Preclinical Cancer Chemoprevention Studies Using Animal Model of Inflammation-Associated Colorectal Carcinogenesis

Abstract
:Abbreviations
| ACF | aberrant crypt foci |
| AD | adenoma |
| ADC | adenocarcinoma |
| AOM | azoxymetheane |
| 5-ASA | 5-aminosalicylic acid |
| CD | Crohn’s disease |
| COX | cyclooxygenase |
| CRC | colorectal cancer |
| DMH | 1,2-dimethylhydrazine |
| DSS | dextran sodium sulfate |
| H&E | hematoxylin and eosin |
| HATs | histone acetylases |
| HCC | hepatocellular carcinoma |
| HDACs | histone decarboxylases |
| HDACIs | histone decarboxylase inhibitors |
| HIF | hypoxia-inducible factor |
| HMG-CoA | 3-hydroxy-3-methylglutary coenzyme A |
| IBD | inflammatory bowel disease |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| KAD | Kyoto Apc Delta |
| MAM | methylazoxymethanol |
| NF-κB | nuclear factor-kappaB |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| PhIP | 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine |
| PPARs | peroxisome proliferator-activated receptors |
| PSC | primary sclerosing cholangitis |
| RXR | retinoid X receptor |
| TNF | tumor necrosis factor |
| Stat3 | signal transducer and activator of transcription |
| UC | ulcerative colitis |
| UDCA | ursodeoxycholic acid |
| VPA | valproic acid |
1. Introduction
2. Development of an Inflammation-Associated CRC Model
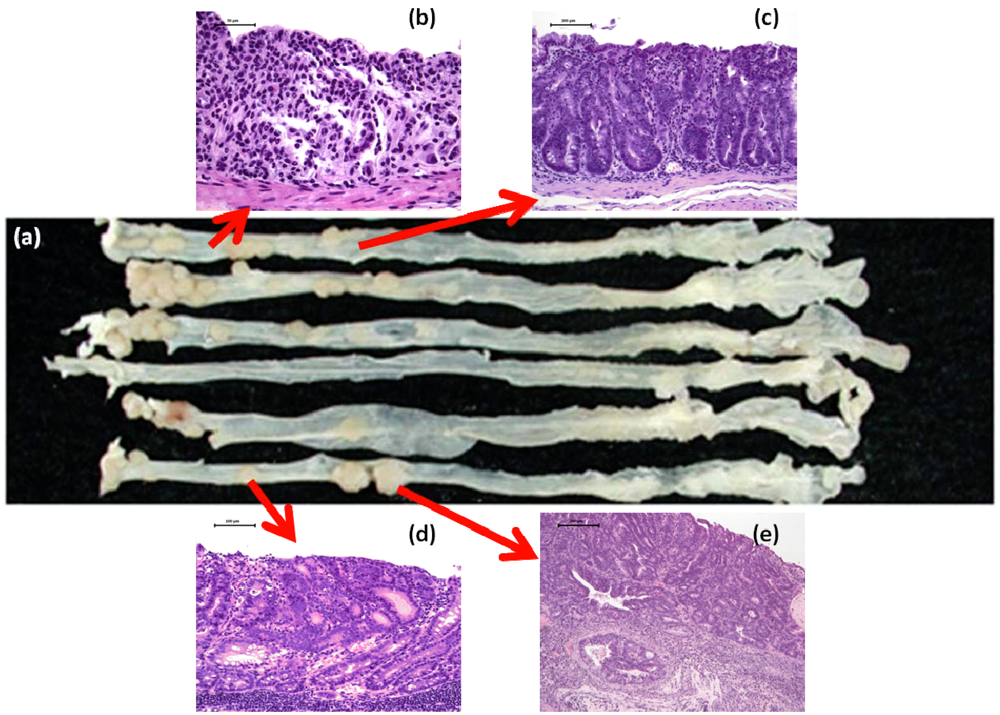
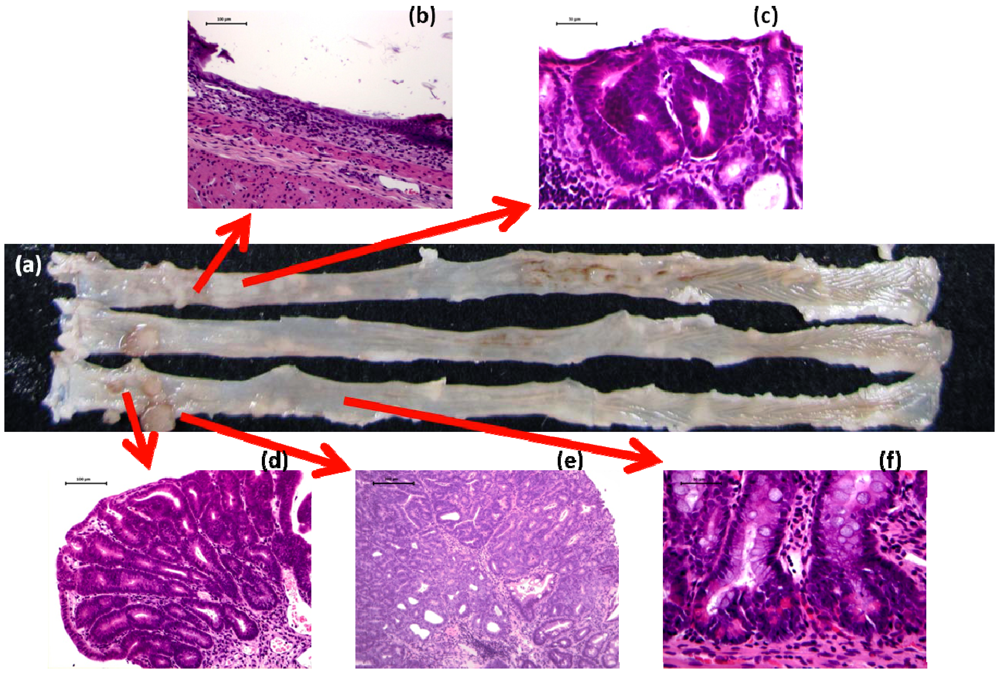
2.1. AOM/DSS Mouse Model
2.2. DMH/DSS and 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)/DSS Mouse Models
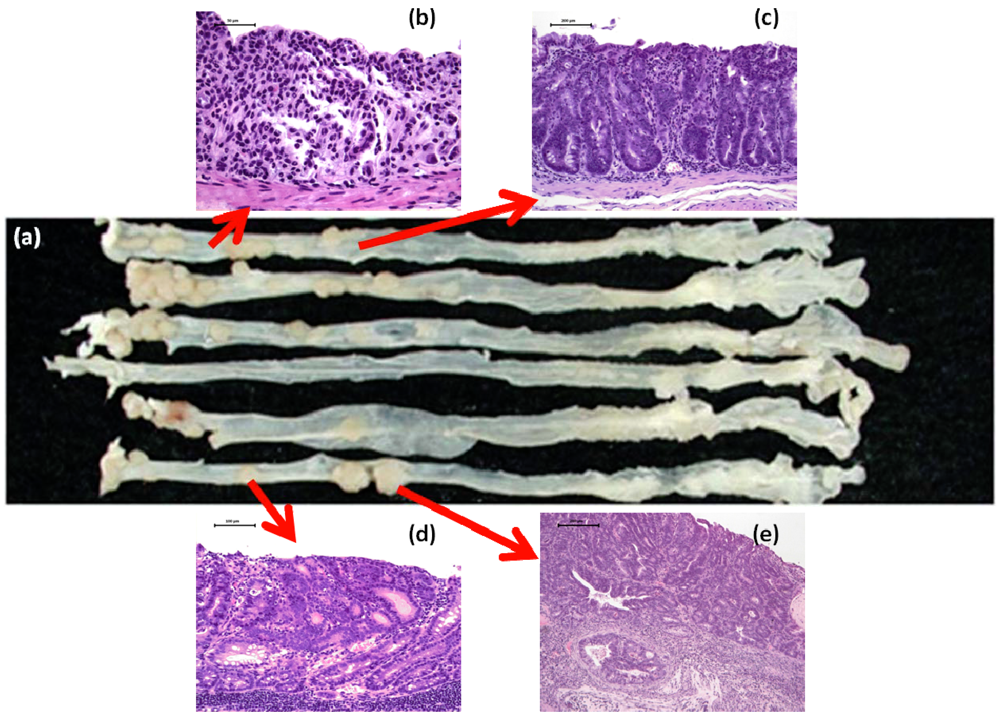
2.3. DSS Promotes CRC Development in ApcMin/+ Mice
2.4. AOM/DSS and DMH/DSS Rat Models
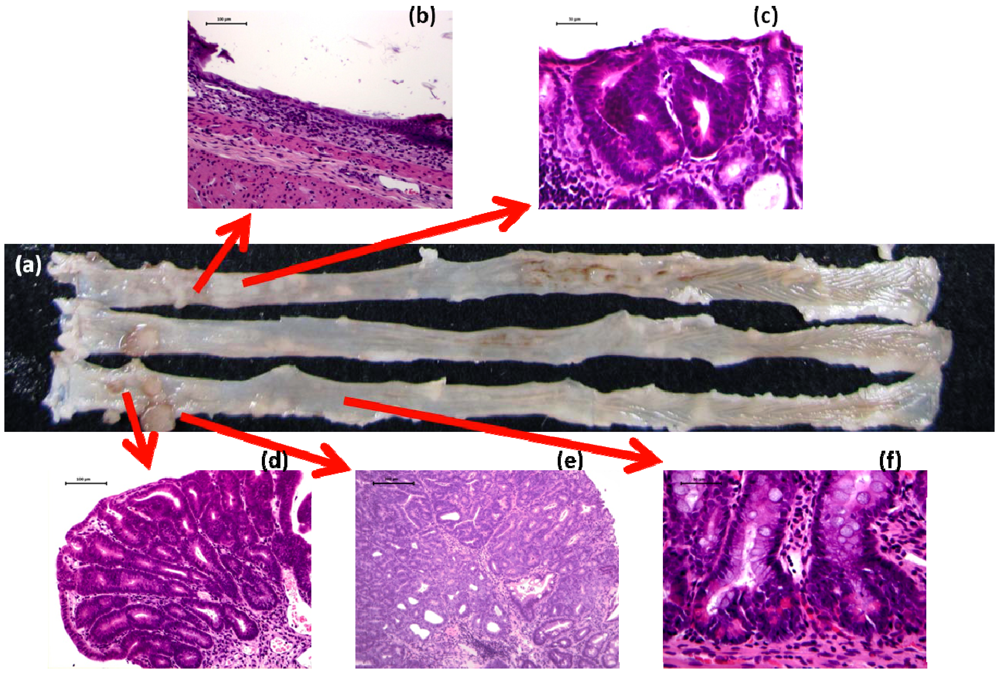
2.5. Detection of Initiators and Promoters in Colorectal Carcinogenesis
3. Exploration of Chemopreventive Agents Using an Inflammation-Associated Colorectal Carcinogenic Model and Elucidation of the Mechanisms
4. Preclinical in Vivo Chemoprevention Studies
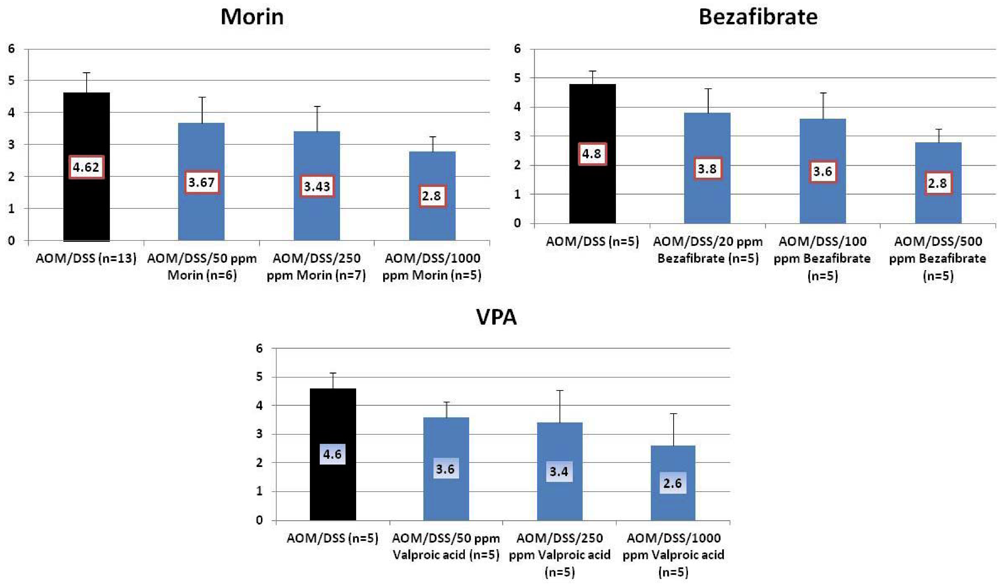
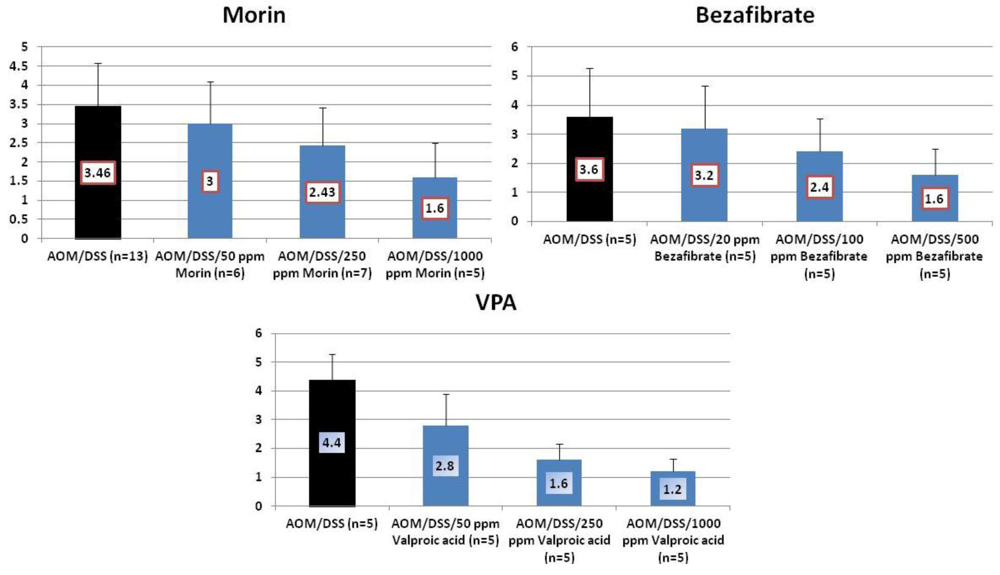
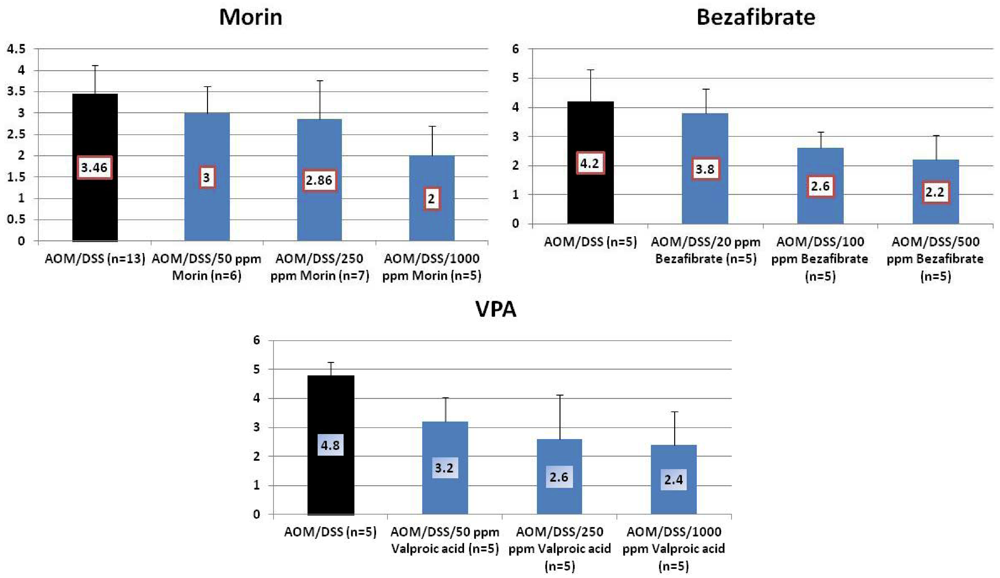
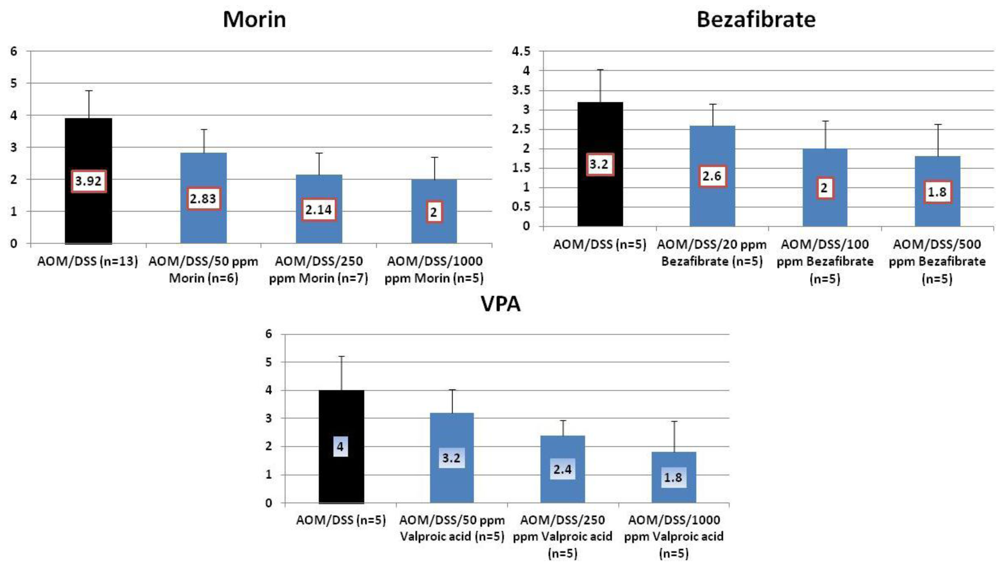
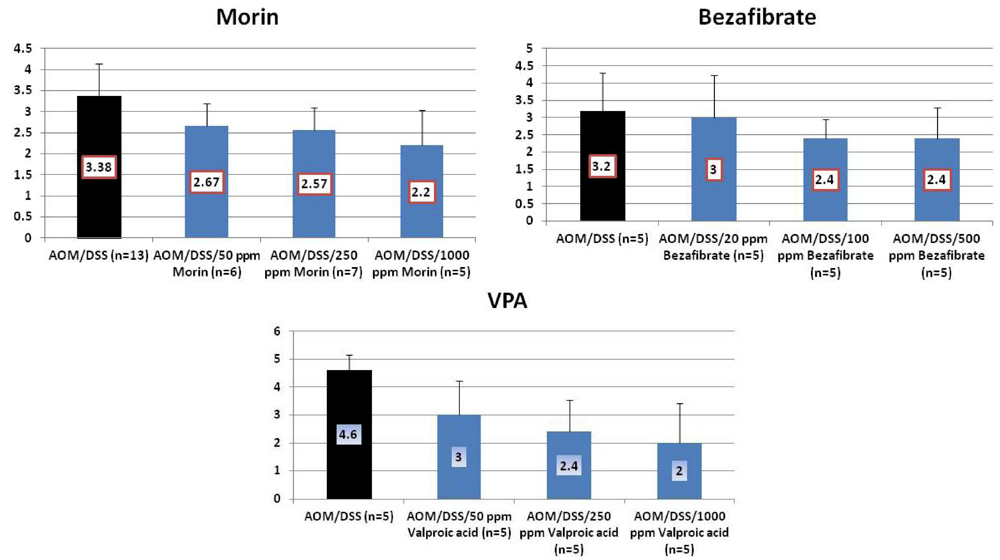
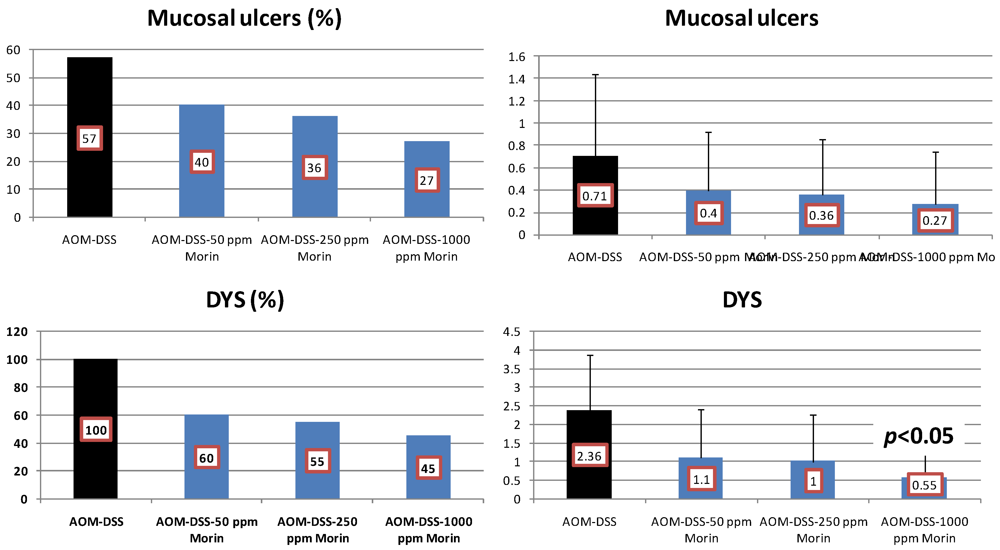
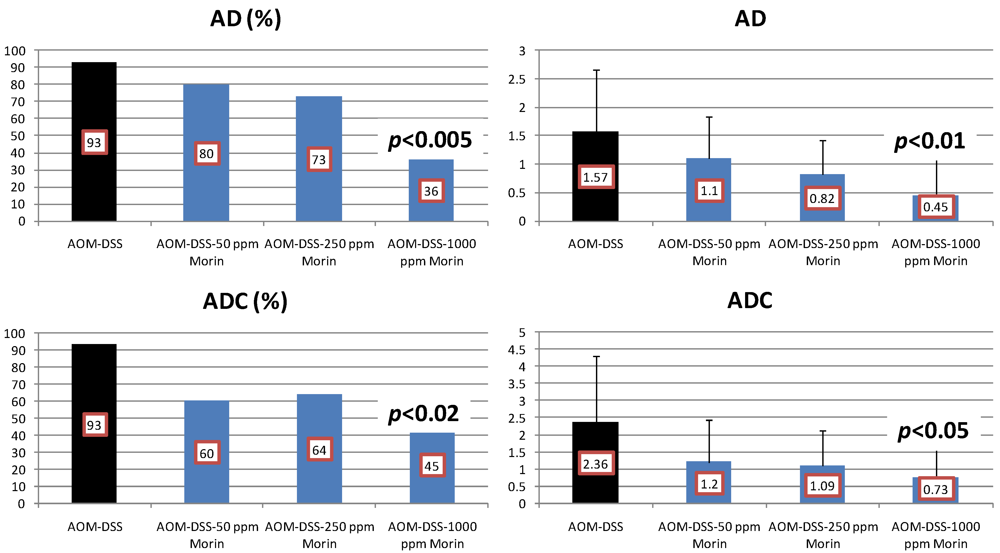
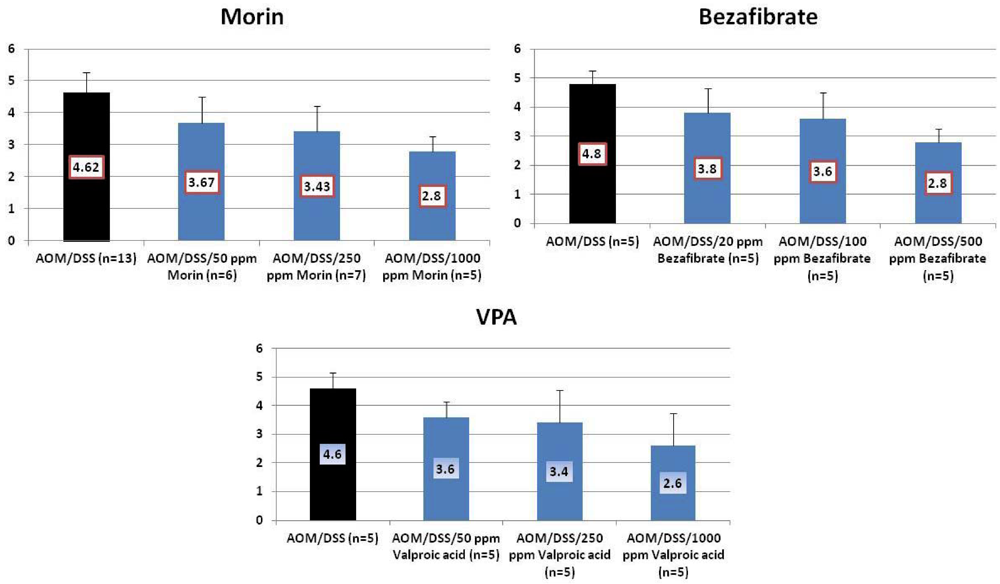
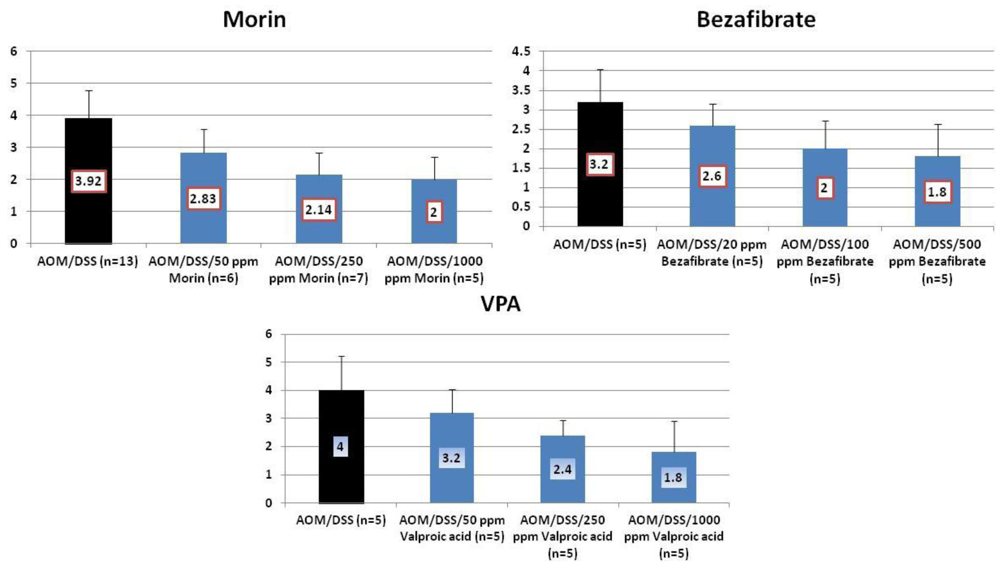
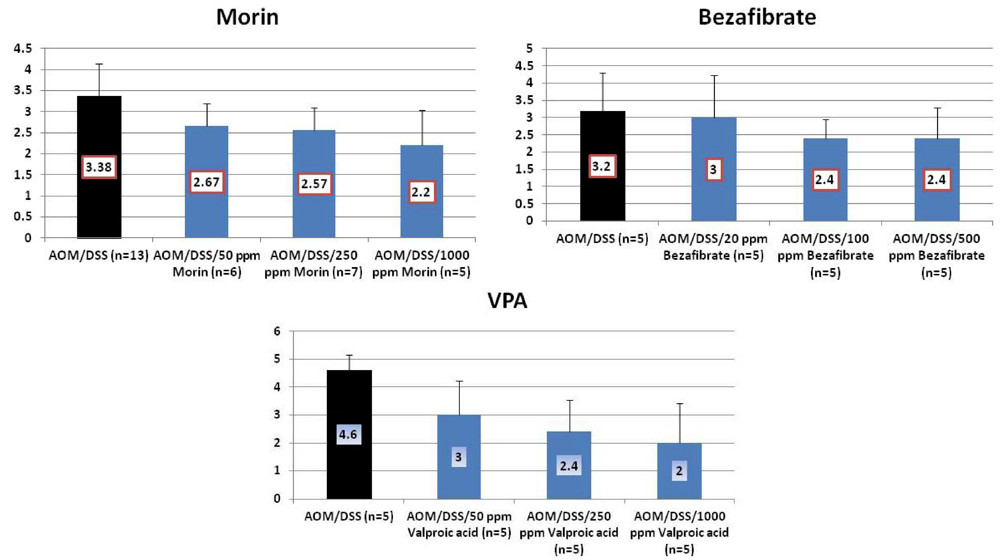
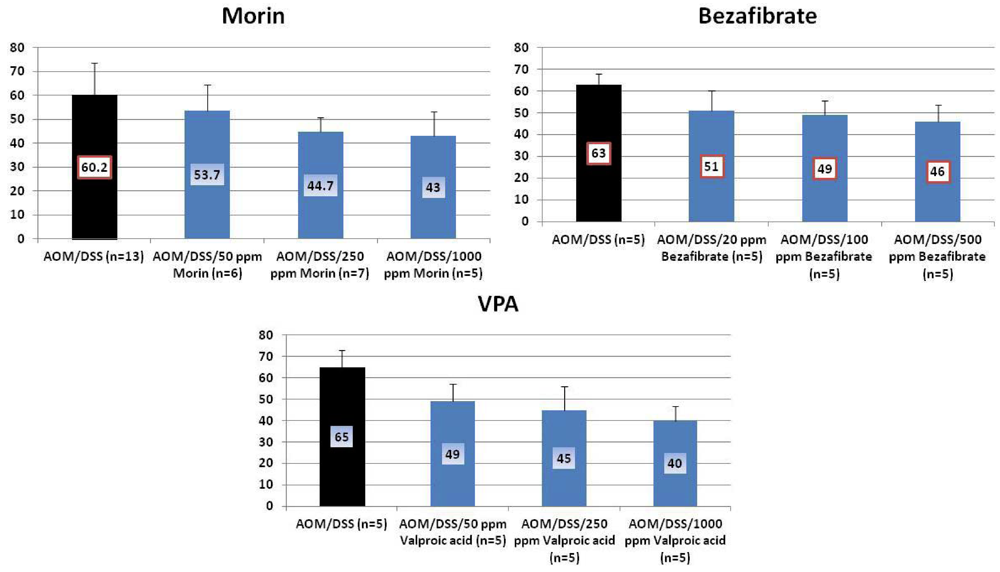
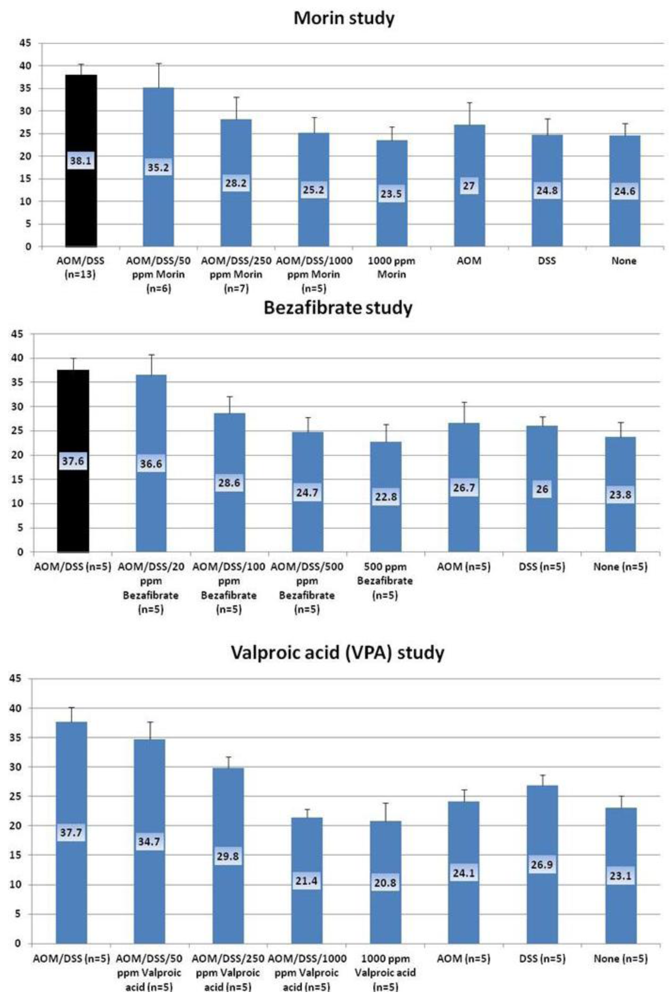
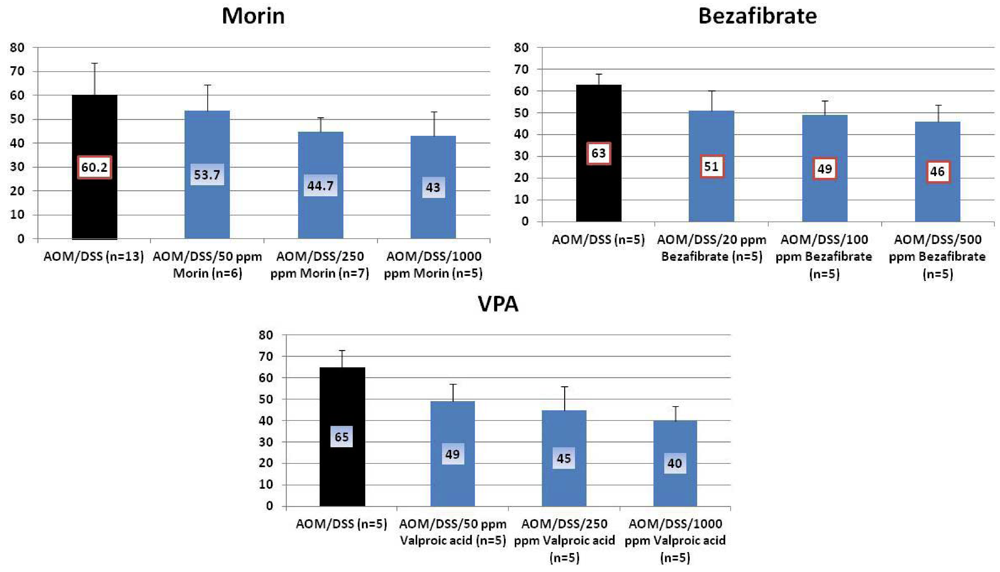
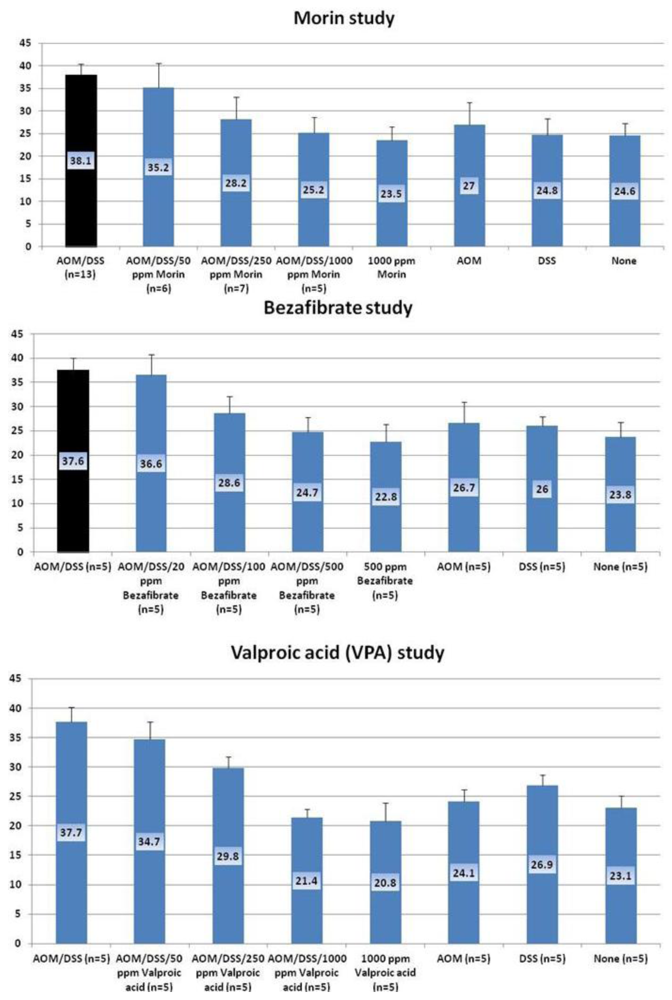
4.1. Morin Study
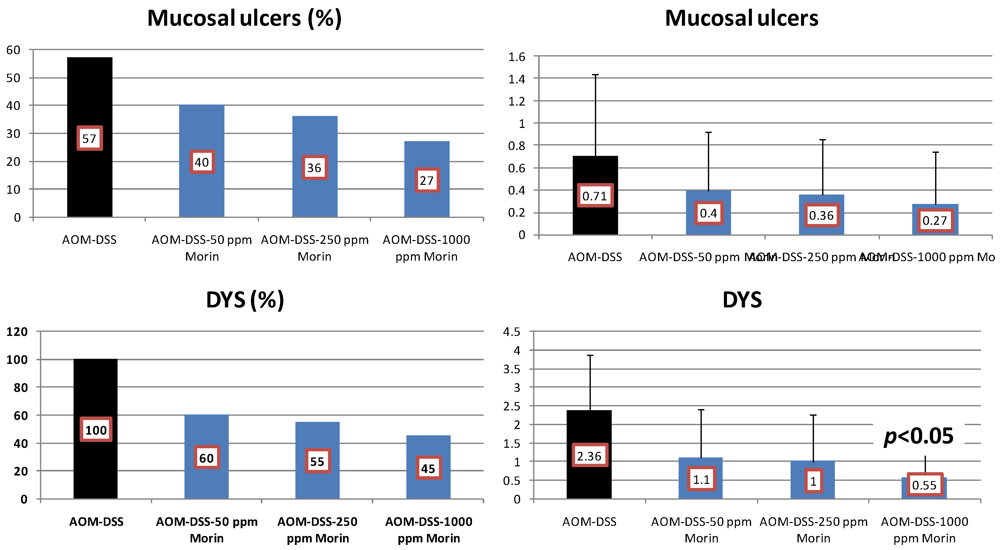
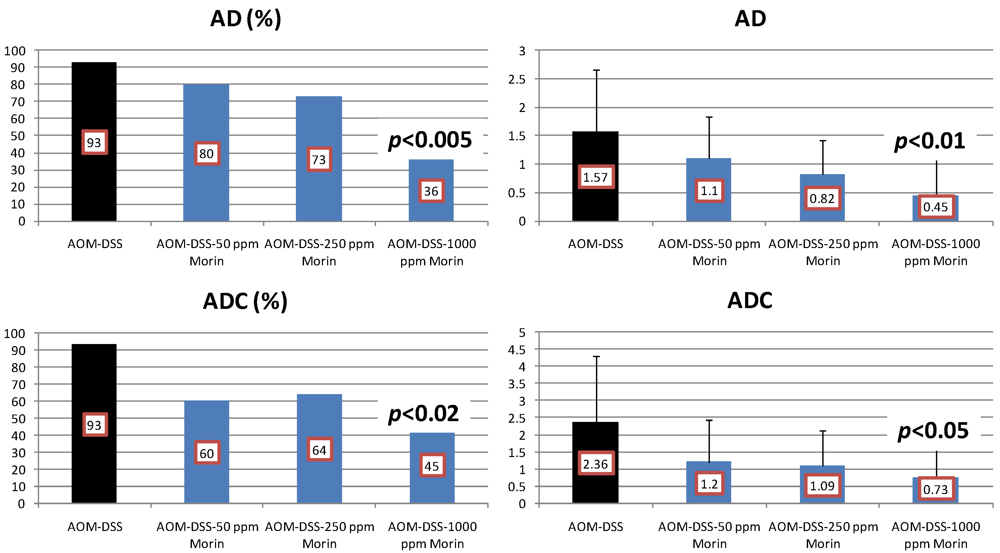
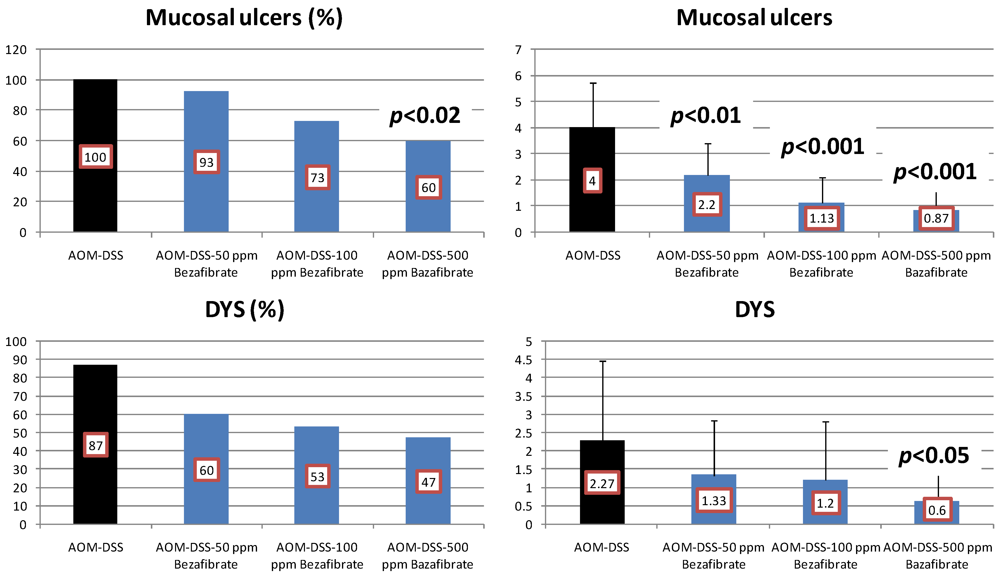
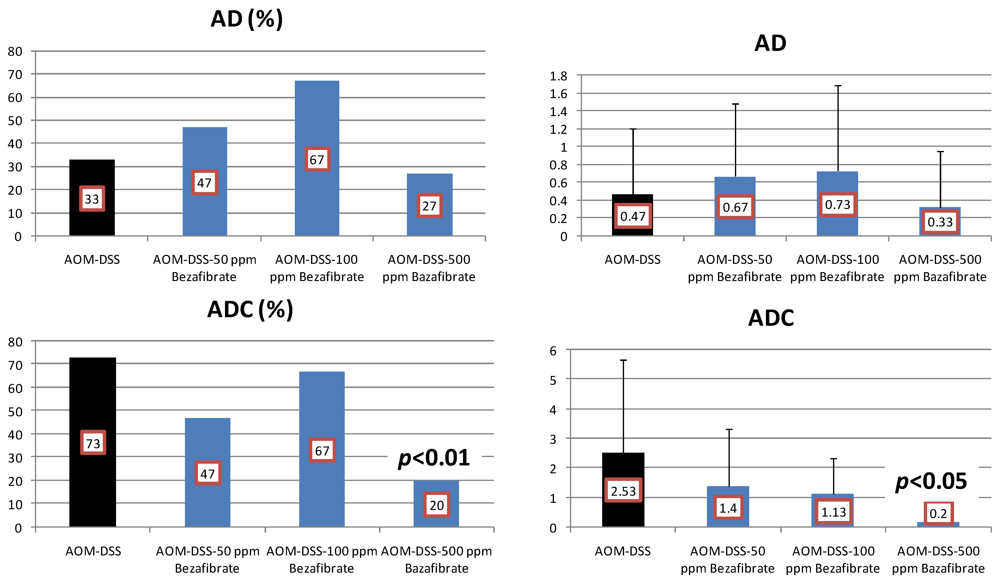
4.2. Bezafibrate Study
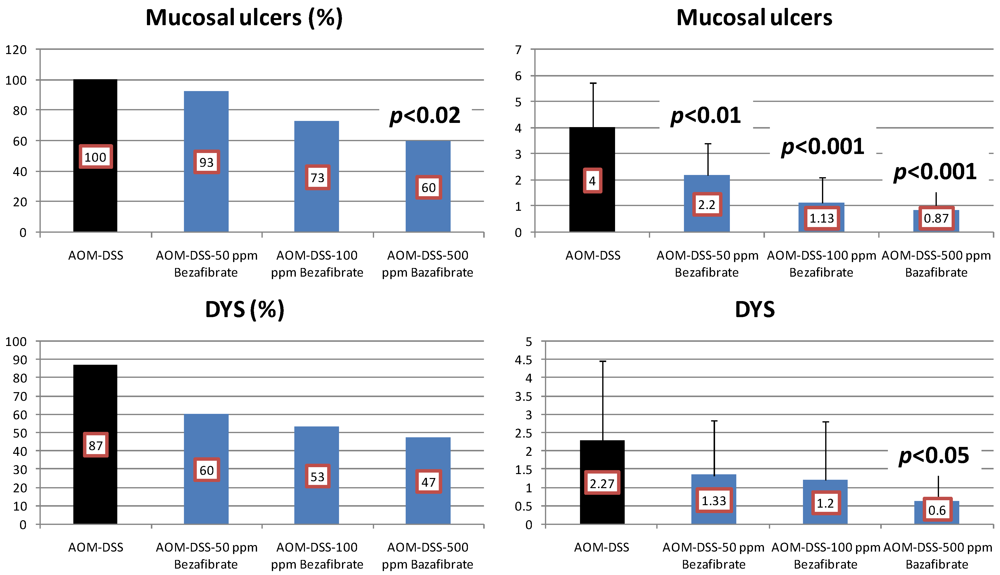
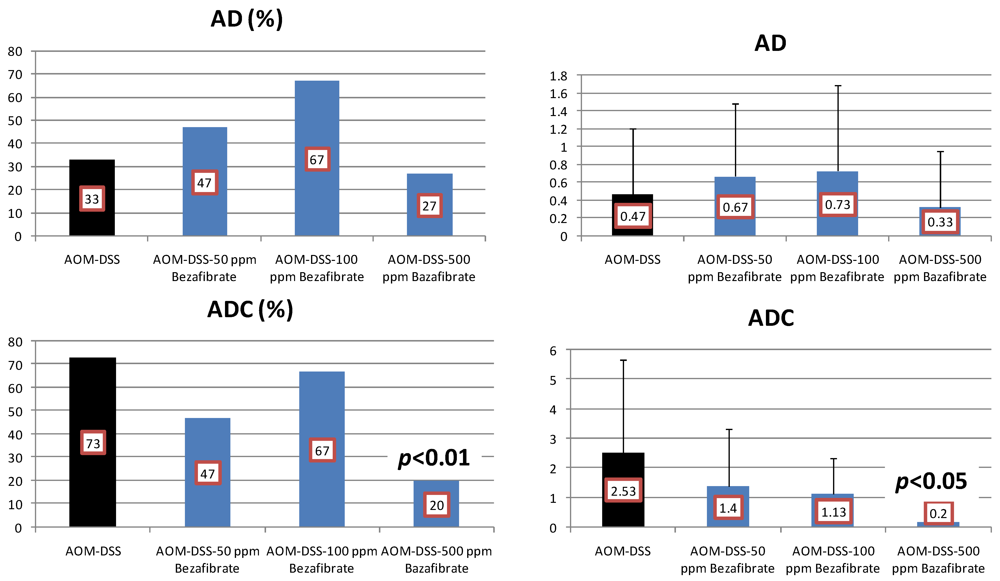
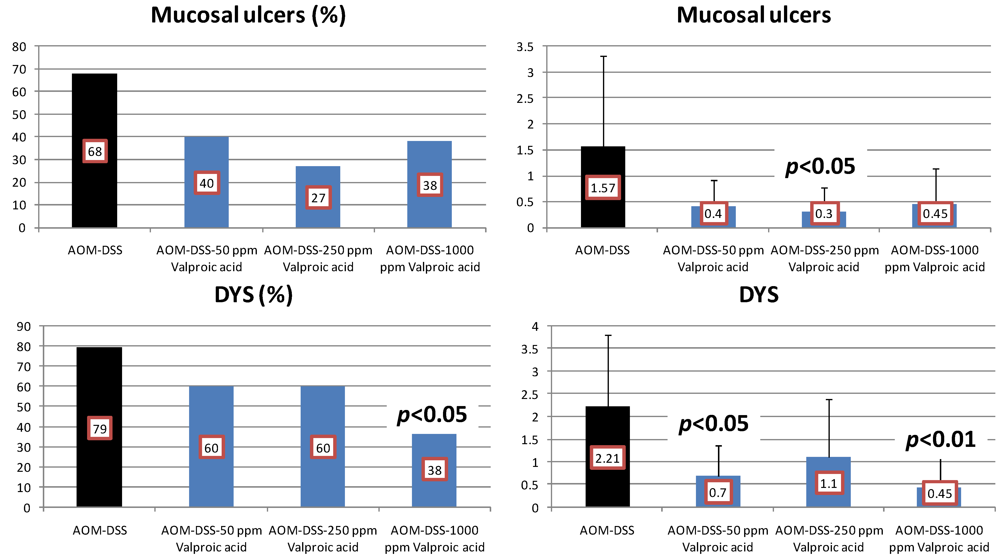
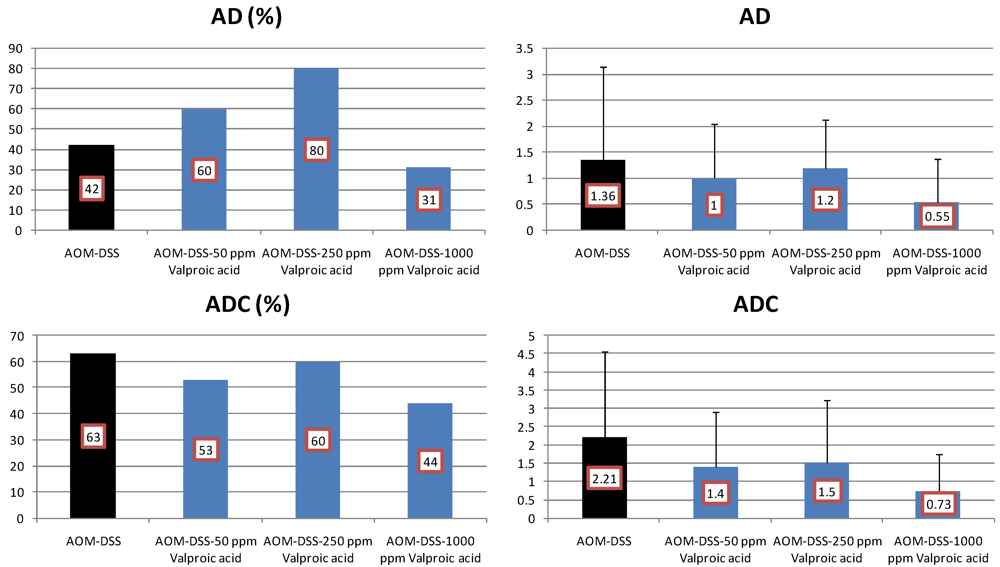
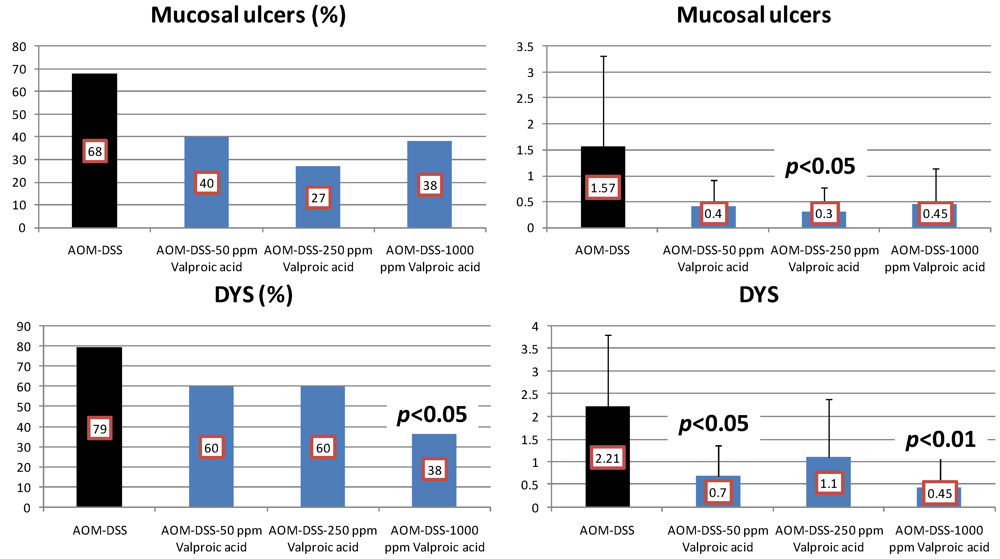
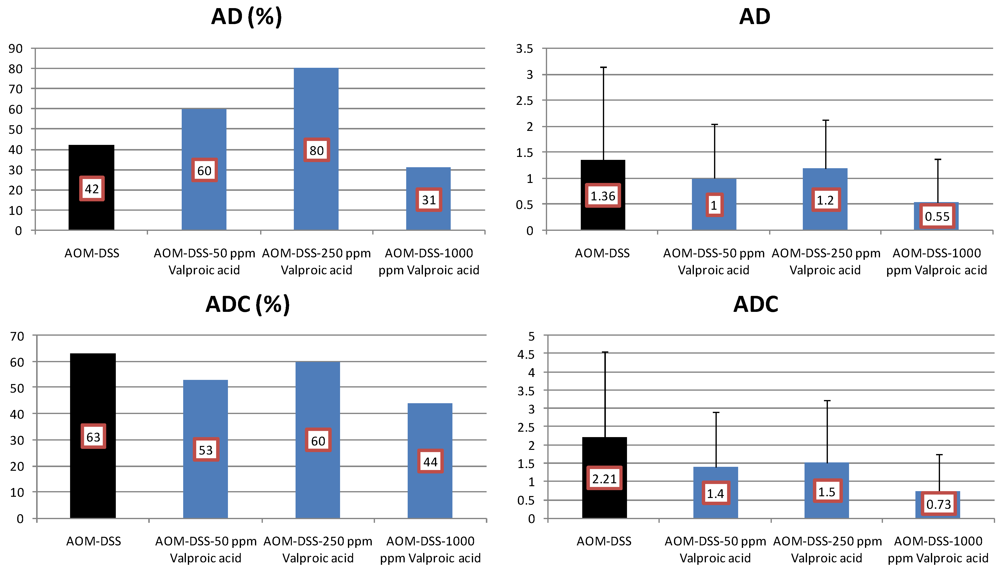
4.3. Valproic Acid (VPA) Study
5. Effects of Morin, Bezafibrate and VPA on Expression of Pro-Inflammatory Cytokines and HIF-1α and Content of Tissue Polyamines in the Inflamed Colon

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene symbol | Primers | |
|---|---|---|
| Rat | Mouse | |
| NF-κB | Forward: 5'-ctggcagctcttctcaaagc-3' | Mm00476361_m1 * |
| Reverse: 5'-ccaggtcatagagaggctcaa-3' | ||
| Tnf-α | Forward: 5'-cgagatgtggaactggcaga-3' | Mm00443258_m1 * |
| Reverse: 5'-ctacgggcttgtcactcga-3' | ||
| IL-1β | Rn00 580432_m1 * | Mm00434228_m1 * |
| Stat3 | Forward: 5'-ttgtgatgcctccttgattgtc-3' | Mm00456961_m1 * |
| Reverse: 5'-atcggaggcttagtgaagaagttc-3' | ||
| Hif-1α | Rn00577560_ml * | Forward: 5'-cctggaaacgagtgaaagga-3'Reverse: 5'-tggtcagctgtggtaatcca-3' |
