Abstract
Oxidative stress induced by reactive oxygen species can result in DNA damage within cells and subsequently increase risk for carcinogenesis. This may be averted by repair of DNA damage through the base or nucleotide excision repair (BER/NER) pathways. PARP, a BER protein, is known for its role in DNA-repair. However, multiple lesions can occur within a small range of DNA, known as oxidative clustered DNA lesions (OCDLs), which are difficult to repair and may lead to the more severe DNA double-strand break (DSB). Inefficient DSB repair can then result in increased mutagenesis and neoplastic transformation. OCDLs occur more frequently within a variety of tumor tissues. Interestingly, PARP is highly expressed in several human cancers. Additionally, chronic inflammation may contribute to tumorigenesis through ROS-induced DNA damage. Furthermore, PARP can modulate inflammation through interaction with NFκB and regulating the expression of inflammatory signaling molecules. Thus, the upregulation of PARP may present a double-edged sword. PARP is needed to repair ROS-induced DNA lesions, but PARP expression may lead to increased inflammation via upregulation of NFκB signaling. Here, we discuss the role of PARP in the repair of oxidative damage versus the formation of OCDLs and speculate on the feasibility of PARP inhibition for the treatment and prevention of cancers by exploiting its role in inflammation.
1. Introduction
Human tissues are exposed to various sources of reactive oxygen species, which can lead to DNA damage including single strand breaks (SSBs) and the more severe, double strand breaks (DSBs). Reactive oxygen species (ROS) are oxygen derivatives that are highly volatile toward cellular components. Sources of the damaging ROS can be both exogenous (UV light, pollution, ionizing radiation) and endogenous (cellular metabolism and inflammation) [1,2]. Cellular homeostasis depends on a delicate balance between damage versus the repair of ROS-induced lesions that occur in lipids, proteins, and DNA. While cellular processes allow for the turnover of lipids and proteins damaged by interaction with ROS, DNA is non-recyclable, and therefore DNA damages must be repaired. Thus, cells have adapted elaborate processes to address DNA lesions caused by ROS, and these lesions are usually fixed by base excision repair (BER) and nucleotide excision repair (NER) pathways upon activation of cell cycle checkpoints [3]. However, ineffective attempts at these processes due to repair pathway deficiencies or clustered DNA lesions can result in cytotoxic or mutagenic effects, and chromosomal instability, all of which may contribute to tumorigenesis [4].
In this review, we examine the possibility of the double-edged sword of the key BER protein PARP-1 (poly-ADP-ribose polymerase) in tumorigenesis by considering its role in ROS-induced DNA damage repair versus its role in inflammation. On one hand, PARP1 upregulation may lead to increased DNA repair of ROS-mediated DNA damage. However, continued activation of PARP-1, especially in situations of chronic inflammation/oxidation, may contribute to the formation of OCDLs and enhance tumorigenesis. Given these two diverging roles of PARP-1, the PARP inhibitors may be used to not only treat cancers but also to prevent cancers that form from the potentiation of inflammation/ROS-induced damage caused by persistent PARP-1 upregulation in response to DNA damage. Thus, we will discuss the possibility of using PARP inhibitors in the treatment of not only familial cancers, but also as part of a combination therapy or preventative reagent in non-familial cancers, and potentially in inflammatory diseases which carry a high risk for cancer development.
2. PARP-1
PARP-1 (also known as ADP-ribosyltransferase-1, or ARTD1) is a member of the poly-ADP-ribose polymerase family, which is a group of 18 enzymes shown to be involved in many processes ranging from DNA repair to cell death [5]. The most well-studied family member, PARP-1, contains three domains including: (1) zinc-finger DNA binding domain; (2) an automodification domain; and (3) a catalytic domain [6]. The catalytic domain is responsible for the enzymatic activity of the protein. This domain functions to catalyze the addition of ADP-ribose chains to target proteins, including itself, from NAD+ donor molecules, resulting in polymer strands in a process known as PARylation. While the PARP family of enzymes is highly conserved, and all contain a catalytic domain signature motif, not all PARP family members have confirmed PARylation activity to date [5]. They are classified as PARP members due solely to the presence of the signature catalytic motif. This review will focus solely on the role of PARP-1.
PARP-1 has been shown to play a role in several nuclear processes including chromatin modification, transcriptional regulation, and DNA damage repair [7]. PARP-1 binds to DNA not only at sites of damage (SSB and DSB), but also DNA crossovers, supercoils and cruciform [8]. The role of PARP-1 in chromatin modification is multifaceted, as studies have demonstrated that PARP-1 can both relax and condense chromatin through interaction with nucleosomes and PARylation of proteins involved in chromatin structure, such as histones H1 and H2B [9,10,11]. Additionally, PARP-1 can regulate transcription through direct interaction with transcription factors, as well as altering their activity via PARylation. For instance, PARP-1 regulates Cox-2, Oct1, E2F-1, and Ap2 through direct interaction, while p53 and RNA polymerases I and II are regulated by PARylation [12,13,14,15,16,17,18]. Additionally, both direct interaction and enzymatic activity by PARP-1 can alter the function of transcription factors, as is the case for NFκB, where PARP-1 binds to and PARylates both p50 and p65 subunits of NFκB [19,20]. The role of PARP-1, either to enhance or repress function of the targets, in each case is unique to the transcription factor and expression level of PARP-1 at a given time.
Perhaps one of the most well known functions of PARP-1 is the role it plays in the sensing and initiation of DNA repair. PARP-1 has been demonstrated to play a role in most forms of DNA repair; including single strand break (base excision repair-BER) and double strand break (homologous recombination [HR] and non-homologous end joining [NHEJ] repair processes [21].
