Implication of Heat Shock Factors in Tumorigenesis: Therapeutical Potential

Abstract
: Heat Shock Factors (HSF) form a family of transcription factors (four in mammals) which were named according to the discovery of their activation by a heat shock. HSFs trigger the expression of genes encoding Heat Shock Proteins (HSPs) that function as molecular chaperones, contributing to establish a cytoprotective state to various proteotoxic stresses and in pathological conditions. Increasing evidence indicates that this ancient transcriptional protective program acts genome-widely and performs unexpected functions in the absence of experimentally defined stress. Indeed, HSFs are able to re-shape cellular pathways controlling longevity, growth, metabolism and development. The most well studied HSF, HSF1, has been found at elevated levels in tumors with high metastatic potential and is associated with poor prognosis. This is partly explained by the above-mentioned cytoprotective (HSP-dependent) function that may enable cancer cells to adapt to the initial oncogenic stress and to support malignant transformation. Nevertheless, HSF1 operates as major multifaceted enhancers of tumorigenesis through, not only the induction of classical heat shock genes, but also of “non-classical” targets. Indeed, in cancer cells, HSF1 regulates genes involved in core cellular functions including proliferation, survival, migration, protein synthesis, signal transduction, and glucose metabolism, making HSF1 a very attractive target in cancer therapy. In this review, we describe the different physiological roles of HSFs as well as the recent discoveries in term of non-cogenic potential of these HSFs, more specifically associated to the activation of “non-classical” HSF target genes. We also present an update on the compounds with potent HSF1-modulating activity of potential interest as anti-cancer therapeutic agents.1. Introduction
The cellular response to proteotoxic stress, historically called “Heat Shock Response” (HSR), is strongly conserved through evolution and involves the activation of transcriptional regulators named Heat Shock Factors (HSFs). The mammalian HSF family consists of four members (HSF1, HSF2, HSF3, and HSF4) and was first characterized as master transcription factors by their abilities to regulate the expression of a set of highly conserved proteins, called Heat Shock Proteins (HSPs). HSPs function mainly as molecular chaperones and display strong cytoprotective effects against stress-induced proteotoxic damage by preventing protein aggregation, targeting misfolded proteins for degradation, or blocking the apoptotic pathway. Beyond the regulation of Hsp genes, HSFs have been involved in the regulation of numerous other genes activated by heat and other stresses [1–4]. In addition, under non stressful conditions, HSFs target a wide spectrum of genes involved in a variety of biological processes including cell maintenance, differentiation and development [2–6]. Last but not least, HSF1 is a conserved regulator of aging by promoting longevity, and has been involved in age-related neurodegeneration (Reviewed in [7–9]).
The universally conserved abilities of HSFs to promote cellular adaptation and survival in response to environmental stress are accompanied by a Janus-like behavior, which can favor oncogenic transformation [10]. The first link between HSR and cancer was provided by the observation that aberrant expression of HSPs is frequently associated with cancer. But, the role of HSF1 in cancer far exceeds the sole regulation of Hsp genes since there is not always a correlation between the high HSF activation and/or expression levels frequently found in cancer cells, and those of HSPs [11–13]. In fact, there is growing evidence that the role of HSFs in tumorigenesis involves the induction of, at least some, classical heat shock genes, as well as many “non-classical” targets. In this review, we present recent discoveries in terms of oncogenic potential of HSFs, and notably those associated with the activation of “non classical” HSF target genes.
2. HSF Structure, Expression and Function
2.1. Structure/Activity
HSF family members (HSF1, HSF2, HSF3, and HSF4) display unique as well as overlapping functions, show tissue-specific patterns of expression, are submitted to numerous post-translational modifications, and interact with many protein partners [14–18]. HSF proteins are composed of functional domains, among which the most conserved is the amino terminal DNA-binding domain (DBD). Upon heat shock, thanks to its oligomerization domain (heptad repeats HR-A/B), adjacent to the DBD, the latent inactive monomeric HSF1 forms a trimer and is subjected to extensive post-translational modifications. In normal conditions, this assembling is prevented by another heptad repeat domain HR-C [19,20], which specifically interacts with HR-A/B domains and maintains HSF1 in a monomeric conformation (Figure 1). The deletion of the HR-C domain results in constitutive trimerization which may explain why HSF4, that lacks this domain, has a constitutive DNA-binding ability [19]. HSF2 and HSF3 are present as a dimer [21] and share the ability to bind DNA sequences, called Heat Shock Elements (HSE), originally defined as succession of several inverted repeats of the pentanucleotide motif NGAAN and recently refined by genome-wide analysis [3]. HSF family members exhibit different binding site preferences in terms of architecture and composition of the HSE, thereby allowing a great diversity in the regulation of specific target genes [4,22,23]. In this respect, HSFs are able to act as both activators and repressors depending on the environmental conditions and the targeting genes [1–3]. In addition, subtle crosstalks exist between HSFs, which, together with the combinatorial possibilities allowed by the different post-translational modifications, multiply the levels of fine-tuning the expression of HSF target genes. Furthermore, in specific conditions, HSF1 and HSF2 can form heterotrimers through which HSF2 transiently modulates the very potent and dominant HSF1 activity on its target genes [24–28].
Upon stress, HSF1 and HSF4 are able to recruit chromatin remodelers SWI/SNF [29,30] and to target stress-specific histone modifications [31]. In human cells, HSF1 was shown to direct the transcription of satellite III DNA in pericentromeric heterochromatic regions of particular human chromosomes that are strikingly characterized by an epigenetical status euchromatin. Therefore, stress-activated HSF1 can trigger the conversion of a partially constitutive heterochromatin status to a transcription competent status [32–34]. Likewise, HSF2 has an epigenetic role, strictly speaking, in the heritability of a decondensed chromatin status on the Hsp70 gene. During mitosis, HSF2 inhibits chromatin condensation through interaction with condensin, in a process called ≪bookmarking≫ [35].