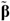 actin actin | Forward: 5'-tcaggtcatcactatcggcaat-3' | Mm00607939_s1* |
| Reverse: 5'-aaagaaagggtgtaaaacgca-3' | ||
6. Conclusions
Acknowledgements
Conflict of interest
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar]
- Tanaka, T.; Suzuki, R. Inflammation and cancer. In Cancer: Disease Progression and Chemoprevention 2007; Tanaka, T., Ed.; Research Signpost: Kerala, India, 2007; pp. 27–44. [Google Scholar]
- Lakatos, P.L.; Lakatos, L. Risk for colorectal cancer in ulcerative colitis: Changes, causes and management strategies. World J. Gastroenterol. 2008, 14, 3937–3947. [Google Scholar] [CrossRef]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G7–G17. [Google Scholar] [CrossRef]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Munkholm, P. Review article: The incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2003, 18, S1–S5. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Murakami, M.; Shimada, R.; Kagami, S. Colitis-related rat colon carcinogenesis induced by 1-hydroxy-anthraquinone and methylazoxymethanol acetate (Review). Oncol. Rep. 2000, 7, 501–508. [Google Scholar]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef]
- Sung, J.J.; Lau, J.Y.; Goh, K.L.; Leung, W.K. Increasing incidence of colorectal cancer in Asia: Implications for screening. Lancet Oncol. 2005, 6, 871–876. [Google Scholar] [CrossRef]
- Tanaka, T.; Oyama, T.; Yasui, Y. Dietary supplements and colorectal cancer. Curr. Topics Neutraceut. Res. 2008, 6, 165–188. [Google Scholar]
- Tanaka, T.; Sugie, S. Inhibition of colon carcinogenesis by dietary non-nutritive compounds. J. Toxicol. Pathol. 2007, 20, 215–235. [Google Scholar] [CrossRef]
- Yasui, Y.; Kim, M.; Oyama, T.; Tanaka, T. Colorectal carcinogenesis and suppression of tumor development by inhibition of enzymes and molecular targets. Curr. Enzym. Inhib. 2009, 5, 1–26. [Google Scholar] [CrossRef]
- Rosenberg, D.W.; Giardina, C.; Tanaka, T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 2009, 30, 183–196. [Google Scholar]
- Tanaka, T. Colorectal carcinogenesis: Review of human and experimental animal studies. J. Carcinog. 2009, 8, 5. [Google Scholar] [CrossRef]
- Tanaka, T. Development of an inflammation-associated colorectal cancer model and its application for research on carcinogenesis and chemoprevention. Int. J. Inflamm. 2012, 2012, 658786. [Google Scholar]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef]
- Tanaka, T.; Yasui, Y.; Ishigamori-Suzuki, R.; Oyama, T. Citrus compounds inhibit inflammation- and obesity-related colon carcinogenesis in mice. Nutr. Cancer 2008, 60, S70–S80. [Google Scholar] [CrossRef]
- Levine, J.S.; Burakoff, R. Chemoprophylaxis of colorectal cancer in inflammatory bowel disease: Current concepts. Inflamm. Bowel Dis. 2007, 13, 1293–1298. [Google Scholar] [CrossRef]
- Zisman, T.L.; Rubin, D.T. Colorectal cancer and dysplasia in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 2662–2669. [Google Scholar] [CrossRef]
- Das, D.; Arber, N.; Jankowski, J.A. Chemoprevention of colorectal cancer. Digestion 2007, 76, 51–67. [Google Scholar] [CrossRef]
- Pardi, D.S.; Loftus, E.V., Jr.; Kremers, W.K.; Keach, J.; Lindor, K.D. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 2003, 124, 889–893. [Google Scholar]
- Tung, B.Y.; Emond, M.J.; Haggitt, R.C.; Bronner, M.P.; Kimmey, M.B.; Kowdley, K.V.; Brentnall, T.A. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann. Intern. Med. 2001, 134, 89–95. [Google Scholar]
- Andrews, J.M.; Travis, S.P.; Gibson, P.R.; Gasche, C. Systematic review: Does concurrent therapy with 5-ASA and immunomodulators in inflammatory bowel disease improve outcomes? Aliment. Pharmacol. Ther. 2009, 29, 459–469. [Google Scholar] [CrossRef]