The mechanisms by which PARP-1 contributes to HR and NHEJ are not as well defined as the role in BER/SSB repair. DSB are sensed by the Mre11-Rad50-Nbs1 complex, which binds to the lesion site on the DNA. Subsequently, ATM is phosphorylated and the repair pathway is initiated [22]. The repair pathway choice is determined by cell cycle stage, structure of the DNA break, and availability of homologous DNA [23]. It has been shown that PARP-1 interacts with several proteins involved in the DSB repair pathways including NBS1, Mre11, Ku80, DNA-PKcs and ATM [24,25,26,27]. More specifically, knockdown and chemical inhibition of PARP-1 revealed the interaction between ATM and PARP-1 is important in the kinetics of phosphorylation of downstream signaling molecules including p53 and H2AX, where inhibition of PARP-1 delays ATM activity [27]. Additionally, it has been shown that PARP-1 and PARP-2 may detect and recruit the protein Mre11, initiating the repair of stalled replication forks upon treatment with hydroxyurea [28]. Furthermore, PARP−/−/ATM−/− and PARP−/−/Ku80−/− mice display embryonic lethality associated with genomic instability [29,30]. While specific mechanisms are not yet elucidated, together these studies suggest PARP-1 is an integral player in the DSB repair pathways.
The role of PARP-1 in the repair of SSB/BER is better defined; however, the exact details are still being investigated. BER and SSB repair serve to correct small lesions that are acquired through a variety of insults including ROS damage or subjection to methylating reagents and irradiation [3]. In order to repair these lesions, the cell first transforms the lesion to a single strand break intermediate via the action of DNA glycosylases and APE1 [31]. This SSB intermediate then recruits PARP-1 to the damage site. Enzymatic activity of PARP-1 works to PARylate itself and other substrates to further recruit repair proteins to the breakage site. Several proteins involved in the BER/SSB repair process have been identified to interact with PARP-1 including XRCC1, DNA polymerase β, PCNA and others [31,32,33,34]. Initially PARP-1 was identified as having a role in BER when PARP-1−/− mice demonstrated increased sensitivity to ionizing radiation and oxidative damages, and cells derived from these mice were hypersensitive to alkylating reagents [35]. Furthermore, BER deficiency due to PARP-1 knockdown was more specifically shown when these cells demonstrated difficulty in DNA strand break sealing after treatment with the alkylating agent methylmethanesulfonate [36]. Additionally, other work has shown that PARP-1 deficiency decreases short and long-patch repair of abasic sites [37]. Interestingly, the role of PARP-1 in ROS-induced damage may be countered by involvement in inflammatory signaling. This then presents a double-edged sword of PARP-1 signaling, where it is participating in DNA repair, but may also induce DNA damage through inflammation if chronically activated for repair processes.
2.1. PARP-1 in Inflammation
While most well known for its role in base excision repair, PARP-1 also serves to regulate other signaling pathways, such as cell death and inflammation. PARP-1 inhibition has been shown to be beneficial in a milieu of animal models of inflammatory diseases, such as diabetes, asthma and atherosclerosis [38]. The role of PARP-1 in inflammatory signaling is becoming more evident as the focus of study shifts from solely examining its involvement in DNA repair to other processes including mechanisms of inflammation.
PARP has been shown to contribute to inflammatory processes through a variety of means, including the regulation of transcription factors, cytokines, adhesion factors and inflammatory mediators [39,40]. The well-known and thoroughly described transcription factor, NFκB was one of the first mediators of inflammation to be identified as a target for PARP activity [41]. NFκB has been shown to be a binding partner and also to be PARylated by PARP-1, as previously discussed. PARP-1 is known to be a co-activator of NFκB [41,42]. These studies established in PARP−/− mice and cell lines, that NFκB activity was greatly abrogated independent of upstream activation of NFκB [41,42]. PARP−/− mice were protected from LPS-induced endotoxic shock, due to a strong downregulation of downstream signaling components that propagate the inflammatory signal, including inducible nitric oxide synthase (iNOS) and TNFα [42]. Recently, this has been further confirmed as it has been shown that systemic inflammation serves to upregulate several members of the PARP family in mouse hippocampus, including PARP-1,-3, -9, -12 and -14 [43]. Additionally, the levels of PARylation were increased, and importantly, levels of iNOS were upregulated. PARP inhibitor served to protect cells against increased PARylation and increased iNOS gene expression which is an integral player in ROS/NOS-induced stress in systemic inflammatory response [43]. In both of these cases, knockout and inhibition of PARP served to downregulate the inflammatory response by abrogating NFκB signaling.
The role of PARP-induced inflammation is also multifaceted. It serves as a co-factor for NFκB, and also serves to switch cell death pathways from the well-controlled and highly regulated apoptosis to the more inflammatory necrosis in the event of high levels of ROS species [44,45,46]. In the inflammatory response, the cellular environment is subjected to increased DNA insults carried out by inflammation-induced ROS. This then can lead to the upregulation of PARP-1; as expression of PARP-1 is dramatically increased with DNA strand breaks [47]. The upregulation of PARP-1, and subsequent elevated PARP-1 activity, can deplete cellular levels of ATP and NAD, forcing the cell into necrosis [44,45]. While BER/SSB processes are upregulated, the increase in PARP-1 will also allow PARP-1 to serve as a co-activator of NFκB. If the delicate balance of insult-repair ratio is disturbed, rapid energy depletion and necrotic cell death can occur, resulting in increased inflammatory signaling [45]. By serving as both an NFκB coactivator and pushing ROS-exposed cells to necrosis, PARP-1 functions to further propagate the inflammatory signal. Combined with the regulation of cytokines and inflammatory mediators such as iNOS, it is clear that PARP-1 is a potentially substantial contributor to inflammation.