2.2. Expression and Physiological Functions
Besides their role in stress response, HSFs assume specific and non-overlapping functions which enlighten their role in cancer. They regulate proliferation, asymmetric division, differentiation, migration, and survival by either activating or repressing their target genes (Table 1).
- -
HSF1. The ubiquitously expressed HSF1, known as a master controller of the cellular-response to different stress conditions, is a maternal factor, stored in the oocyte, and essential for oogenesis and preimplantation development through the regulation of Hsp90α gene expression [36–38]; reviewed in [5]. It is also required for the development and maintenance of tissues including the adult brain [39–41] germ cells [42–44], ciliated cells [45], and immune cells [46,47], regulating both Hsp and non-Hsp target genes.
- -
HSF2. As mentioned above, HSF2 is able to finely tune the HSR mediated by HSF1. However, its role in stress is more striking upon proteasome inhibition, because it regulates the expression of proteasome subunits [6]. In the context of differentiation, as in stress, a pool of nuclear HSF2 can associate with HSF1 and modulate the expression of heat-shock proteins [24,26] as well as noncoding satellite III RNA in nuclear stress bodies (unique subnuclear organelles which form in response to heat shock) [27]. Interestingly, HSF2 is mainly associated with brain development and gametogenesis [48–50] where it displays a spatiotemporal expression pattern [5,51]. In the developing brain cortex, HSF2 regulates multiple aspects of neuronal migration through gene expression, like p35, a crucial activator of the serine/threonine kinase, Cdk5. In spermatogenesis, HSF2 cell-specific expression is negatively regulated by microRNA miR18, which belongs to the Oncomir-1 cluster associated with tumorigenesis [52]. HSF2 and HSF1 can form heterotrimers in testis [27] and share common target genes during spermatogenesis, in particular sex chromosomal multicopy genes [15,53]. HSF2 binding to its target genes during spermatogenesis is associated with histone H4 acetylation [53]. The inactivation of both HSF1 and HSF2 leads to completely male sterility, reinforcing their intertwined functions, which also rely on global chromatin remodeling.
- -
HSF3. Initially only identified in the chicken (cHSF3), this ubiquitously expressed factor has been recently identified in the mouse (mHSF3). In human, HSF3 might be present as a pseudogene, since no transcripts have been detected [54]. In contrast to others HSFs, HSF3 does not display the same regulatory role in different species [55]. Whereas cHSF3 is the major HSF in response to heat shock and therefore the main inducer of HSPs, mHSF3 does not induce classical hsp genes, but instead induces the expression of other stress-responsive genes, raising the possibility that it could regulate distinct sets of genes in development or longevity [18].
- -
HSF4. Mainly expressed in the human heart, brain, skeletal muscle, and pancreas [56], HSF4 has been shown to be required for the development and maintenance of sensory organs [5,57,58]. During this process, HSF4 either cooperates or competes with HSF1 for common target genes, including members of the Fibroblast Growth Factor (FGF) family, in a cell-specific manner in lens and olfactory epithelium [45,59]. Recently, it has been reported that HSF4 binds to more flexible consensus HSE, which are not found in the promoter proximal regions of Hsp genes, but are preferentially located in introns, exons and distal regions. HSF4 binding is associated with reduced histone H3K9 methylation. Among the different targets of HSF4 after a heat shock, 33% are non-classical heat-shock genes [4]. Substantial proportions of these regions are also bound by HSF1, reinforcing the concept of their mutual interactions.
Overall, not only HSF1, but also other HSF family members, play significant roles in the induction of non-classical genes in response to heat shock or in developmental conditions. In this respect, HSFs regulate new target genes which include proteostasis genes (proteasome subunits), growth factor genes (FGF, Leukemia Inhibitory Factor (LIF)), and genes that are directly or indirectly involved in cytoskeleton dynamics (Hsp90 and cortical actin in eggs [37,38]; p35/p39/Cdk5 [50]; bIV tubulin in ciliary beating activity [60]; Bfsp, lens specific intermediate filaments [61]). HSFs also direct the establishment of epigenetic marks and might impact genome structure, i.e. chromatin condensation state in spermatogenesis; histone methylation or acetylation status and likely retrotransposons [4,53,62].
3. HSFs in Cancers
3.1. Multiple Roles of HSF1 in Cancer
3.1.1. Role of HSF1 in the Initiation and Maintenance of Transformed Phenotypes
HSF1 has been involved in the etiology of cancer by its multiple effects in facilitating transformation and tumor invasiveness in response to diverse oncogenic stimuli [10]. In this respect, Hsf1−/− mice show lower incidence of tumors induced by mutations of the RAS oncogene or a hot spot mutation in the tumor suppressor p53, and they show improved survival [10]. HSF1 is also crucial for cell transformation and tumorigenesis induced by the human epidermal growth factor receptor-2 (HER2), an oncogene responsible for breast tumors aggressiveness. Knockdown of HSF1 leads to growth arrest and senescence of HER-2-expressing cells [63]. Moreover, HSF1 is required for the maintenance of the transformed phenotype in: (i) established oncogenic cell lines; (ii) breast cells lines with progressive oncogenic states (i.e., primary human mammary epithelial (PHME) cells; immortalized human mammary epithelial (HME) cells; fully transformed and tumorigenic HME cells (HMLER); [64]); (iii) a collection of breast cell lines derived from spontaneous human tumors, with various p53 status (MCF-7 or various mutant alleles BT-474, MDAMB-231, and T47D); (iv) malignant cells of diverse histological origins either derived from human tumors (HeLa (cervix), PC-3 (prostate), and S462 and 90-8 (peripheral nerve sheath) or derived by in vitro transformation (293T; kidney). Furthermore, in immortalized MEFs, HSF1 is essential for basal and EGF-induced migration, a process crucial for tumor invasion and metastasis [65].