- Velayos, F.S.; Terdiman, J.P.; Walsh, J.M. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: A systematic review and metaanalysis of observational studies. Am. J. Gastroenterol. 2005, 100, 1345–1353. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Blanchard, J.F.; Metge, C.; Yogendran, M. Does the use of 5-aminosalicylates in inflammatory bowel disease prevent the development of colorectal cancer? Am. J. Gastroenterol. 2003, 98, 2784–2788. [Google Scholar]
- Bernstein, C.N.; Eaden, J.; Steinhart, A.H.; Munkholm, P.; Gordon, P.H. Cancer prevention in inflammatory bowel disease and the chemoprophylactic potential of 5-aminosalicylic acid. Inflamm. Bowel Dis. 2002, 8, 356–361. [Google Scholar] [CrossRef]
- Kohno, H.; Suzuki, R.; Yasui, Y.; Miyamoto, S.; Wakabayashi, K.; Tanaka, T. Ursodeoxycholic acid versus sulfasalazine in colitis-related colon carcinogenesis in mice. Clin. Cancer Res. 2007, 13, 2519–2525. [Google Scholar]
- Tanaka, T.; Shnimizu, M.; Moriwaki, H. Cancer chemoprevention by carotenoids. Molecules 2012, 17, 3202–3242. [Google Scholar]
- Takahashi, M.; Wakabayashi, K. Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci. 2004, 95, 475–480. [Google Scholar] [CrossRef]
- Boivin, G.P.; Washington, K.; Yang, K.; Ward, J.M.; Pretlow, T.P.; Russell, R.; Besselsen, D.G.; Godfrey, V.L.; Doetschman, T.; Dove, W.F.; et al. Pathology of mouse models of intestinal cancer: Consensus report and recommendations. Gastroenterology 2003, 124, 762–777. [Google Scholar]
- Clapper, M.L.; Cooper, H.S.; Chang, W.C. Dextran sulfate sodium-induced colitis-associated neoplasia: A promising model for the development of chemopreventive interventions. Acta Pharmacol. Sin. 2007, 28, 1450–1459. [Google Scholar] [CrossRef]
- Suzuki, R.; Miyamoto, S.; Yasui, Y.; Sugie, S.; Tanaka, T. Global gene expression analysis of the mouse colonic mucosa treated with azoxymethane and dextran sodium sulfate. BMC Cancer 2007, 7, 84. [Google Scholar] [CrossRef]
- Oyama, T.; Yasui, Y.; Sugie, S.; Koketsu, M.; Watanabe, K.; Tanaka, T. Dietary tricin suppresses inflammation-related colon carcinogenesis in male Crj: CD-1 mice. Cancer Prev. Res. (Phila) 2009, 2, 1031–1038. [Google Scholar] [CrossRef]
- Yasui, Y.; Tanaka, T. Protein expression analysis of inflammation-related colon carcinogenesis. J. Carcinog. 2009, 8, 10. [Google Scholar] [CrossRef]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 1993, 69, 238–249. [Google Scholar]
- Murthy, S.N.; Cooper, H.S.; Shim, H.; Shah, R.S.; Ibrahim, S.A.; Sedergran, D.J. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig. Dis. Sci. 1993, 38, 1722–1734. [Google Scholar] [CrossRef]
- Suzuki, R.; Kohno, H.; Sugie, S.; Nakagama, H.; Tanaka, T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis 2006, 27, 162–169. [Google Scholar]
- Suzuki, R.; Kohno, H.; Sugie, S.; Tanaka, T. Sequential observations on the occurrence of preneoplastic and neoplastic lesions in mouse colon treated with azoxymethane and dextran sodium sulfate. Cancer Sci. 2004, 95, 721–727. [Google Scholar] [CrossRef]
- Suzuki, R.; Kohno, H.; Sugie, S.; Tanaka, T. Dose-dependent promoting effect of dextran sodium sulfate on mouse colon carcinogenesis initiated with azoxymethane. Histol. Histopathol. 2005, 20, 483–492. [Google Scholar]
- Lefebvre, A.M.; Chen, I.; Desreumaux, P.; Najib, J.; Fruchart, J.C.; Geboes, K.; Briggs, M.; Heyman, R.; Auwerx, J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat. Med. 1998, 4, 1053–1057. [Google Scholar]
- Saez, E.; Tontonoz, P.; Nelson, M.C.; Alvarez, J.G.; Ming, U.T.; Baird, S.M.; Thomazy, V.A.; Evans, R.M. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat. Med. 1998, 4, 1058–1061. [Google Scholar] [CrossRef]