In support of the pro-inflammatory functions of PARP-1, PARP inhibitors have been demonstrated to be an effective treatment of many inflammatory-related diseases in preclinical models. The wide array of inflammatory conditions that have shown improvement by PARP inhibition ranges from chronic diseases such as diabetic neuropathy and atherosclerosis to more acute events such as sunburn-related inflammation and cisplatin-induced kidney inflammation [48,49,50,51,52,53,54]. These inflammatory states are wide in breadth and demonstrate that PARP may contribute to several levels of regulation in a wide range of inflammatory processes and stimuli. These mechanisms are yet to be fully determined and provide subject for further investigation. An overview of the multiple cellular processes regulated by PARP-1 is depicted in Figure 1.
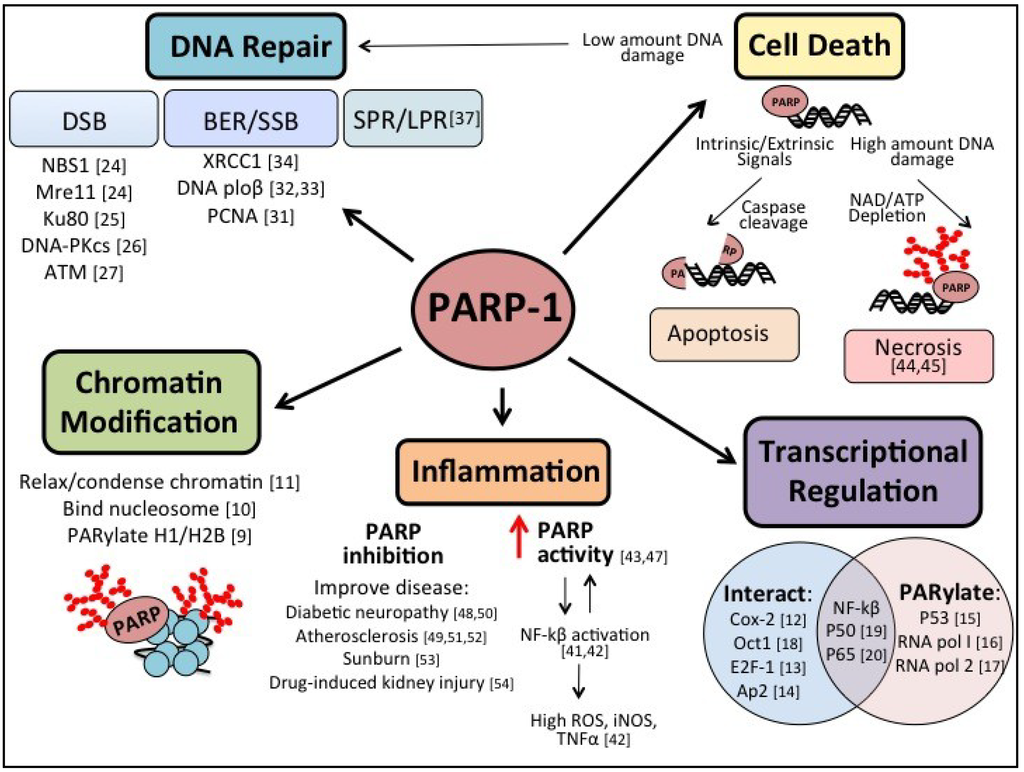
Figure 1.
Schematic delineating the multifaceted nature of Poly(ADP) Ribose Polymerase (PARP): DNA repair, Chromatin Modification, Inflammation, Transcriptional Regulation, and Cell Death.
Figure 1.
Schematic delineating the multifaceted nature of Poly(ADP) Ribose Polymerase (PARP): DNA repair, Chromatin Modification, Inflammation, Transcriptional Regulation, and Cell Death.
2.2. PARP-1 in Cancer
In the updated version of the landmark description of the hallmarks of cancer, Hanahan and Weinberg describe two “enabling characteristics” that allow acquisition of the traditional cancer hallmarks such as resisting cell death and inducing angiogenesis. These enabling characteristics are: (1) genome instability and mutation and (2) tumor-promoting inflammation [55]. Interestingly, PARP-1 sits at the nexus of both of these enabling characteristics. As previously discussed, PARP-1 plays a large role in genome maintenance (DNA repair, chromatin remodeling, transcription factor regulation), and also contributes to the propagation of the inflammatory phenotype. Clinically, the focus of PARP-1 is as a target for the treatment of familial cancers, such as BRCA1/2 deficient breast and ovarian tumors [56]. However, based on its many functions, PARP inhibition may be applicable as a mode of therapy beyond familial cancers.
The majority of the literature to this point has not addressed the possibility of PARP as a pro-tumorigenic factor. This interesting paradigm may be related to its interaction with NFκB, its role in inflammation and subsequent ROS generation, or its pro-survival functions. Mice deficient in PARP have shown decreased susceptibility to skin cancer due to reduced NFκB signaling [57]. In an in vitro model of melanoma, PARP-1 driven NFκB signaling led to an increase in the secretion of pro-metastatic cytokines during senescence, termed the PARP-1 NFκB-Associated Secretome, or PNAS. Inhibition of PARP or NFκB in this system abrogates the proinvasive phenotype conferred by PARP-1/NFκB signaling [58]. The extracellular signal-activated kinase, or Erk, has been shown to modulate PARP-1 activity, including the phosphorylation of PARP-1 and the PARylation of NFκB (p65) in an inflammatory response model. This suggests that Erk may also regulate the PARP-1-dependent activation of NFκB [40], further points to a role for this interaction in cancer, as dysregulated Erk pathways are thought to be a present in an estimated one-third of cancers [59]. Additionally, in the mouse model where NFκB signaling was disrupted, a reduction in mammary tumor formation was observed, indicating NFκB signaling as a requirement for tumorigenesis for HER2+ tumors [60]. Interestingly, our laboratory has also shown HER2+ breast cancer cells are sensitive to PARP inhibition independent of HR repair mechanisms but rather due to suppression of NFκB signaling. This further suggests PARP and NFκB together play a crucial role in carcinogenesis [61] and provides more evidence for the broader utility of PARP inhibitors in cancer therapy.
