar] [CrossRef]
- Shi, G.H.; Ye, D.W.; Yao, X.D.; Zhang, S.L.; Dai, B.; Zhang, H.L.; Shen, Y.J.; Zhu, Y.; Zhu, Y.P.; Xiao, W.J.; et al. Involvement of microrna-21 in mediating chemo-resistance to docetaxel in androgen-independent prostate cancer pc3 cells. Acta Pharmacol. Sin. 2010, 31, 867–873. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, M.; Stribinskis, V.; Klinge, C.M.; Ramos, K.S.; Colburn, N.H.; Li, Y. Microrna-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008, 27, 4373–4379. [Google Scholar] [CrossRef]
- Peng, X.; Guo, W.; Liu, T.; Wang, X.; Tu, X.; Xiong, D.; Chen, S.; Lai, Y.; Du, H.; Chen, G.; et al. Identification of mirs-143 and -145 that is associated with bone metastasis of prostate cancer and involved in the regulation of emt. PLoS One 2011, 6, e20341. [Google Scholar]
- Kong, D.; Li, Y.; Wang, Z.; Banerjee, S.; Ahmad, A.; Kim, H.R.; Sarkar, F.H. Mir-200 regulates pdgf-d-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 2009, 27, 1712–1721. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The mir-200 family and mir-205 regulate epithelial to mesenchymal transition by targeting zeb1 and sip1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Liep, J.; Rabien, A.; Jung, K. Feedback networks between micrornas and epigenetic modifications in urological tumors. Epigenetics 2012, 7, 315–325. [Google Scholar] [CrossRef]
- Saito, Y.; Jones, P.A. Epigenetic activation of tumor suppressor micrornas in human cancer cells. Cell Cycle 2006, 5, 2220–2222. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microrna-1 and microrna-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- Duursma, A.M.; Kedde, M.; Schrier, M.; le Sage, C.; Agami, R. Mir-148 targets human dnmt3b protein coding region. RNA 2008, 14, 872–877. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by micrornas: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar]
- Thorne, J.L.; Maguire, O.; Doig, C.L.; Battaglia, S.; Fehr, L.; Sucheston, L.E.; Heinaniemi, M.; O’Neill, L.P.; McCabe, C.J.; Turner, B.M.; et al. Epigenetic control of a vdr-governed feed-forward loop that regulates p21(waf1/cip1) expression and function in non-malignant prostate cells. Nucleic Acids Res. 2011, 39, 2045–2056. [Google Scholar] [CrossRef]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. Microrna-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly dnmt3a and 3b and indirectly dnmt1. Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. Microrna-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3a and 3b. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef]
- Majid, S.; Dar, A.A.; Saini, S.; Shahryari, V.; Arora, S.; Zaman, M.S.; Chang, I.; Yamamura, S.; Tanaka, Y.; Chiyomaru, T.; et al. Mirna-34b inhibits prostate cancer through demethylation, active chromatin modifications, and akt pathways. Clin. Cancer Res. 2013, 19, 73–84. [Google Scholar] [CrossRef]
- Cao, P.; Deng, Z.; Wan, M.; Huang, W.; Cramer, S.D.; Xu, J.; Lei, M.; Sui, G. Microrna-101 negatively regulates ezh2 and its expression is modulated by androgen receptor and hif-1alpha/hif-1beta. Mol. Cancer 2010, 9, 108. [Google Scholar] [CrossRef]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. Mir-449a targets hdac-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef]
- Gong, A.Y.; Eischeid, A.N.; Xiao, J.; Zhao, J.; Chen, D.; Wang, Z.Y.; Young, C.Y.; Chen, X.M. Mir-17-5p targets the p300/cbp-associated factor and modulates androgen receptor transcriptional activity in cultured prostate cancer cells. BMC Cancer 2012, 12, 492. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The polycomb complex prc2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microrna-101 leads to overexpression of histone methyltransferase ezh2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef]
- Cao, Q.; Mani, R.S.; Ateeq, B.; Dhanasekaran, S.M.; Asangani, I.A.; Prensner, J.R.; Kim, J.H.; Brenner, J.C.; Jing, X.; Cao, X.; et al. Coordinated regulation of polycomb group complexes through micrornas in cancer. Cancer Cell 2011, 20, 187–199. [Google Scholar] [CrossRef]