- Sarraf, P.; Mueller, E.; Jones, D.; King, F.J.; DeAngelo, D.J.; Partridge, J.B.; Holden, S.A.; Chen, L.B.; Singer, S.; Fletcher, C.; et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat. Med. 1998, 4, 1046–1052. [Google Scholar]
- Hirono, I.; Kuhara, K.; Hosaka, S.; Tomizawa, S.; Golberg, L. Induction of intestinal tumors in rats by dextran sulfate sodium. J. Natl. Cancer Inst. 1981, 66, 579–583. [Google Scholar]
- Mori, H.; Ohbayashi, F.; Hirono, I.; Shimada, T.; Williams, G.M. Absence of genotoxicity of the carcinogenic sulfated polysaccharides carrageenan and dextran sulfate in mammalian DNA repair and bacterial mutagenicity assays. Nutr. Cancer 1984, 6, 92–97. [Google Scholar]
- Alrawi, S.J.; Schiff, M.; Carroll, R.E.; Dayton, M.; Gibbs, J.F.; Kulavlat, M.; Tan, D.; Berman, K.; Stoler, D.L.; Anderson, G.R. Aberrant crypt foci. Anticancer Res. 2006, 26, 107–119. [Google Scholar]
- Bird, R.P. Role of aberrant crypt foci in understanding the pathogenesis of colon cancer. Cancer Lett. 1995, 93, 55–71. [Google Scholar] [CrossRef]
- Gupta, A.K.; Pretlow, T.P.; Schoen, R.E. Aberrant crypt foci: What we know and what we need to know. Clin. Gastroenterol. Hepatol. 2007, 5, 526–533. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Yoshitani, S.; Takashima, S.; Okumura, A.; Murakami, A.; Hosokawa, M. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res. 2001, 61, 2424–2428. [Google Scholar]
- Kohno, H.; Suzuki, R.; Sugie, S.; Tanaka, T. Beta-Catenin mutations in a mouse model of inflammation-related colon carcinogenesis induced by 1,2-dimethylhydrazine and dextran sodium sulfate. Cancer Sci. 2005, 96, 69–76. [Google Scholar] [CrossRef]
- Tanaka, T.; Suzuki, R.; Kohno, H.; Sugie, S.; Takahashi, M.; Wakabayashi, K. Colonic adenocarcinomas rapidly induced by the combined treatment with 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and dextran sodium sulfate in male ICR mice possess beta-catenin gene mutations and increases immunoreactivity for beta-catenin, cyclooxygenase-2 and inducible nitric oxide synthase. Carcinogenesis 2005, 26, 229–238. [Google Scholar]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Hata, K.; Sugie, S.; Niho, N.; Sakano, K.; Takahashi, M.; Wakabayashi, K. Dextran sodium sulfate strongly promotes colorectal carcinogenesis in Apc(Min/+) mice: Inflammatory stimuli by dextran sodium sulfate results in development of multiple colonic neoplasms. Int. J. Cancer 2006, 118, 25–34. [Google Scholar] [CrossRef]
- Yamada, Y.; Hata, K.; Hirose, Y.; Hara, A.; Sugie, S.; Kuno, T.; Yoshimi, N.; Tanaka, T.; Mori, H. Microadenomatous lesions involving loss of Apc heterozygosity in the colon of adult Apc(Min/+) mice. Cancer Res. 2002, 62, 6367–6370. [Google Scholar]
- Tanaka, T.; Yasui, Y.; Tanaka, M.; Oyama, T.; Rahman, K.M. Melatonin suppresses AOM/DSS-induced large bowel oncogenesis in rats. Chem. Biol. Interact. 2009, 177, 128–136. [Google Scholar] [CrossRef]
- Toyoda-Hokaiwado, N.; Yasui, Y.; Muramatsu, M.; Masumura, K.; Takamune, M.; Yamada, M.; Ohta, T.; Tanaka, T.; Nohmi, T. Chemopreventive effects of silymarin against 1,2-dimethylhydrazine plus dextran sodium sulfate-induced inflammation-associated carcinogenicity and genotoxicity in the colon of gpt delta rats. Carcinogenesis 2011. [Google Scholar] [CrossRef]
- Yoshimi, K.; Tanaka, T.; Takizawa, A.; Kato, M.; Hirabayashi, M.; Mashimo, T.; Serikawa, T.; Kuramoto, T. Enhanced colitis-associated colon carcinogenesis in a novel Apc mutant rat. Cancer Sci. 2009, 100, 2022–2027. [Google Scholar] [CrossRef]
- Toyoda-Hokaiwado, N.; Yasui, Y.; Muramatsu, M.; Masumura, K.; Takamune, M.; Yamada, M.; Ohta, T.; Tanaka, T.; Nohmi, T. Chemopreventive effects of silymarin against 1,2-dimethylhydrazine plus dextran sodium sulfate-induced inflammation-associated carcinogenicity and genotoxicity in the colon of gpt delta rats. Carcinogenesis 2011, 32, 1512–1517. [Google Scholar]