HSF1 is often elevated in human cancer cells compare to normal cells. However, somatic mutations in Hsf1 have not yet been identified in human cancers, and overexpression of HSF1 does not lead to transformation, as mutant RAS does. Here, HSF1 does not display typical oncogenic features per se but represents a mechanistic and therapeutic challenging target. It is possible that the core function pathways regulated by HSF1 in normal cells might be massively mobilized in cancer cells, like signal transduction, ribosome biogenesis, translation and glucose metabolism [10]. Indeed, cancer cells growth and development highly depend on normal cellular functions governed by genes, which are not typical cancer related genes, and through which HSF1 can promote tumorigenesis.
The unique HSF present in fission yeast cells is essential for normal growth and drives the transcription of target genes that encode proteins with a broad range of biological functions, including protein folding and degradation, energy generation, protein trafficking, maintenance of cell integrity, small molecules transport, cell signaling and transcription [66,67]. Conversely, in murine cells, HSF1 is more dispensable for cell growth and survival, although Hsf1−/− mice show defects in postnatal growth and placenta formation, suggesting that in normal cells HSF1, while a major actor in the heat shock response, is also important for other cellular process [68].
In normal mammalian cells, HSF1 is known to regulate Hsp genes, but also a wide range of non-Hsp genes upon stress or differentiation [2,3]. In aggressive cancer cells, HSF1 is expressed at high levels, which could amplify its activity and broaden the spectrum of its targets. By analogy to what happens in yeast, HSF1 could regulate central functions of the cell biology, in addition to the already known HSP expression. However, although the levels of HSPs (especially HSP27, HSP70 and HSP90) are elevated in different types of cancers, there is not always a correlation between HSF1 constitutive activation and HSP expression. In this way, genetic knockdown of HSF1 fails to induce a decrease in the levels of HSPs in some cancer cell lines [69]. It is believed that the elevated expression of HSF1 in cancer cells does not lead to a kind of global “heat shock response”, but only specific members of the HSP family are induced in a tumor-dependent manner. That explains why HSP expression in cancer cells displays considerable variation, depending on the HSP member and the tumor characteristics. For instance, in HER-2 (human epidermal growth factor receptor-2 oncogene) expressing cells, in aggressive breast cancer models, HSF1 knockdown is accompanied by a specific down-regulation of HSP27 and HSP70 expression [63].
3.1.2. HSF1 and Prostate Cancers
HSF1 expression has been shown to be elevated in prostate carcinoma compared to its normal counterpart [70]. An elevated protein expression of HSF1 and some HSPs was reported in the aggressively malignant cell lines DU145 and CA-HPV-10. However, no difference in the RNA level was detected suggesting that HSP induction was not mediated by HSF1-dependent transcription. In addition, growth of PC3 cells in vivo, as tumor xenografts, was accompanied by a marked decrease in HSPs expression (HSP27, HSP70, HSP60, HSP90) whereas HSF level was not modulated. In a PC3 metastatic variant (PC3M), a strong increase in HSF1 mRNA was observed, as well as an increase in HSF1 protein and nuclear localization [71]. Overexpression of HSF1 in non-metastatic PC-3 cells was accompanied by an upregulation of HSP27 at the protein level, but HSP70 and HSP90 were not affected, once again pointing out that the elevated expression of HSPs in cancer cells does not always depend on HSF1. Interestingly, in prostate cancer tissue, HSP27 upregulation, but not HSP70 or HSP90, was significantly associated with clinicopathological factors [72]. In prostate cancer cells, HSF1 also influences the development of an aneuploid state and mitotic progression [73].
3.1.3. HSF1 and Breast Cancers
HSF1 may partly participate in breast cancer progression by inducing specific HSPs, mainly HSP27 [74]. In breast cancer, the highly malignant factor heregulin beta-1 (HRGβ1) is a secreted factor, which binds to c-ErbB-3 and -4 receptors and provokes the recruitment of c-erbB-2 and receptors heterodimerization. The binding of HRGβ1 to the cell surface induces an increase in HSF1 levels that results in anchorage-independent growth and protection from cisplatin-induced apoptosis mediated by HRGβ1 [74]. One aspect of this process relies on the inhibition of GSK3, a kinase that antagonized HSF1 activation. Part of the anti-apoptotic effects of HSF1 seems to operate via activation of the Hsp70 promoter and therefore likely involves HSPs. Remarkably a crosstalk between the beta-catenin/Wnt pathway and the heat shock cascade has been identified in breast cancer tumors with high metastatic potential. Indeed, the formation of a complex between beta-catenin, HSP27 and HSF1 has been detected in breast cancer biopsies. The striking presence of beta-catenin in the cytoplasm (and not only at the membrane) co-expressed with HSP27 and with HSF1 being also nuclear could have some clinical relevance in terms of prognosis [75]. Notably, HSF1 plays an additional role in the etiology of breast cancer. Unlike normal cells, cancer cells preferentially catabolize glucose by glycolysis, thereby producing high levels of lactic acid. For instance, Zhao et al. have shown that ErbB2 promotes glycolysis in a HSF1-dependent manner. Overexpression of ErbB2 increases the expression of HSF1, which binds to lactate dehydrogenase A (LDH-A) promoter and activates its transcription [76], allowing the metabolization of pyruvate to lactate.