While these studies are not conclusive evidence that PARP-1 plays a role in tumorigenesis, it is worth considering that PARP-1 regulated signaling may contribute to the initiation or progression of tumors. Interestingly, PARP-1 is highly expressed in a variety of cancers, including breast and hepatocellular carcinoma (HCC), and is correlated with poor prognosis in early breast cancers [62,63,64]. In studies of breast tumors, PARP-1 expression was correlated with higher grade and estrogen receptor (ER) negative status, suggesting that PARP-1 was over-expressed in more aggressive tumor subtypes [63,64]. In the HCC study by Shimizu et al., it was found that PARP-1 expression was significantly increased in HCC samples as compared to uninvolved liver tissue, and that PARP-1 expression was also increased in cirrhosis tissues, further implicating PARP-1 in the inflammatory response and in the susceptibility to tumorigenesis [62]. Recently, it was also shown that PARP-1 expression as well as PARylation levels were increased in non-small cell lung cancer cell lines resistant to cisplatin, as compared to their cisplatin-susceptible counterparts, indicating PARP-1 expression and activity may be correlated with tumor resistance to therapy [65]. In addition, PARP-1 and its interaction with the cell cycle control protein p21 (CDKN1A) may also contribute to tumorigenesis and the tumor phenotype. Mechanisms published by Cazzalini et al. [66] demonstrate that p21 interacts directly with PARP-1 in the DNA repair process, and knockdown of p21 lead to increased PARP-1 PARylation activity. p21 is often downregulated in cancers due to its regulation by p53 (reviewed in [67]), and based on this study could lead to increased PARP-1 activity. Along these lines, downregulated p21 and subsequent increased PARP-1 activity may provide an additional mechanism through which PARP-1 may play a role in tumorigenesis.
Together, these studies present an interesting perspective on PARP-1 as a tumorigenic factor that is yet to be fully explored. Is chronic activation of PARP-1 in highly oxidative environments responsible for the increased inflammation leading to genome instability and cancer, or are the DNA damages causing an upregulation of PARP-1? One potential mechanism through which PARP-1 and the inflammation-DNA damage feedback loop could be propagated is through the development and attempted repair of oxidative clustered DNA lesions.
3. Oxidative Clustered DNA Lesions (OCDLs)
Cells and tissues are constantly exposed to oxidative stresses, which can result in DNA damages that are usually repaired by the base excision repair process, or BER. If the balance between repair mechanisms versus ROS oxidation is perturbed, such as in environments where chronic inflammation or highly metabolic tissues reside, accumulation of these types of DNA damage can lead to a phenomenon known as oxidative clustered DNA lesions, or OCDLs.
OCDLs are DNA lesions characterized by two or more bistranded DNA lesions within a 10 base-pair sequence [68]. OCDLs were introduced originally as locally multiply damaged sites (LMDS) in 1981 by Ward et al. [69], and have more recently garnered attention due to the possible role of OCDL’s in tumorigenesis and inflammatory disease [70]. The OCDLs can contain a variety of DNA lesions including apurinic-apyramidinic sites, SSBs, and oxidized bases. Endogenous and low levels of exogenous DNA lesions are usually repaired efficiently by BER (which involves PARP-1 activity). However, OCDLs are hypothesized to present more of a challenge to fix, due to the inability of the repair machinery to access the damaged sites within such as small area of DNA [69,71]. Therefore, inefficient repair may lead to the more serious DSB or alternatively, enhanced mutagenicity, with one study showing that 10% of non-double strand break clusters are converted to DSB in NHEJ-deficient CHO cells [72,73]. While lesions greater than 3 bp apart on opposing strands have been shown to result in a DSB upon repair, opposing lesions less than 3 bp apart have been demonstrated to inhibit lesion repair due to the formation of a SSB intermediate which interferes with and/or decreases repair efficiency [74]. In vitro work in a bacterial plasmid model has demonstrated that attempted repair of opposing oxidative DNA lesions results in a significantly higher mutation rate within 5,6-dihydrothymine (DHT) and of 8-oxo-7,8-dihydroguanine (8-oxoG) clustered lesions [75]. Additionally, other work in a similar model demonstrated that two closely opposed 8-oxoG lesions could result in higher mutation rates as the BER mechanisms are incapable of effectively repairing both lesions [74,76,77].
3.1. OCLDs and Cancer
Endogenous or spontaneous OCDLs occur normally within cells of humans and animals in the range of 200–500 clusters/Gbp [78,79,80]; however, several tumor types have been shown to harbor an increased level of OCDLs [81]. This suggests that there may be correlation between a higher risk of tumorigenesis in tissues and accumulated OCDLs. Additionally, oxidative stress is thought to contribute to tumorigenesis as many chronic inflammatory states lead to increase risk of cancer, such as H. pylori infections, hepatitis, and ulcerative colitis [1]. This is due in part to the large production of free radicals in the inflammatory environment leading to increased oxidative DNA damage and genome instability [82]. Decreased clusters were found in cells treated with selenium, a known anti-oxidant, and has been shown to protect against oxidative-induced DNA damage and cancer [79,83]. Functionally, OCDLs may contribute to tumorigenesis as they have been shown to display increased risk for mutagenicity and genetic instability due to the inherent challenges associated with their repair [76,84,85]. Furthermore, IR-induced lesions were shown to persist in mouse tissue 20 weeks post treatment, suggesting these lesions are long lasting and difficult to repair, which could lead to an increased risk of mutagenicity, and tumorigenesis [86].