- Kohno, H.; Totsuka, Y.; Yasui, Y.; Suzuki, R.; Sugie, S.; Wakabayashi, K.; Tanaka, T. Tumor-initiating potency of a novel heterocyclic amine, aminophenylnorharman in mouse colonic carcinogenesis model. Int. J. Cancer 2007, 121, 1659–1664. [Google Scholar] [CrossRef]
- Hata, K.; Tanaka, T.; Kohno, H.; Suzuki, R.; Qiang, S.H.; Kuno, T.; Hirose, Y.; Hara, A.; Mori, H. Lack of enhancing effects of degraded lambda-carrageenan on the development of beta-catenin-accumulated crypts in male DBA/2J mice initiated with azoxymethane. Cancer Lett. 2006, 238, 69–75. [Google Scholar] [CrossRef]
- Kohno, H.; Suzuki, R.; Curini, M.; Epifano, F.; Maltese, F.; Gonzales, S.P.; Tanaka, T. Dietary administration with prenyloxycoumarins, auraptene and collinin, inhibits colitis-related colon carcinogenesis in mice. Int. J. Cancer 2006, 118, 2936–2942. [Google Scholar] [CrossRef]
- Kim, M.; Murakami, A.; Miyamoto, S.; Tanaka, T.; Ohigashi, H. The modifying effects of green tea polyphenols on acute colitis and inflammation-associated colon carcinogenesis in male ICR mice. Biofactors 2010, 36, 43–51. [Google Scholar]
- Kohno, H.; Suzuki, R.; Sugie, S.; Tanaka, T. Suppression of colitis-related mouse colon carcinogenesis by a COX-2 inhibitor and PPAR ligands. BMC Cancer 2005, 5, 46. [Google Scholar] [CrossRef]
- Kohno, H.; Takahashi, M.; Yasui, Y.; Suzuki, R.; Miyamoto, S.; Kamanaka, Y.; Naka, M.; Maruyama, T.; Wakabayashi, K.; Tanaka, T. A specific inducible nitric oxide synthase inhibitor, ONO-1714 attenuates inflammation-related large bowel carcinogenesis in male Apc(Min/+) mice. Int. J. Cancer 2007, 121, 506–513. [Google Scholar] [CrossRef]
- Yasui, Y.; Suzuki, R.; Miyamoto, S.; Tsukamoto, T.; Sugie, S.; Kohno, H.; Tanaka, T. A lipophilic statin, pitavastatin, suppresses inflammation-associated mouse colon carcinogenesis. Int. J. Cancer 2007, 121, 2331–2339. [Google Scholar] [CrossRef]
- Chen, Y.C.; Shen, S.C.; Chow, J.M.; Ko, C.H.; Tseng, S.W. Flavone inhibition of tumor growth via apoptosis in vitro and in vivo. Int. J. Oncol. 2004, 25, 661–670. [Google Scholar]
- Galvez, J.; Coelho, G.; Crespo, M.E.; Cruz, T.; Rodriguez-Cabezas, M.E.; Concha, A.; Gonzalez, M.; Zarzuelo, A. Intestinal anti-inflammatory activity of morin on chronic experimental colitis in the rat. Aliment. Pharmacol. Ther. 2001, 15, 2027–2039. [Google Scholar] [CrossRef]
- Ocete, M.A.; Galvez, J.; Crespo, M.E.; Cruz, T.; Gonzalez, M.; Torres, M.I.; Zarzuelo, A. Effects of morin on an experimental model of acute colitis in rats. Pharmacology 1998, 57, 261–270. [Google Scholar] [CrossRef]
- Tanaka, T.; Kawabata, K.; Honjo, S.; Kohno, H.; Murakami, M.; Shimada, R.; Matsunaga, K.; Yamada, Y.; Shimizu, M. Inhibition of azoxymethane-induced aberrant crypt foci in rats by natural compounds, caffeine, quercetin and morin. Oncol. Rep. 1999, 6, 1333–1340. [Google Scholar]
- Kawabata, K.; Tanaka, T.; Honjo, S.; Kakumoto, M.; Hara, A.; Makita, H.; Tatematsu, N.; Ushida, J.; Tsuda, H.; Mori, H. Chemopreventive effect of dietary flavonoid morin on chemically induced rat tongue carcinogenesis. Int. J. Cancer 1999, 83, 381–386. [Google Scholar] [CrossRef]
- Fang, S.H.; Hou, Y.C.; Chang, W.C.; Hsiu, S.L.; Chao, P.D.; Chiang, B.L. Morin sulfates/glucuronides exert anti-inflammatory activity on activated macrophages and decreased the incidence of septic shock. Life Sci. 2003, 74, 743–756. [Google Scholar] [CrossRef]
- Tanaka, T.; Kawabata, K.; Kakumoto, M.; Hara, A.; Murakami, A.; Kuki, W.; Takahashi, Y.; Yonei, H.; Maeda, M.; Ota, T.; et al. Citrus auraptene exerts dose-dependent chemopreventive activity in rat large bowel tumorigenesis: The inhibition correlates with suppression of cell proliferation and lipid peroxidation and with induction of phase II drug-metabolizing enzymes. Cancer Res. 1998, 58, 2550–2556. [Google Scholar]