3.1.4. HSF1 and Gastro-Intestinal Cancers
The search for activation of signal transduction pathways in sporadic colorectal cancers (CRC) revealed an increase in the mRNA levels for heat shock and NFκB pathway genes [77]. Namely, an increase of Hsf1 mRNA in 86% of patients was reported in sporadic colorectal cancer. In that context, HSF1 upregulation in patients was accompanied by an elevated expression of HSP27, HSP90 but also of iNOS (NO synthase; 63%). In addition, HSF1, through the induction of the HSP70 co-chaperone BAG-3, which stabilizes the level of anti-apoptotic Bcl-2 family members, facilitates colon cancer cell survival during pro-apoptotic stress [78]. The oncogenesis of hereditary CRC cancers is believed to involve four signal transduction pathways: (a) the APC-β-catenin-TCF-myc (Wnt) pathway; (b) the microsatellite unstable pathway; (c) the p53 pathway; and (d) the estrogen receptor hypermethylation pathway [79,80]. Although no data are currently available on the potential role of HSF1 in hereditary CRC, at least two of these pathways (Wnt and p53) are known to be affected by HSF1 [77,79,80].
In gastro-intestinal cancer, the level of HSPs is often upregulated with a high variability and without evident correlation with HSF1 level [81]. Instead, the increase observed in HSF1 expression in gastro-intestinal cancer tissues, drives the repression of the pro-apoptotic protein, X-linked inhibitor of apoptosis protein (XIAP)-associated factor-1 (XAF-1) [82]. Thus, HSF1 can impair the apoptotic pathway in cancer cells, at least partly, in a HSP independent manner.
Interestingly, the protein TC1 (a positive regulator of the signaling Wnt/beta-catenin via inhibition of Chibby), which is upregulated in an aggressive subtype of gastric cancers, correlates with poor prognosis and has been reported to induce a heat shock response in cancer cells by favoring HSF1 expression. Moreover, HSF1 and TC1 mutually activate each other [83,84].
3.2. Role of HSF2 and HSF4 in Glioma and Neuroblastoma
HSP70, HSC70 and HSP90 are found expressed in the tumor parenchyma of all high-grade and most low-grade gliomas, including oligodendrogliomas [85]. A recent work from Mustafa et al., has reported an overexpression of HSF2 (and a modestly elevated level of HSF1) in different stages of glial tumorigenesis, compared to normal brain. The potential involvement of HSF2 in glioma is interesting considering that HSF2 is involved in central nervous system development [5].
Concerning HSF4, while no differences in its expression were found in low-grade glioma and normal brain, it was significantly downregulated in glioblastoma [86].
Neuroblastoma (NB) and Ewing's sarcoma (ES) represent the most common extracranial solid tumors of neuroectodermal origin of childhood. While NB and ES cells globally showed very similar protein expression patterns, GRP78, GRP75, HSC70, HSP70, HSP47, HSP90α, and HSP27 are markedly more expressed in NB cell lines. To date, there is no data related to HSF expression or activity in this type of cancer although it would be worth studying since HSP profile is largely upregulated.
In addition, in human neuroblastoma, the dual-specificity phosphatase 26 (DUSP26) is overexpressed. DUSP26 has been described to inhibit HSF4 phosphorylation induced by Mitogen Anctivated Protein Kinases (MAPK), thereby negatively affecting its ability to bind DNA [87]. Therefore, the negative control exerted by DUSP26 on HSF4, altogether with the inverse correlation in the expression of these two proteins in neuroblastoma, may contribute to repress HSF4 activity in neuroblastoma and its potential crosstalk with HSF1.
3.3. Potential Role for HSF3
The expression of HSF3 in the chicken is regulated by the proto-oncogene c-Myb, thus favoring HSP expression, as well as cell proliferation. Moreover, Tanikawa et al. reported that a mutated form of p53 is able to inhibit c-Myb mediated cHSF3 transcription allowing tumor progression [88]. Since in humans HSF3 is not expressed, it would be worth studying whether a relation between other HSFs and c-Myb is maintained.
In summary, the principal proteins involved in cancer that have been shown to affect HSF expression or activity are indicated in Table 2.