3.2. PARP-1-Friend or Foe of OCDLs?
The potential roles of PARP-1 and OCLDs in tumorigenesis provide a thought-provoking paradigm where in “normal” conditions, ROS induces DNA damage, which is then repaired via the BER process that involves PARP-1 recruitment and enzymatic activity. However, in highly oxidative environments or conditions of chronic inflammation, the sustained upregulation of PARP-1 from the repair process may lead to more inflammation and ROS generation, contributing to the formation of OCDLs. This introduces an interesting question. Is PARP-1 responsible for the increased inflammation leading to genome instability (i.e., through potentiating OCDLs), or are the DNA damages causing an upregulation of PARP-1, and thereby permitting inflammation to influence the cellular environment? This resulting inflammatory feedback loop propagates the phenotype that may be contributing to tumorigenesis. There has been no work to our knowledge that addresses these questions as of yet, but it does present an intriguing area for new research. Additionally, it may provide a feasible avenue for the novel use of PARP-1 inhibitors to prevent cancer outside that of BRCA-associated tumors.
While clinical trials have been conducted for the use of PARP-1 inhibitors in the treatment of cancer, the focus of PARP-1 inhibition was concentrated on its use to obtain synthetic lethality in familial cancers, such as those with a BRCA1/2 mutated phenotypes [87,88,89]. However, this potential connection to OCDLs and inflammation may provide a wider therapeutic application for PARP-1 inhibitors as a potential preventative agent in cancers highly associated with inflammatory phenotypes. By treating with PARP-1 inhibitors, one could reduce the inflammatory process, and thereby also decrease the induction of oxidative induced lesions (like OCDLs) that may lead to cancers.
The principle behind PARP inhibitor treatment of familial cancers deficient in homologous recombination (HR) repair is that by inhibiting the BER pathway in conjunction with endogenous failure of the HR pathway, the cell will be incapable of repairing DNA damage leading to cell death. However, our lab has demonstrated that PARP-1 inhibition also kills HER2+ breast cancer cells despite proficient HR repair, indicating PARP-1 may be an effective treatment for non-familial, sporadic cancers as well as familial cancers [61]. Interestingly, we found that susceptibility of non-familial cancers to PARP-1 inhibition may be through altered NFκB signaling [61]. While the mechanisms behind this are not yet determined, it does provide evidence that the applications of PARP-1 inhibitors in the treatment of cancers may be further extended than originally thought. Therefore, it may be possible to use of PARP-1 inhibitors to not only treat familial cancers, but also to treat and even prevent cancers of non-familial origin, especially cancers associated with chronic inflammation. Chronic PARP-1 activation in tissues undergoing high metabolic activity or tissues subjected to continual ROS-induced stress may be at increased risk for developing cancer driven by PARP-1 signaling, which may be the case for the study in hepatocellular carcinoma mentioned previously which show elevated PARP-1 levels in cirrhotic patients [62]. Together this points to PARP inhibition as an attractive target not only for familial cancers, but also as a treatment, and potentially a preventative agent, for cancers in tissues or organs experiencing chronic ROS-induced damage or high levels of metabolism. A schematic of this potential divergent role of PARP-1 is shown in Figure 2.
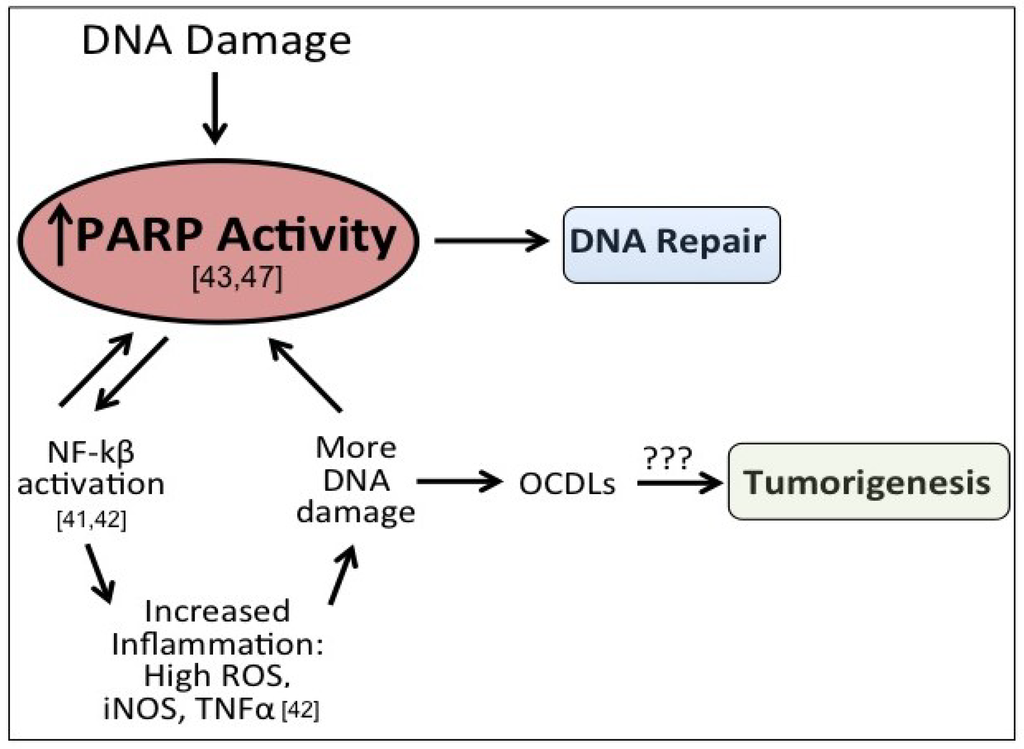
Figure 2.
Potential role of elevated PARP-1 in tumorigenesis. After DNA damage, PARP-1 activates DNA repair. However, PARP-1 also acts a co-activator of NFkB signaling, which can propogate inflammatory signaling and lead to more DNA damage, including the formation of oxidatively-clustered DNA lesions (OCDLs). The formation of OCDLs have been shown to be elevated in numerous tumor types. PARP-1 activity could potentially be beneficial or harmful in the repair of ROS-induced DNA lesions.
Figure 2.