- Yasui, Y.; Hosokawa, M.; Mikami, N.; Miyashita, K.; Tanaka, T. Dietary astaxanthin inhibits colitis and colitis-associated colon carcinogenesis in mice via modulation of the inflammatory cytokines. Chemico. Biol. Interact. 2011, 193, 79–87. [Google Scholar] [CrossRef]
- Manna, S.K.; Aggarwal, R.S.; Sethi, G.; Aggarwal, B.B.; Ramesh, G.T. Morin (3,5,7,2',4'-Pentahydroxyflavone) abolishes nuclear factor-kappaB activation induced by various carcinogens and inflammatory stimuli, leading to suppression of nuclear factor-kappaB-regulated gene expression and up-regulation of apoptosis. Clin. Cancer Res. 2007, 13, 2290–2297. [Google Scholar] [CrossRef]
- Giovannucci, E. Insulin and colon cancer. Cancer Causes Control 1995, 6, 164–179. [Google Scholar] [CrossRef]
- Mutoh, M.; Niho, N.; Wakabayashi, K. Concomitant suppression of hyperlipidemia and intestinal polyp formation by increasing lipoprotein lipase activity in Apc-deficient mice. Biol. Chem. 2006, 387, 381–385. [Google Scholar] [CrossRef]
- Tabuchi, M.; Kitayama, J.; Nagawa, H. Hypertriglyceridemia is positively correlated with the development of colorectal tubular adenoma in Japanese men. World J. Gastroenterol. 2006, 12, 1261–1264. [Google Scholar]
- Matthiessen, M.W.; Pedersen, G.; Albrektsen, T.; Adamsen, S.; Fleckner, J.; Brynskov, J. Peroxisome proliferator-activated receptor expression and activation in normal human colonic epithelial cells and tubular adenomas. Scand. J. Gastroenterol. 2005, 40, 198–205. [Google Scholar]
- Niho, N.; Takahashi, M.; Kitamura, T.; Shoji, Y.; Itoh, M.; Noda, T.; Sugimura, T.; Wakabayashi, K. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res. 2003, 63, 6090–6095. [Google Scholar]
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnes, C.M.; Fannon, M.; Laforme, A.M.; Chaponis, D.M.; Folkman, J.; Kieran, M.W. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990. [Google Scholar]
- Xiao, S.; Anderson, S.P.; Swanson, C.; Bahnemann, R.; Voss, K.A.; Stauber, A.J.; Corton, J.C. Activation of peroxisome proliferator-activated receptor alpha enhances apoptosis in the mouse liver. Toxicol. Sci. 2006, 92, 368–377. [Google Scholar] [CrossRef]
- Bruce, W.R.; Wolever, T.M.; Giacca, A. Mechanisms linking diet and colorectal cancer: The possible role of insulin resistance. Nutr. Cancer 2000, 37, 19–26. [Google Scholar] [CrossRef]
- Colangelo, L.A.; Gapstur, S.M.; Gann, P.H.; Dyer, A.R.; Liu, K. Colorectal cancer mortality and factors related to the insulin resistance syndrome. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 385–391. [Google Scholar]
- Cowey, S.; Hardy, R.W. The metabolic syndrome: A high-risk state for cancer? Am. J. Pathol. 2006, 169, 1505–1522. [Google Scholar] [CrossRef]
- Giovannucci, E. Insulin, insulin-like growth factors and colon cancer: A review of the evidence. J. Nutr. 2001, 131, 3109S–3120S. [Google Scholar]
- Thompson, E.A. PPARgamma physiology and pathology in gastrointestinal epithelial cells. Mol. Cells 2007, 24, 167–176. [Google Scholar]
- Yasui, Y.; Kim, M.; Tanaka, T. PPAR Ligands for Cancer Chemoprevention. PPAR Res. 2008, 2008, 548919. [Google Scholar]
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From orphan receptors to drug discovery. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef]
- Peters, J.M.; Aoyama, T.; Burns, A.M.; Gonzalez, F.J. Bezafibrate is a dual ligand for PPARalpha and PPARbeta: Studies using null mice. Biochim. Biophys. Acta 2003, 1632, 80–89. [Google Scholar]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef]