4. HSFs and Cancer Related Targets
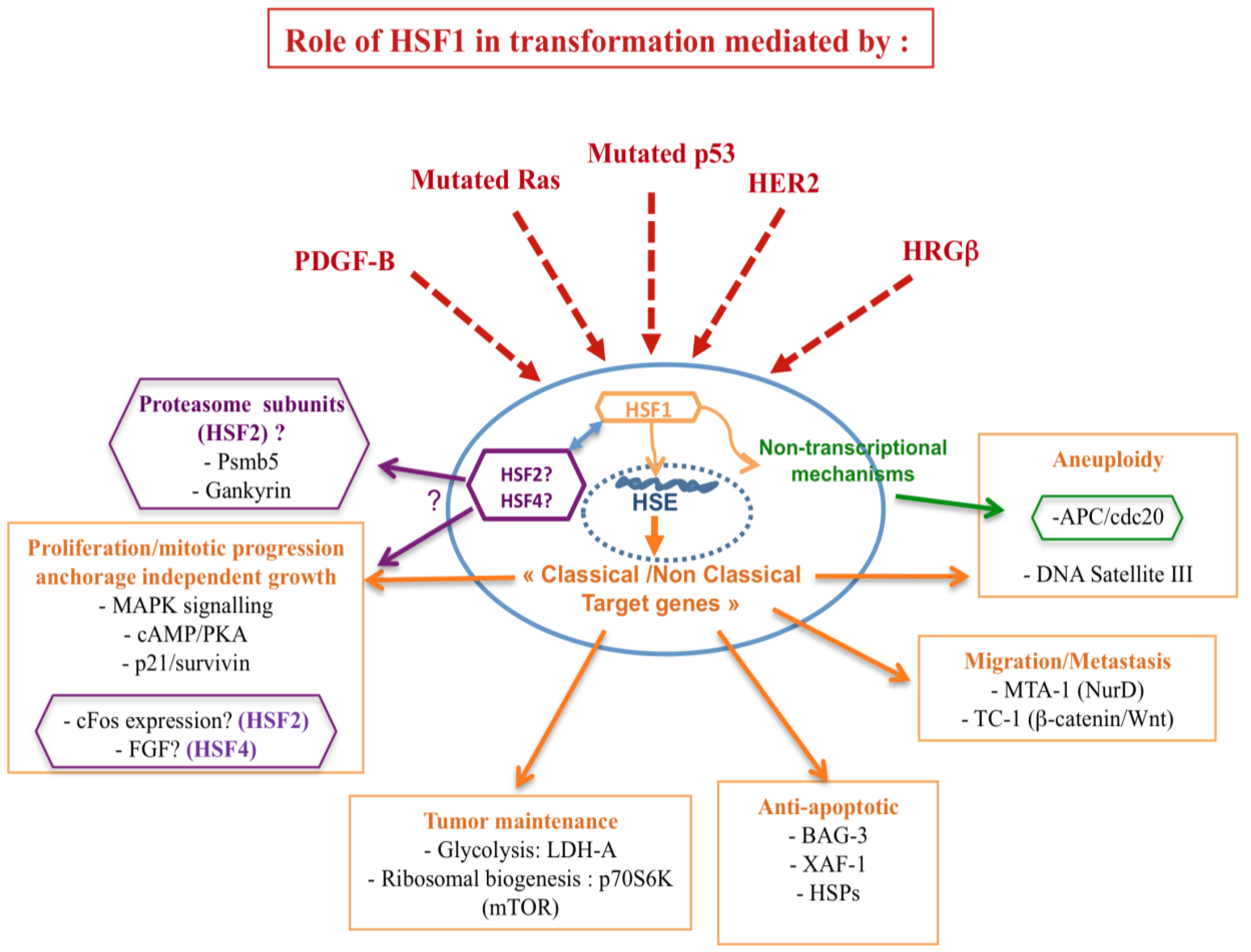
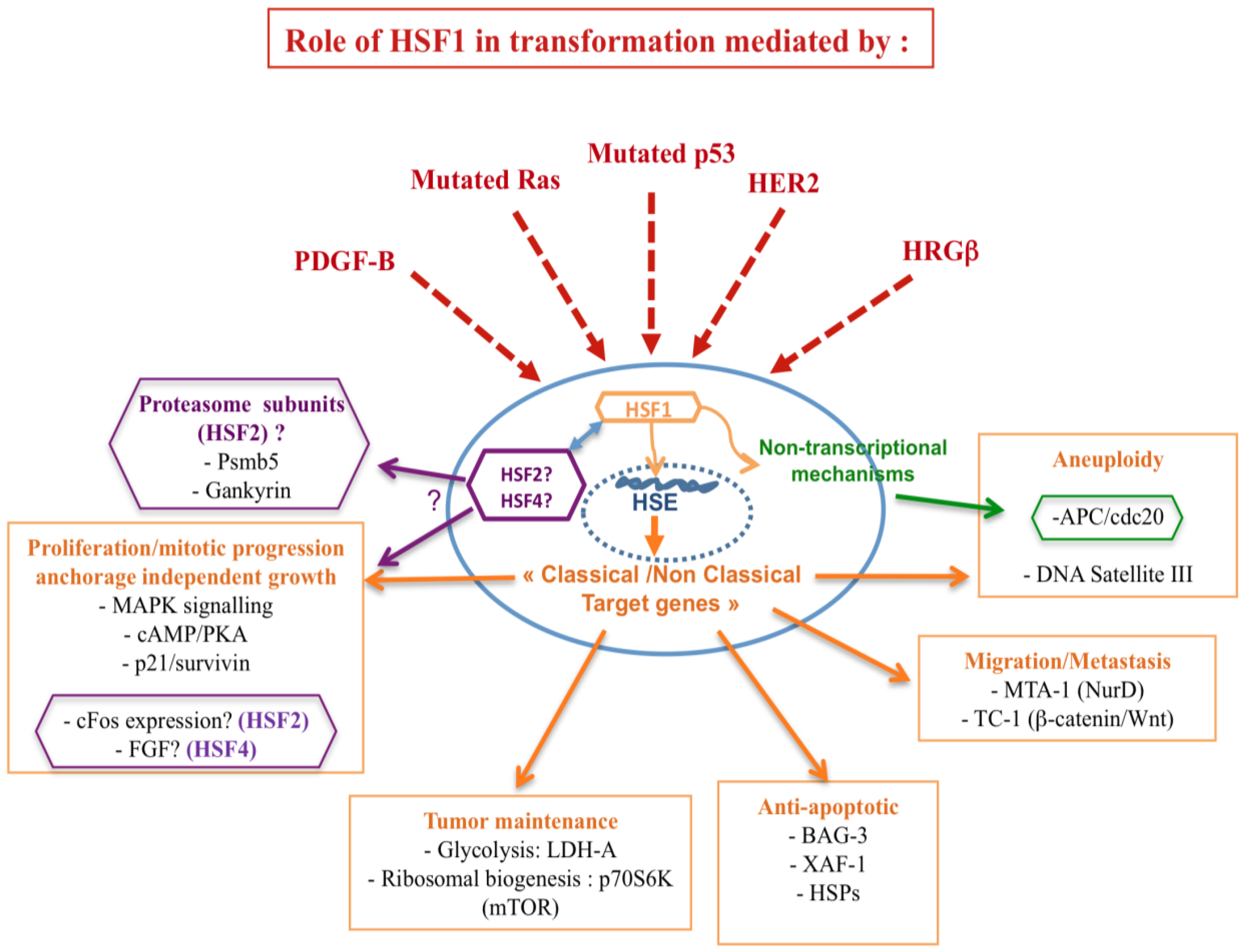
In light of the involvement of HSFs in mediating tumorigenesis, convincing evidence refers to the impact of HSF1 on the tumor suppressor p53 and the oncogene RAS. Herein, p53 and Ras, pivotal integrators of signaling pathways and key regulators of cell fate decisions, are the two most altered genes in human cancers. Moreover, HSF1 seems to participate to the metastatic potential of the cancer cell through cooperation with different metastatic related genes, such as the prometastatic co-repressor gene MTA1. It is noteworthy that HSF1 possesses additional properties in cancer, such as the enhancement of pro-malignant signaling pathways, involving PKA and TOR [10]. In addition, HSFs act via targets involved in the establishment of genomic instability and epigenetic mechanisms (Figure 2).