- Cohen, E.E.; Zhu, H.; Lingen, M.W.; Martin, L.E.; Kuo, W.L.; Choi, E.A.; Kocherginsky, M.; Parker, J.S.; Chung, C.H.; Rosner, M.R. A feed-forward loop involving protein kinase calpha and micrornas regulates tumor cell cycle. Cancer Res. 2009, 69, 65–74. [Google Scholar] [CrossRef]
- Brosh, R.; Shalgi, R.; Liran, A.; Landan, G.; Korotayev, K.; Nguyen, G.H.; Enerly, E.; Johnsen, H.; Buganim, Y.; Solomon, H.; et al. P53-repressed mirnas are involved with e2f in a feed-forward loop promoting proliferation. Mol. Syst. Biol. 2008, 4, 229. [Google Scholar]
- Polioudakis, D.; Bhinge, A.A.; Killion, P.J.; Lee, B.K.; Abell, N.S.; Iyer, V.R. A myc-microrna network promotes exit from quiescence by suppressing the interferon response and cell-cycle arrest genes. Nucleic Acids Res. 2013, 41, 2239–2254. [Google Scholar] [CrossRef]
- Hassan, M.Q.; Maeda, Y.; Taipaleenmaki, H.; Zhang, W.; Jafferji, M.; Gordon, J.A.; Li, Z.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; et al. Mir-218 directs a wnt signaling circuit to promote differentiation of osteoblasts and osteomimicry of metastatic cancer cells. J. Biol. Chem. 2012, 287, 42084–42092. [Google Scholar] [CrossRef]
- McInnes, N.; Sadlon, T.J.; Brown, C.Y.; Pederson, S.; Beyer, M.; Schultze, J.L.; McColl, S.; Goodall, G.J.; Barry, S.C. Foxp3 and foxp3-regulated micrornas suppress satb1 in breast cancer cells. Oncogene 2012, 31, 1045–1054. [Google Scholar] [CrossRef]
- Da Costa Martins, P.A.; Salic, K.; Gladka, M.M.; Armand, A.S.; Leptidis, S.; el Azzouzi, H.; Hansen, A.; Coenen-de Roo, C.J.; Bierhuizen, M.F.; van der Nagel, R.; et al. Microrna-199b targets the nuclear kinase dyrk1a in an auto-amplification loop promoting calcineurin/nfat signalling. Nat. Cell Biol. 2010, 12, 1220–1227. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and hdac2 expression is associated with shorter psa relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef]
- Nagy, Z.; Tora, L. Distinct gcn5/pcaf-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 2007, 26, 5341–5357. [Google Scholar] [CrossRef]
- Dudziec, E.; Miah, S.; Choudhry, H.M.; Owen, H.C.; Blizard, S.; Glover, M.; Hamdy, F.C.; Catto, J.W. Hypermethylation of cpg islands and shores around specific micrornas and mirtrons is associated with the phenotype and presence of bladder cancer. Clin. Cancer Res. 2011, 17, 1287–1296. [Google Scholar] [CrossRef]
- Gandellini, P.; Folini, M.; Longoni, N.; Pennati, M.; Binda, M.; Colecchia, M.; Salvioni, R.; Supino, R.; Moretti, R.; Limonta, P.; et al. Mir-205 exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase cepsilon. Cancer Res. 2009, 69, 2287–2295. [Google Scholar] [CrossRef]
- Iorio, M.V.; Croce, C.M. Micrornas in cancer: Small molecules with a huge impact. J. Clin. Oncol. 2009, 27, 5848–5856. [Google Scholar] [CrossRef]
- Bhatnagar, N.; Li, X.; Padi, S.K.; Zhang, Q.; Tang, M.S.; Guo, B. Downregulation of mir-205 and mir-31 confers resistance to chemotherapy-induced apoptosis in prostate cancer cells. Cell Death Dis. 2010, 1, e105. [Google Scholar] [CrossRef]
- Vrba, L.; Jensen, T.J.; Garbe, J.C.; Heimark, R.L.; Cress, A.E.; Dickinson, S.; Stampfer, M.R.; Futscher, B.W. Role for DNA methylation in the regulation of mir-200c and mir-141 expression in normal and cancer cells. PLoS One 2010, 5, e8697. [Google Scholar] [CrossRef]
- Hermeking, H. The mir-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef]
- Lodygin, D.; Tarasov, V.; Epanchintsev, A.; Berking, C.; Knyazeva, T.; Korner, H.; Knyazev, P.; Diebold, J.; Hermeking, H. Inactivation of mir-34a by aberrant cpg methylation in multiple types of cancer. Cell Cycle 2008, 7, 2591–2600. [Google Scholar] [CrossRef]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microrna component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of mir-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microrna mir-34a inhibits prostate cancer stem cells and metastasis by directly repressing cd44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- Rauhala, H.E.; Jalava, S.E.; Isotalo, J.; Bracken, H.; Lehmusvaara, S.; Tammela, T.L.; Oja, H.; Visakorpi, T. Mir-193b is an epigenetically regulated putative tumor suppressor in prostate cancer. Int. J. Cancer 2010, 127, 1363–1372. [Google Scholar] [CrossRef]