- Robillard, R.; Fontaine, C.; Chinetti, G.; Fruchart, J.C.; Staels, B. Fibrates. Handb. Exp. Pharmacol. 2005, 170, 389–406. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z.; Schwammenthal, E.; Adler, Y.; Goldenberg, I.; Leor, J.; Boyko, V.; Mandelzweig, L.; Behar, S. Peroxisome proliferator-activated receptor ligand bezafibrate for prevention of type 2 diabetes mellitus in patients with coronary artery disease. Circulation 2004, 109, 2197–2202. [Google Scholar]
- Tenenbaum, A.; Boyko, V.; Fisman, E.Z.; Goldenberg, I.; Adler, Y.; Feinberg, M.S.; Motro, M.; Tanne, D.; Shemesh, J.; Schwammenthal, E.; et al. Does the lipid-lowering peroxisome proliferator-activated receptors ligand bezafibrate prevent colon cancer in patients with coronary artery disease? Cardiovasc. Diabetol. 2008, 7, 18. [Google Scholar] [CrossRef]
- Cang, S.; Ma, Y.; Liu, D. New clinical developments in histone deacetylase inhibitors for epigenetic therapy of cancer. J. Hematol. Oncol. 2009, 2, 22. [Google Scholar] [CrossRef]
- Tanji, N.; Ozawa, A.; Kikugawa, T.; Miura, N.; Sasaki, T.; Azuma, K.; Yokoyama, M. Potential of histone deacetylase inhibitors for bladder cancer treatment. Expert Rev. Anticancer Ther. 2011, 11, 959–965. [Google Scholar] [CrossRef]
- Wanczyk, M.; Roszczenko, K.; Marcinkiewicz, K.; Bojarczuk, K.; Kowara, M.; Winiarska, M. HDACi—Going through the mechanisms. Front. Biosci. 2011, 16, 340–359. [Google Scholar] [CrossRef]
- Blaheta, R.A.; Cinatl, J., Jr. Anti-tumor mechanisms of valproate: A novel role for an old drug. Med. Res. Rev. 2002, 22, 492–511. [Google Scholar] [CrossRef]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Milutinovic, S.; D’Alessio, A.C.; Detich, N.; Szyf, M. Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis 2007, 28, 560–571. [Google Scholar]
- Kuendgen, A.; Schmid, M.; Schlenk, R.; Knipp, S.; Hildebrandt, B.; Steidl, C.; Germing, U.; Haas, R.; Dohner, H.; Gattermann, N. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer 2006, 106, 112–119. [Google Scholar]
- Tatebe, H.; Shimizu, M.; Shirakami, Y.; Sakai, H.; Yasuda, Y.; Tsurumi, H.; Moriwaki, H. Acyclic retinoid synergises with valproic acid to inhibit growth in human hepatocellular carcinoma cells. Cancer Lett. 2009, 285, 210–217. [Google Scholar] [CrossRef]
- Drexler, H.C.; Euler, M. Synergistic apoptosis induction by proteasome and histone deacetylase inhibitors is dependent on protein synthesis. Apoptosis 2005, 10, 743–758. [Google Scholar] [CrossRef]
- Emanuele, S.; Lauricella, M.; Carlisi, D.; Vassallo, B.; D’Anneo, A.; di Fazio, P.; Vento, R.; Tesoriere, G. SAHA induces apoptosis in hepatoma cells and synergistically interacts with the proteasome inhibitor Bortezomib. Apoptosis 2007, 12, 1327–1338. [Google Scholar]
- Fandy, T.E.; Shankar, S.; Ross, D.D.; Sausville, E.; Srivastava, R.K. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia 2005, 7, 646–657. [Google Scholar] [CrossRef]
- Yu, C.; Rahmani, M.; Conrad, D.; Subler, M.; Dent, P.; Grant, S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood 2003, 102, 3765–3774. [Google Scholar] [CrossRef]
- Chavez-Blanco, A.; Segura-Pacheco, B.; Perez-Cardenas, E.; Taja-Chayeb, L.; Cetina, L.; Candelaria, M.; Cantu, D.; Gonzalez-Fierro, A.; Garcia-Lopez, P.; Zambrano, P.; et al. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A phase I study. Mol. Cancer 2005, 4, 22. [Google Scholar]
- Liu, T.; Kuljaca, S.; Tee, A.; Marshall, G.M. Histone deacetylase inhibitors: Multifunctional anticancer agents. Cancer Treat. Rev. 2006, 32, 157–165. [Google Scholar] [CrossRef]
- Mahlknecht, U.; Hoelzer, D. Histone acetylation modifiers in the pathogenesis of malignant disease. Mol. Med. 2000, 6, 623–644. [Google Scholar]