4.1. HSF1, Tumor Suppressors, and Oncogenes
4.1.1. Tumor Suppressor p53
The first direct in vivo evidence of HSF1 implication in the development of spontaneous tumors arises from p53−/− mice. The rapid tumor evolution observed in mice lacking p53 mainly results in lymphoma, while p53−/−Hsf1−/− mice rarely develop lymphomas, but rather succumb to other types of cancers, like testicular carcinoma [89]. Tumor suppressor p53 is frequently inactivated by genetic mutations in different cancers [90]. In this context, Dai et al. have shown that HSF1 deficiency dramatically reduces spontaneous tumor formation in mice carrying a common, dominant-negative mutation of the p53 gene, whereas Hsf1+/+ and Hsf1+/− mice bearing dominant-negative mutation of p53 develop a broad spectrum of tumor types (sarcomas, lymphoma, carcinomas; [10]). This was confirmed by Hsf1 knock down, using specific siRNAs, in different mouse and human cell systems [10]. These different results observed in these complementary works [10,89] may be explained by differences in the models used, involving distinct mouse genetic background, and the use of clinically relevant p53 mutation versus p53 null strategy.
Dysfunction in the proteasome pathway can lead to many disorders including cancers [91], and proteasome inhibitors are currently used in therapeutical approaches [92]. Recently, Lecomte et al. have reported that some proteasome subunits, such as Psmb5 and Gankyrin, were significantly downregulated in Hsf2−/− MEFs [6]. Interestingly, Gankyrin is an oncoprotein involved in p53 degradation, thus suggesting a potential role of HSF2 in the regulation of the tumor suppressor p53.
4.1.2. Oncogene RAS and HSF1
Compared with wild-type MEFs, Hsf1−/− MEFs are refractory to proliferation and resistant to focus formation, driven by oncogenic H-RASV12D or by the proto-oncogene PDGF-B, which are both mitogenic signal transducers [10]. In contrast, expression of c-MYC and LTA (regulator of cell-cycle progression, which are not expected to increase proliferation in already-immortalized cells, but predisposing cells to apoptosis) does not induce proliferation in immortalized Hsf1+/+ MEFs. Accordingly, Hsf1−/− MEFs show no enhanced survival in response to RAS and PDGF/B expression, but reduced survival in response to c-MYC and LTA expression [10].
4.1.3. Oncogene HER2 and HSF1
HSF1 is also required for cell transformation and tumorigenesis induced by the oncogene HER2 (Human Epidermal growth factor Receptor-2), which is responsible for aggressive breast tumors. Indeed, knockdown of Hsf1 prevents neoplastic transformation (foci formation or tumor growth in xenografts) induced by HER2 expression in untransformed human mammary epithelial MCF-10A cells. This suggests that the proliferation of HER2-expressing cells is critically dependent on HSF1 [63]. Strikingly, this anti-tumorigenic effect of HSF1 downregulation was associated with HER2-induced upregulation of the cyclin-dependent kinase inhibitor p21, a major regulator of senescence in cancer cells [93], and accompanied by a decrease in the mitotic regulator survivin, which is also involved in growth arrest and senescence. Survivin is a member of the inhibitors of apoptosis (IAPs) family and a critical regulator of mitosis by modulation of aurora B kinase [94]. Its decrease is partially controlled by both HSF1 and p21 [63]. In addition, HSF1-mediated control of breast cancer cell senescence was due, at least partly, to the regulation of HSP70 and HSP27 expression.
4.1.4. Loss of Hormonal Control, Epigenetic and Metastasis-Related Targets of HSF1
The potential role of HSF1 in cancer metastasis was first reported in prostate cancer where HSF1 overexpression was correlated with aggressiveness of the tumors [70,71]. Since then, studies have shown that HSF1 is able to mediate tumorigenic effects through the recruitment of the prometastatic co-repressor gene MTA1 (a component of the NuRD complex containing the histone deacetylases HDAC1 and HDAC2) [95]. MTA1 is expressed in numerous human cancers, including breast, prostate and gastro-intestinal cancers and its expression correlates with tumor aggressiveness and metastasis [96–98]. Herein, MTA1 protein expression is higher in hormone-refractory metastatic prostate cancer compared to clinically localized disease and benign prostatic tissues [97].
Breast cancer cell can also escape from hormonal control, leading to higher metastatic potential. Khaleque et al., have reported that HSF1 binds to the corepressor MTA1 in human cultured breast cancer cells and carcinoma samples. The HSF1-MTA1 complex, strongly induced by the transforming ligand heregulin (a ligand for erbB receptor from the EGF family), assembles on the chromatin of breast cancer cells and mainly binds to the promoter of estrogen responsive genes [95]. Thus, HSF1 induces repression of estrogen-dependent gene transcription, an effect linked to cancer invasiveness.