Potential role of elevated PARP-1 in tumorigenesis. After DNA damage, PARP-1 activates DNA repair. However, PARP-1 also acts a co-activator of NFkB signaling, which can propogate inflammatory signaling and lead to more DNA damage, including the formation of oxidatively-clustered DNA lesions (OCDLs). The formation of OCDLs have been shown to be elevated in numerous tumor types. PARP-1 activity could potentially be beneficial or harmful in the repair of ROS-induced DNA lesions.
4. Conclusions
In summary, PARP-1 sits at a critical axis. PARP-1 activity not only assists in repair of ROS-induced DNA lesions, but also can contribute to further damage by promoting inflammation. As a result, upregulated PARP-1 signaling may lead to increased genetic instability and carcinogenesis. Here we have discussed the potential for using PARP-1 inhibition as treatment for not only familial cancers, but also cancers associated with chronic inflammation and high metabolism, which result in high levels of ROS. Therefore, a broader utility of PARP inhibitors likely exists, and further investigation is warranted for future clinical trials in situations of high oxidative stress such that tumor initiation may be prevented.
Acknowledgments
This work was supported by grants from the Department of Defense (ESY), the Sidney Kimmel Foundation (ESY), the American Association of Cancer Research (ESY), the Breast Cancer Research Foundation of Alabama (BRCFA) (ESY), and an NRSA T32 postdoctoral training grant (AFS).
Conflict of Interest
The authors declare no conflicts of interest.
References
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer. 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. BioEssays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Kameshita, I.; Matsuda, Z.; Taniguchi, T.; Shizuta, Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J. Biol. Chem. 1984, 259, 4770–4776. [Google Scholar]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef]
- Kraus, W.L.; Lis, J.T. PARP goes transcription. Cell 2003, 113, 677–683. [Google Scholar] [CrossRef]
- Huletsky, A.; de Murcia, G.; Muller, S.; Hengartner, M.; Menard, L.; Lamarre, D.; Poirier, G.G. The effect of poly(ADP-ribosyl)ation on native and H1-depleted chromatin. A role of poly(ADP-ribosyl)ation on core nucleosome structure. J. Biol. Chem. 1989, 264, 8878–8886. [Google Scholar]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef]
- Lin, Y.; Tang, X.; Zhu, Y.; Shu, T.; Han, X. Identification of PARP-1 as one of the transcription factors binding to the repressor element in the promoter region of COX-2. Arch. Biochem. Biophys. 2011, 505, 123–129. [Google Scholar] [CrossRef]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Luo, R.; Samara, R.; Espinoza, L.A.; Hassa, P.O.; Hottiger, M.O.; Smulson, M.E. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene 2003, 22, 8460–8471. [Google Scholar] [CrossRef]
- Kannan, P.; Yu, Y.; Wankhade, S.; Tainsky, M.A. PolyADP-ribose polymerase is a coactivator for AP-2-mediated transcriptional activation. Nucleic Acids Res. 1999, 27, 866–874. [Google Scholar] [CrossRef]
- Wesierska-Gadek, J.; Schmid, G. Poly(ADP-ribose) polymerase-1 regulates the stability of the wild-type p53 protein. Cell. Mol. Biol. Lett. 2001, 6, 117–140. [Google Scholar]
- Muller, W.E.; Zahn, R.K. Poly ADP-ribosylation of DNA-dependent RNA polymerase I from quail oviduct. Dependence on progesterone stimulation. Mol. Cell. Biochem. 1976, 12, 147–159. [Google Scholar] [CrossRef]
- Taniguchi, T.; Suzuki, S.; Shizuta, Y. Poly (ADP-ribosyl)ation of RNA polymerase II from wheat germ. Biochem. Biophys. Res. Commun. 1985, 127, 526–532. [Google Scholar] [CrossRef]
- Nie, J.; Sakamoto, S.; Song, D.; Qu, Z.; Ota, K.; Taniguchi, T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS Lett. 1998, 424, 27–32. [Google Scholar] [CrossRef]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar]
- Kameoka, M.; Ota, K.; Tetsuka, T.; Tanaka, Y.; Itaya, A.; Okamoto, T.; Yoshihara, K. Evidence for regulation of NF-kappaB by poly(ADP-ribose) polymerase. Biochem. J. 2000, 346, 641–649. [Google Scholar] [CrossRef]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef]
- Shrivastav, M.; de Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–208. [Google Scholar]
- Galande, S.; Kohwi-Shigematsu, T. Poly(ADP-ribose) polymerase and Ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J. Biol. Chem. 1999, 274, 20521–20528. [Google Scholar] [CrossRef]
- Ariumi, Y.; Masutani, M.; Copeland, T.D.; Mimori, T.; Sugimura, T.; Shimotohno, K.; Ueda, K.; Hatanaka, M.; Noda, M. Suppression of the poly(ADP-ribose) polymerase activity by DNA-dependent protein kinase in vitro. Oncogene 1999, 18, 4616–4625. [Google Scholar] [CrossRef]
- Haince, J.F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef]
- Menisser-de Murcia, J.; Mark, M.; Wendling, O.; Wynshaw-Boris, A.; de Murcia, G. Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol. Cell. Biol. 2001, 21, 1828–1832. [Google Scholar] [CrossRef]
- Henrie, M.S.; Kurimasa, A.; Burma, S.; Menissier-de Murcia, J.; de Murcia, G.; Li, G.C.; Chen, D.J. Lethality in PARP-1/Ku80 double mutant mice reveals physiological synergy during early embryogenesis. DNA Repair 2003, 2, 151–158. [Google Scholar] [CrossRef]