- Sachdeva, M.; Mo, Y.Y. Mir-145-mediated suppression of cell growth, invasion and metastasis. Am. J. Transl. Res. 2010, 2, 170–180. [Google Scholar]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. P53 represses c-myc through induction of the tumor suppressor mir-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar]
- Sachdeva, M.; Mo, Y.Y. Microrna-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res. 2010, 70, 378–387. [Google Scholar] [CrossRef]
- Zaman, M.S.; Chen, Y.; Deng, G.; Shahryari, V.; Suh, S.O.; Saini, S.; Majid, S.; Liu, J.; Khatri, G.; Tanaka, Y.; et al. The functional significance of microrna-145 in prostate cancer. Br. J. Cancer 2010, 103, 256–264. [Google Scholar] [CrossRef]
- Suh, S.O.; Chen, Y.; Zaman, M.S.; Hirata, H.; Yamamura, S.; Shahryari, V.; Liu, J.; Tabatabai, Z.L.; Kakar, S.; Deng, G.; et al. Microrna-145 is regulated by DNA methylation and p53 gene mutation in prostate cancer. Carcinogenesis 2011, 32, 772–778. [Google Scholar] [CrossRef]
- Hulf, T.; Sibbritt, T.; Wiklund, E.D.; Bert, S.; Strbenac, D.; Statham, A.L.; Robinson, M.D.; Clark, S.J. Discovery pipeline for epigenetically deregulated mirnas in cancer: Integration of primary mirna transcription. BMC Genomics 2011, 12, 54. [Google Scholar] [CrossRef]
- Iorio, M.V.; Visone, R.; di Leva, G.; Donati, V.; Petrocca, F.; Casalini, P.; Taccioli, C.; Volinia, S.; Liu, C.G.; Alder, H.; et al. Microrna signatures in human ovarian cancer. Cancer Res. 2007, 67, 8699–8707. [Google Scholar] [CrossRef]
- Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Kassir, Z.; Moreno, C.S.; Young, A.N.; Varma, V.; Speed, T.P.; Cowley, M.; et al. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (lres) reduces transcriptional plasticity. Nat. Cell Biol. 2010, 12, 235–246. [Google Scholar]
- Cookson, M.S.; Aus, G.; Burnett, A.L.; Canby-Hagino, E.D.; D’Amico, A.V.; Dmochowski, R.R.; Eton, D.T.; Forman, J.D.; Goldenberg, S.L.; Hernandez, J.; et al. Variation in the definition of biochemical recurrence in patients treated for localized prostate cancer: The american urological association prostate guidelines for localized prostate cancer update panel report and recommendations for a standard in the reporting of surgical outcomes. J. Urol. 2007, 177, 540–545. [Google Scholar] [CrossRef]
- Goering, W.; Kloth, M.; Schulz, W.A. DNA methylation changes in prostate cancer. Methods Mol. Biol. 2012, 863, 47–66. [Google Scholar] [CrossRef]
- Perry, A.S.; Watson, R.W.; Lawler, M.; Hollywood, D. The epigenome as a therapeutic target in prostate cancer. Nat. Rev. Urol. 2010, 7, 668–680. [Google Scholar] [CrossRef]
- Hoque, M.O.; Topaloglu, O.; Begum, S.; Henrique, R.; Rosenbaum, E.; van Criekinge, W.; Westra, W.H.; Sidransky, D. Quantitative methylation-specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J. Clin. Oncol. 2005, 23, 6569–6575. [Google Scholar] [CrossRef]
- Gonzalgo, M.L.; Pavlovich, C.P.; Lee, S.M.; Nelson, W.G. Prostate cancer detection by gstp1 methylation analysis of postbiopsy urine specimens. Clin. Cancer Res. 2003, 9, 2673–2677. [Google Scholar]
- Jeronimo, C.; Usadel, H.; Henrique, R.; Silva, C.; Oliveira, J.; Lopes, C.; Sidransky, D. Quantitative gstp1 hypermethylation in bodily fluids of patients with prostate cancer. Urology 2002, 60, 1131–1135. [Google Scholar] [CrossRef]
- Roupret, M.; Hupertan, V.; Yates, D.R.; Catto, J.W.; Rehman, I.; Meuth, M.; Ricci, S.; Lacave, R.; Cancel-Tassin, G.; de la Taille, A.; et al. Molecular detection of localized prostate cancer using quantitative methylation-specific pcr on urinary cells obtained following prostate massage. Clin. Cancer Res. 2007, 13, 1720–1725. [Google Scholar] [CrossRef]