- Hallas, J.; Friis, S.; Bjerrum, L.; Stovring, H.; Narverud, S.F.; Heyerdahl, T.; Gronbaek, K.; Andersen, M. Cancer risk in long-term users of valproate: A population-based case-control study. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 1714–1719. [Google Scholar] [CrossRef]
- Chen, C.L.; Sung, J.; Cohen, M.; Chowdhury, W.H.; Sachs, M.D.; Li, Y.; Lakshmanan, Y.; Yung, B.Y.; Lupold, S.E.; Rodriguez, R. Valproic acid inhibits invasiveness in bladder cancer but not in prostate cancer cells. J. Pharmacol. Exp. Ther. 2006, 319, 533–542. [Google Scholar] [CrossRef]
- Xia, Q.; Sung, J.; Chowdhury, W.; Chen, C.L.; Hoti, N.; Shabbeer, S.; Carducci, M.; Rodriguez, R. Chronic administration of valproic acid inhibits prostate cancer cell growth in vitro and in vivo. Cancer Res. 2006, 66, 7237–7244. [Google Scholar] [CrossRef]
- Blumberg, R.S. Inflammation in the intestinal tract: Pathogenesis and treatment. Dig. Dis. 2009, 27, 455–464. [Google Scholar] [CrossRef]
- Kim, M.; Miyamoto, S.; Yasui, Y.; Oyama, T.; Murakami, A.; Tanaka, T. Zerumbone, a tropical ginger sesquiterpene, inhibits colon and lung carcinogenesis in mice. Int. J. Cancer 2009, 124, 264–271. [Google Scholar] [CrossRef]
- Tanaka, T.; de Azevedo, M.B.; Duran, N.; Alderete, J.B.; Epifano, F.; Genovese, S.; Tanaka, M.; Curini, M. Colorectal cancer chemoprevention by 2 beta-cyclodextrin inclusion compounds of auraptene and 4'-geranyloxyferulic acid. Int. J. Cancer 2010, 126, 830–840. [Google Scholar]
- Tanaka, T.; Sugiura, H.; Inaba, R.; Nishikawa, A.; Murakami, A.; Koshimizu, K.; Ohigashi, H. Immunomodulatory action of citrus auraptene on macrophage functions and cytokine production of lymphocytes in female BALB/c mice. Carcinogenesis 1999, 20, 1471–1476. [Google Scholar] [CrossRef]
- Shimizu, M.; Shirakami, Y.; Sakai, H.; Yasuda, Y.; Kubota, M.; Adachi, S.; Tsurumi, H.; Hara, Y.; Moriwaki, H. (−)-Epigallocatechin gallate inhibits growth and activation of the VEGF/VEGFR axis in human colorectal cancer cells. Chem. Biol. Interact. 2010, 185, 247–252. [Google Scholar] [CrossRef]
- Ben-Shoshan, M.; Amir, S.; Dang, D.T.; Dang, L.H.; Weisman, Y.; Mabjeesh, N.J. 1alpha,25-dihydroxyvitamin D3 (Calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells. Mol. Cancer Ther. 2007, 6, 1433–1439. [Google Scholar] [CrossRef]
- Paz, E.A.; Garcia-Huidobro, J.; Ignatenkos, N.A. Polyamines in cancer. Adv. Clin. Chem. 2011, 54, 45–70. [Google Scholar] [CrossRef]
- Kohno, H.; Tanaka, T.; Kawabata, K.; Hirose, Y.; Sugie, S.; Tsuda, H.; Mori, H. Silymarin, a naturally occurring polyphenolic antioxidant flavonoid, inhibits azoxymethane-induced colon carcinogenesis in male F344 rats. Int. J. Cancer 2002, 101, 461–468. [Google Scholar] [CrossRef]
- Tanaka, T.; Kawabata, K.; Kakumoto, M.; Makita, H.; Ushida, J.; Honjo, S.; Hara, A.; Tsuda, H.; Mori, H. Modifying effects of a flavonoid morin on azoxymethane-induced large bowel tumorigenesis in rats. Carcinogenesis 1999, 20, 1477–1484. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tanaka, T. Preclinical Cancer Chemoprevention Studies Using Animal Model of Inflammation-Associated Colorectal Carcinogenesis. Cancers 2012, 4, 673-700. https://doi.org/10.3390/cancers4030673
Tanaka T. Preclinical Cancer Chemoprevention Studies Using Animal Model of Inflammation-Associated Colorectal Carcinogenesis. Cancers. 2012; 4(3):673-700. https://doi.org/10.3390/cancers4030673
Chicago/Turabian StyleTanaka, Takuji. 2012. "Preclinical Cancer Chemoprevention Studies Using Animal Model of Inflammation-Associated Colorectal Carcinogenesis" Cancers 4, no. 3: 673-700. https://doi.org/10.3390/cancers4030673
APA StyleTanaka, T. (2012). Preclinical Cancer Chemoprevention Studies Using Animal Model of Inflammation-Associated Colorectal Carcinogenesis. Cancers, 4(3), 673-700. https://doi.org/10.3390/cancers4030673