Migration properties in correlation with metastasis potential were also shown to be controlled by HSF1. Mouse embryonic cells derived from Hsf1−/− mice are deficient in both basal and EGF-mediated migration. This default in migration, which is partly due to the downregulation of EGF receptor and likely involves Ras, is associated with an impairment of the MAPK signaling pathway [65].
4.1.5. Cdc20/APC Genomic Instability and HSF1
As described above, HSFs have been described to modulate the structure of chromatin and to establish some epigenetic marks (see introduction). In particular HSF1, in association with HSF2, was shown to steer the transcription of the human DNA satellite III in the centromeric regions, establishing an epigenetic status resembling euchromatin [14,99,100]. The involvement of HSF2 in the expression of satellite III DNA was further confirmed [27]. Remarkably, transcription of satellite III DNA is associated with aneuploidy in several cancers such as ovarian cancers, melanoma or myeloma [101–103] and thus could be related to HSF status.
Accordingly, Lee et al. have recently shown that HSF1, via its regulatory domain, when overexpressed in radiation induced-fibrosarcoma cells, interacts with cdc20, the co-activator of anaphase-promoting complex (APC), and thus, inhibits the mitotic exit and the ubiquitination activity of APC on two key anaphase inhibitors, Cyclin B1 and securin. As a consequence, this non-transcriptional HSF1 effect leads to aneuploidy and multinucleated cells associated with micronuclei and genomic alteration [104]. In prostate carcinoma cells, a dominant negative construct of HSF1 dramatically alters the DNA content of PC3 cells and inhibits aneuploidy and Cyclin B1 stabilization [73]. Further, HSF1-mediated aneuploidy was facilitated in p53-defective cells. This phenomenon was associated to an increase in HSF1 activity through phosphorylation by a Polo-like kinase, as well as cdc20/HSF1 interaction [105].
Altogether, HSF1 activity is hijacked in a pleiotropic manner by a large diversity of cancer cells and oncogenes to favor tumor initiation and progression. In sharp contrast, it is noteworthy that HSF1 knockout has a minimal effect on the proliferation/survival of normal primary human cells.
4.2. Potential Tumorigenic Functions for HSF2 and HSF4
In contrast to HSF1, there is little direct evidence on the involvement of HSF2 and HSF4 in tumorigenesis [106]. Although they can play a role indirectly by their ability to modulate HSF1, several works pointed out their regulatory role in cancer related genes. During mitosis, HSF2 has been shown to bind and activate, not only the HSE promoter of the different Hsps (bookmarking), but also the promoter of the proto-oncogene c-Fos. The protein c-Fos is often up-regulated in tumor cells and is well-known for its oncogenic activity [107]. Thereby, it is tempting to conclude that HSF2 might regulate the oncogenic potential of c-Fos [108].
In addition, HSF2 is a labile protein whose degradation was recently shown to be regulated by anaphase-promoting complex/cyclosome (APC/C), a ubiquitin E3 ligase, which drives the degradation of cell cycle regulators in cycling cells by associating with the coactivators Cdc20 and Cdh1 [28]. Therefore, APC/C, by its effect on HSF2, could be involved in a cell cycle-dependent manner in the modulation of HSF1 activity, which could have profound effects on cancer progression.
Finally, the oncomiR miR18 has been shown to regulate the expression of HSF2 during spermatogenesis [52]. It could be possible that miR18 or other miRNA could regulate HSF2 expression in cancer cells, which, by modulating HSF1, could influence cancer initiation or progression.
HSF4 regulates normal cell proliferation and differentiation during mouse lens development, and belongs to the first group in the hierarchy of genes involved in this process [5,59]. Interestingly, HSF4, through its effect repressing the expression of the Fibroblast Growth factor-7 (FGF-7) gene which encodes a specific mitogen for epithelial cells, is also important for governing ocular surface morphogenesis. Upregulation of FGF-7 has been reported to be associated with many human neoplastic tumors of epithelial origin including pancreatic, breast and gastric cancer, thus underlying the oncogenic role of FGF-7 [109–111]. Most importantly, Chimaka et al., have demonstrated that overexpression of FGF-7 in Keratin 12-rtTA/tet-O-FGF-7 double transgenic mouse model, in which FGF-7 expression in corneal epithelium is driven by doxycycline treatment, induces epithelial hyperplasia that mimic the human ocular surface squamous neoplasia (OSSN) [112]. In light of this work and the observation that FGF-7 is upregulated in Hsf4−/− lens epithelial cells, a likely scenario is that HSF4 may interfere with the tumorigenic function of FGF-7 [4].
5. The Inhibition of HSFs in Cancer Therapy
5.1. Targeting HSF1
HSF1 knockdown experiments in cancer cells demonstrate the interest of blocking this transcription factor in cancer therapy. A study using different human HSF1 targeted shRNAi constructs was performed using a collection of breast cancer cell lines that differed in their p53 status, HER2 expression, estrogen sensitivity and metastatic potential. All cancer cells were strongly affected by the HSF1–inhibitory hairpins [10], Further, Rossi et al., have determined that the ideal size target for siRNA mediated HSF1 silencing is 322–340 nucleotides. By generating a pSUPER-HSF1 vector, able to potently suppress HSF1 gene, they dramatically increased the sensitivity to hyperthermochemotherapy (combination of a Cisplatin treatment with heat-shock), leading to massive apoptosis of Hela cervical cancer cells [113]. These approaches, albeit attractive, are still not established for clinical use. In this respect, an expanding array of small, drug-like compounds is currently available, some with potent HSF1-modulating activity in organisms, but only a few of them are in clinical trials (see below).