- Ellinger, J.; Haan, K.; Heukamp, L.C.; Kahl, P.; Buttner, R.; Muller, S.C.; von Ruecker, A.; Bastian, P.J. Cpg island hypermethylation in cell-free serum DNA identifies patients with localized prostate cancer. Prostate 2008, 68, 42–49. [Google Scholar] [CrossRef]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Rogenhofer, S.; Heukamp, L.C.; Gutgemann, I.; Walter, B.; Hofstadter, F.; Buttner, R.; Muller, S.C.; et al. Global levels of histone modifications predict prostate cancer recurrence. Prostate 2010, 70, 61–69. [Google Scholar] [CrossRef]
- Deligezer, U.; Yaman, F.; Darendeliler, E.; Dizdar, Y.; Holdenrieder, S.; Kovancilar, M.; Dalay, N. Post-treatment circulating plasma bmp6 mrna and h3k27 methylation levels discriminate metastatic prostate cancer from localized disease. Clin. Chim. Acta 2010, 411, 1452–1456. [Google Scholar] [CrossRef]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. Micrornas in body fluids-the mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of micrornas in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef]
- El-Hefnawy, T.; Raja, S.; Kelly, L.; Bigbee, W.L.; Kirkwood, J.M.; Luketich, J.D.; Godfrey, T.E. Characterization of amplifiable, circulating rna in plasma and its potential as a tool for cancer diagnostics. Clin. Chem. 2004, 50, 564–573. [Google Scholar] [CrossRef]
- Selth, L.A.; Tilley, W.D.; Butler, L.M. Circulating micrornas: Macro-utility as markers of prostate cancer? Endocr. Relat. Cancer 2012, 19, R99–R113. [Google Scholar] [CrossRef]
- Zhang, H.L.; Yang, L.F.; Zhu, Y.; Yao, X.D.; Zhang, S.L.; Dai, B.; Zhu, Y.P.; Shen, Y.J.; Shi, G.H.; Ye, D.W. Serum mirna-21: Elevated levels in patients with metastatic hormone-refractory prostate cancer and potential predictive factor for the efficacy of docetaxel-based chemotherapy. Prostate 2011, 71, 326–331. [Google Scholar] [CrossRef]
- Selth, L.A.; Townley, S.; Gillis, J.L.; Ochnik, A.M.; Murti, K.; Macfarlane, R.J.; Chi, K.N.; Marshall, V.R.; Tilley, W.D.; Butler, L.M. Discovery of circulating micrornas associated with human prostate cancer using a mouse model of disease. Int. J. Cancer 2012, 131, 652–661. [Google Scholar] [CrossRef]
- Yaman Agaoglu, F.; Kovancilar, M.; Dizdar, Y.; Darendeliler, E.; Holdenrieder, S.; Dalay, N.; Gezer, U. Investigation of mir-21, mir-141, and mir-221 in blood circulation of patients with prostate cancer. Tumour Biol. 2011, 32, 583–588. [Google Scholar] [CrossRef]
- Mahn, R.; Heukamp, L.C.; Rogenhofer, S.; von Ruecker, A.; Muller, S.C.; Ellinger, J. Circulating micrornas (mirna) in serum of patients with prostate cancer. Urology 2011, 77, 1265.e9–1265.e16. [Google Scholar] [CrossRef]
- Bryant, R.J.; Pawlowski, T.; Catto, J.W.; Marsden, G.; Vessella, R.L.; Rhees, B.; Kuslich, C.; Visakorpi, T.; Hamdy, F.C. Changes in circulating microrna levels associated with prostate cancer. Br. J. Cancer 2012, 106, 768–774. [Google Scholar] [CrossRef]
- Brase, J.C.; Johannes, M.; Schlomm, T.; Falth, M.; Haese, A.; Steuber, T.; Beissbarth, T.; Kuner, R.; Sultmann, H. Circulating mirnas are correlated with tumor progression in prostate cancer. Int. J. Cancer 2011, 128, 608–616. [Google Scholar] [CrossRef]
- Regulome Explorer. Available online: http://explorer.cancerregulome.org/ (accessed on 16 July 2013).
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Singh, P.K.; Campbell, M.J. The Interactions of microRNA and Epigenetic Modifications in Prostate Cancer. Cancers 2013, 5, 998-1019. https://doi.org/10.3390/cancers5030998
Singh PK, Campbell MJ. The Interactions of microRNA and Epigenetic Modifications in Prostate Cancer. Cancers. 2013; 5(3):998-1019. https://doi.org/10.3390/cancers5030998
Chicago/Turabian StyleSingh, Prashant Kumar, and Moray J. Campbell. 2013. "The Interactions of microRNA and Epigenetic Modifications in Prostate Cancer" Cancers 5, no. 3: 998-1019. https://doi.org/10.3390/cancers5030998
APA StyleSingh, P. K., & Campbell, M. J. (2013). The Interactions of microRNA and Epigenetic Modifications in Prostate Cancer. Cancers, 5(3), 998-1019. https://doi.org/10.3390/cancers5030998