- Srivastava, D.K.; Berg, B.J.; Prasad, R.; Molina, J.T.; Beard, W.A.; Tomkinson, A.E.; Wilson, S.H. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J. Biol. Chem. 1998, 273, 21203–21209. [Google Scholar]
- Liu, Y.; Prasad, R.; Beard, W.A.; Kedar, P.S.; Hou, E.W.; Shock, D.D.; Wilson, S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem. 2007, 282, 13532–13541. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Kim, K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 1995, 269, 699–702. [Google Scholar]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998, 18, 3563–3571. [Google Scholar]
- De Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef]
- Trucco, C.; Oliver, F.J.; de Murcia, G.; Menissier-de Murcia, J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998, 26, 2644–2649. [Google Scholar] [CrossRef]
- Dantzer, F.; de La Rubia, G.; Menissier-De Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- Giansanti, V.; Dona, F.; Tillhon, M.; Scovassi, A.I. PARP inhibitors: New tools to protect from inflammation. Biochem. Pharmacol. 2010, 80, 1869–1877. [Google Scholar] [CrossRef]
- Bai, P.; Virag, L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 2012, 586, 3771–3777. [Google Scholar] [CrossRef]
- Liu, L.; Ke, Y.; Jiang, X.; He, F.; Pan, L.; Xu, L.; Zeng, X.; Ba, X. Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-kappaB transcription in murine macrophages. Hum. Immunol. 2012, 73, 439–447. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol. Chem. 1999, 380, 953–959. [Google Scholar]
- Oliver, F.J.; Menissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef]
- Czapski, G.A.; Adamczyk, A.; Strosznajder, R.P.; Strosznajder, J.B. Expression and activity of PARP family members in the hippocampus during systemic inflammation: Their role in the regulation of prooxidative genes. Neurochem. Int. 2013, 62, 664–673. [Google Scholar] [CrossRef]
- Filipovic, D.M.; Meng, X.; Reeves, W.B. Inhibition of PARP prevents oxidant-induced necrosis but not apoptosis in LLC-PK1 cells. Am. J. Physiol. 1999, 277, F428–F436. [Google Scholar]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef]
- Tentori, L.; Balduzzi, A.; Portarena, I.; Levati, L.; Vernole, P.; Gold, B.; Bonmassar, E.; Graziani, G. Poly (ADP-ribose) polymerase inhibitor increases apoptosis and reduces necrosis induced by a DNA minor groove binding methyl sulfonate ester. Cell Death Differ. 2001, 8, 817–828. [Google Scholar] [CrossRef]
- De Murcia, G.; de Murcia, J.M. Poly(ADP-ribose) polymerase: A molecular nick-sensor. Trends Biochem. Sci. 1994, 19, 172–176. [Google Scholar] [CrossRef]
- Lupachyk, S.; Shevalye, H.; Maksimchyk, Y.; Drel, V.R.; Obrosova, I.G. PARP inhibition alleviates diabetes-induced systemic oxidative stress and neural tissue 4-hydroxynonenal adduct accumulation: Correlation with peripheral nerve function. Free Radic. Biol. Med. 2011, 50, 1400–1409. [Google Scholar] [CrossRef]
- Oumouna-Benachour, K.; Hans, C.P.; Suzuki, Y.; Naura, A.; Datta, R.; Belmadani, S.; Fallon, K.; Woods, C.; Boulares, A.H. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: Effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation 2007, 115, 2442–2450. [Google Scholar] [CrossRef]
- Szabo, C.; Biser, A.; Benko, R.; Bottinger, E.; Susztak, K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes 2006, 55, 3004–3012. [Google Scholar] [CrossRef]
- Benko, R.; Pacher, P.; Vaslin, A.; Kollai, M.; Szabo, C. Restoration of the endothelial function in the aortic rings of apolipoprotein E deficient mice by pharmacological inhibition of the nuclear enzyme poly(ADP-ribose) polymerase. Life Sci. 2004, 75, 1255–1261. [Google Scholar] [CrossRef]
- Hans, C.P.; Zerfaoui, M.; Naura, A.S.; Troxclair, D.; Strong, J.P.; Matrougui, K.; Boulares, A.H. Thieno[2,3-c]isoquinolin-5-one, a potent poly(ADP-ribose) polymerase inhibitor, promotes atherosclerotic plaque regression in high-fat diet-fed apolipoprotein E-deficient mice: Effects on inflammatory markers and lipid content. J. Pharmacol. Exp. Ther. 2009, 329, 150–158. [Google Scholar] [CrossRef]
- Farkas, B.; Magyarlaki, M.; Csete, B.; Nemeth, J.; Rabloczky, G.; Bernath, S.; Literáti Nagy, P.; Sümegi, B. Reduction of acute photodamage in skin by topical application of a novel PARP inhibitor. Biochem. Pharmacol. 2002, 63, 921–932. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Horvath, B.; Kechrid, M.; Tanchian, G.; Rajesh, M.; Naura, A.S.; Boulares, A.H.; Pacher, P. Poly(ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic. Biol. Med. 2011, 51, 1774–1788. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Weil, M.K.; Chen, A.P. PARP inhibitor treatment in ovarian and breast cancer. Curr. Probl. Cancer 2011, 35, 7–50. [Google Scholar] [CrossRef]
- Martin-Oliva, D.; O’Valle, F.; Munoz-Gamez, J.A.; Valenzuela, M.T.; Nunez, M.I.; Aguilar, M.; Ruiz de Almodóvar, J.M.; Garcia del Moral, R.; Oliver, F.J.; et al. Crosstalk between PARP-1 and NF-kappaB modulates the promotion of skin neoplasia. Oncogene 2004, 23, 5275–5283. [Google Scholar] [CrossRef]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, J.L.; Karin, M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15852–15857. [Google Scholar] [CrossRef]
- Nowsheen, S.; Cooper, T.; Bonner, J.A.; LoBuglio, A.F.; Yang, E.S. HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res. 2012, 72, 4796–4806. [Google Scholar] [CrossRef]
- Shimizu, S.; Nomura, F.; Tomonaga, T.; Sunaga, M.; Noda, M.; Ebara, M.; Saisho, H. Expression of poly(ADP-ribose) polymerase in human hepatocellular carcinoma and analysis of biopsy specimens obtained under sonographic guidance. Oncol. Rep. 2004, 12, 821–825. [Google Scholar]
- Rojo, F.; Garcia-Parra, J.; Zazo, S.; Tusquets, I.; Ferrer-Lozano, J.; Menendez, S.; Eroles, P.; Chamizo, C.; Servitja, S.; Ramírez-Merino, N.; et al. Nuclear PARP-1 protein overexpression is associated with poor overall survival in early breast cancer. Ann. Oncol. 2012, 23, 1156–1164. [Google Scholar] [CrossRef]
- Domagala, P.; Huzarski, T.; Lubinski, J.; Gugala, K.; Domagala, W. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: Possible implications for PARP-1 inhibitor therapy. Breast Cancer Res. Treat. 2011, 127, 861–869. [Google Scholar] [CrossRef]
- Michels, J.; Vitale, I.; Galluzzi, L.; Adam, J.; Olaussen, K.A.; Kepp, O.; Senovilla, L.; Talhaoui, I.; Guegan, J.; Enot, D.P.; et al. Cisplatin Resistance Associated with PARP Hyperactivation. Cancer Res. 2013, 73, 2271–2280. [Google Scholar] [CrossRef]
- Cazzalini, O.; Dona, F.; Savio, M.; Tillhon, M.; Maccario, C.; Perucca, P.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. p21CDKN1A participates in base excision repair by regulating the activity of poly(ADP-ribose) polymerase-1. DNA Repair 2010, 9, 627–635. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Georgakilas, A.G. Processing of DNA damage clusters in human cells: Current status of knowledge. Mol. Biosyst. 2008, 4, 30–35. [Google Scholar] [CrossRef]
- Ward, J.F. Some biochemical consequences of the spatial distribution of ionizing radiation-produced free radicals. Radiat. Res. 1981, 86, 185–195. [Google Scholar] [CrossRef]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Goodhead, D.T. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar] [CrossRef]
- Gulston, M.; de Lara, C.; Jenner, T.; Davis, E.; O’Neill, P. Processing of clustered DNA damage generates additional double-strand breaks in mammalian cells post-irradiation. Nucleic Acids Res. 2004, 32, 1602–1609. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutation Res. 2011, 711, 123–133. [Google Scholar] [CrossRef]
- Malyarchuk, S.; Youngblood, R.; Landry, A.M.; Quillin, E.; Harrison, L. The mutation frequency of 8-oxo-7,8-dihydroguanine (8-oxodG) situated in a multiply damaged site: Comparison of a single and two closely opposed 8-oxodG in Escherichia coli. DNA Repair 2003, 2, 695–705. [Google Scholar] [CrossRef]
- Shikazono, N.; Pearson, C.; O’Neill, P.; Thacker, J. The roles of specific glycosylases in determining the mutagenic consequences of clustered DNA base damage. Nucleic Acids Res. 2006, 34, 3722–3730. [Google Scholar] [CrossRef]
- Malyarchuk, S.; Brame, K.L.; Youngblood, R.; Shi, R.; Harrison, L. Two clustered 8-oxo-7,8-dihydroguanine (8-oxodG) lesions increase the point mutation frequency of 8-oxodG, but do not result in double strand breaks or deletions in Escherichia coli. Nucleic Acids Res. 2004, 32, 5721–5731. [Google Scholar] [CrossRef]
- Pearson, C.G.; Shikazono, N.; Thacker, J.; O’Neill, P. Enhanced mutagenic potential of 8-oxo-7,8-dihydroguanine when present within a clustered DNA damage site. Nucleic Acids Res. 2004, 32, 263–270. [Google Scholar] [CrossRef]
- Gollapalle, E.; Wang, R.; Adetolu, R.; Tsao, D.; Francisco, D.; Sigounas, G.; Georgakilas, A.G. Detection of oxidative clustered DNA lesions in X-irradiated mouse skin tissues and human MCF-7 breast cancer cells. Radiat. Res. 2007, 167, 207–216. [Google Scholar] [CrossRef]
- Bennett, P.V.; Cuomo, N.L.; Paul, S.; Tafrov, S.T.; Sutherland, B.M. Endogenous DNA damage clusters in human skin, 3-D model, and cultured skin cells. Free Radic. Biol. Med. 2005, 39, 832–839. [Google Scholar] [CrossRef]
- Chastain, P.D., 2nd; Nakamura, J.; Swenberg, J.; Kaufman, D. Nonrandom AP site distribution in highly proliferative cells. FASEB J. 2006, 20, 2612–2614. [Google Scholar] [CrossRef]
- Nowsheen, S.; Wukovich, R.L.; Aziz, K.; Kalogerinis, P.T.; Richardson, C.C.; Panayiotidis, M.I.; Bonner, W.M.; Sedelnikova, O.A.; Georgakilas, A.G. Accumulation of oxidatively induced clustered DNA lesions in human tumor tissues. Mutat. Res. 2009, 674, 131–136. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- El-Bayoumy, K. The protective role of selenium on genetic damage and on cancer. Mutat. Res. 2001, 475, 123–139. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Bennett, P.V.; Wilson, D.M., 3rd; Sutherland, B.M. Processing of bistranded abasic DNA clusters in gamma-irradiated human hematopoietic cells. Nucleic Acids Res. 2004, 32, 5609–5620. [Google Scholar] [CrossRef]
- Singleton, B.K.; Griffin, C.S.; Thacker, J. Clustered DNA damage leads to complex genetic changes in irradiated human cells. Cancer Res. 2002, 62, 6263–6269. [Google Scholar]
- Bock, F.J.; Krumschnabel, G.; Manzl, C.; Peintner, L.; Tanzer, M.C.; Hermann-Kleiter, N.; Baier, G.; Llacuna, L.; Yelamos, J.; Villunger, A. Loss of PIDD limits NF-kappaB activation and cytokine production but not cell survival or transformation after DNA damage. Cell Death Differ. 2013, 20, 546–557. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).