5.2. Chemical Inhibitors of HSF1
Chemical inhibitors of HSF1 activation have been described including quercetin, genistein and the synthetic benzylidene lactams, KNK437. The natural product Stresgenin B has also been reported to inhibit induction of HSPs, but its mode of action is still unknown [114]. Quercetin increases the sensitivity of drug-resistant cancer cells to anti-cancer agents [115], and sensitizes cancer cells to hyperthermia, cisplatin [116] and tiazofurin [117]. HSF1 is strongly depleted following quercetin treatment and this effect appeared more marked in neuroblastoma cells. Moreover, a strict correspondence between the quercetin concentrations necessary to cause both HSP inhibition and doxorubicin sensitizing effect was observed, suggesting that neuroblastoma cell' resistance to doxorubicin treatment might be due to high levels of HSPs, under the control of HSF1. As for quercetin treatment, neuroblastoma cells were the most sensitive to HSF1 silencing effect. Knockdown of HSF1 in these cells strongly increased the anti-proliferative and pro-apoptotic effects induced by cisplatin [118]. Although efficient in cancer experimental models, the clinical use of these inhibitors is limited because of their pleiotropic effects [119].
To date, the most potent inhibitor of the HSF1 transcriptional function is Triptolide, a diterpene triepoxide from the plant Triptergium wilfordii. Triptolide does not interfere with the early events leading to trimer formation, hyperphosphorylation and DNA binding of HSF1 [120]. Albeit toxic at high concentrations, a therapeutic dose of Triptolide has been defined to treat pancreatic cancer xenografts [121]. Nevertheless, the precise mechanism of triptolide action remains to be investigated.
Several cancer treatment approaches, such as HSP90 inhibitors (geldanamycin) and proteasome inhibitors (Bortezomib), induced a proteotoxic stress that activates a pro-survival pathway, which explains the low efficiency of these therapies [122]. Both HSP90 and proteasome inhibition are known to activate HSF1 and HSF2 [21,25,123–125]. From a high-throughput screening of molecular products that can interfere with the heat shock response, two new compounds were identified namely, NZ28 and emunin (emetine derivative). These two molecules exhibit little acute toxicity and allowed a strong sensitization of myeloma cells to proteasome and HSP90 inhibitors, as well as of prostate carcinoma cells to proteasome inhibitors [126]. Their molecular action is not yet known but seems to involve post-transcriptional events downstream HSF1. Similarly, by a high content target based screening, Au et al., have identified another small molecule inhibitor of HSF1 transcriptional activity [127]. This molecule substantially hampers granules formation in heat-shocked Hela cells and significantly inhibits HSF1 phosphorylation. This effect is associated with a reduction in HSP70 and HSP90 expression. Recently, a malaria drug, quinacrine (QC) has been reported to prevent the induction of HSF1-dependent transcription of hsp70 gene in a relative selective manner. The combinational treatment with a HSP90 inhibitor (17-DMAG) results in suppression of tumor growth in mouse syngeneic models [122].
5.3. Viral Approaches
Recently, an interesting anti-cancer approach has been developed based on the fact that HSF1 overexpression enhances the oncolytic effect of a replicative adenovirus. E1B55kD deleted oncolityc adenovirus, Adel55, was designed to achieve cancer specific cytotoxicity. A construct bearing Adel55-cHSF1 for tumor gene therapy, demonstrated that it favored oncolysis and viral replication by increasing its burst in breast cancer cells and SW620 xenografts [128]. The future will tell of the feasibility of such a viral therapy in cancer patients.
5.4. Targeting other HSFs
It has recently been reported that, in Hsf2 knockout cells, the proteasome activity is lower compared to normal counterparts. This inhibitory effect is due to the regulatory role of HSF2 on the transcription of some proteasome subunits. Therapeutic use of proteasome inhibitors participates to the selection of the resistant clones, which are associated to the induction of beta5 and beta2 proteasome subunits expression [129,130]. Since in Hsf2−/− MEFs one of these two subunits is downregulated, this suggests that inhibition of HSF2 could be beneficial for the sensitization of cancer cells to proteasome inhibitors. More importantly, HSF2 regulates Gankyrin expression that is responsible for p53 tumor suppressor degradation [6]. Targeting HSF2 could stabilize p53 and favor apoptosis. Therefore, these observations suggest that specific HSF2 inhibitors could reduce the chemoresistance to proteasome inhibition that is so frequently observed in clinical studies. This pioneer study defines a new cancer research axe focused on HSF2 that definitely deserves further investigation.
6. Concluding Remarks
HSF1 expression is likely to be crucial for the “non-oncogene addiction” and the stressed phenotype of cancer cells. These stresses include proteotoxic and oxidative stress, frequent spontaneous DNA damage and aneuploidy [69]. HSF1 by itself does not act as a classical oncogene or tumor suppressor. Neither enforced overexpression nor knockout directly drives transformation. HSF1 acts as a major multifaceted enhancer of tumorigenesis by regulating diverse core cellular functions that include proliferation, survival, protein synthesis and glucose metabolism, and therefore, is an attractive potential target in cancer therapy. However, some considerations must be taken when using HSF1 inhibitors because, although they seem beneficial in cancer treatment, they might, in parallel, accelerate neurodegenerative processes and aging. Indeed, HSF1, partly through the induction of chaperones that inhibit protein aggregation like HSP27, displays a protective effect against neurodegenerative diseases [131,132]. In this respect, therapeutic induction of HSF1-mediated stress response by non-toxic agents, like HSP90 inhibitors and Celastroloids, is currently being explored in Huntington disease [131,132]. It should also be mentioned here that hyperthermia, a strategy in cancer that could be considered as the opposite of HSF inhibition, has proved its therapeutical value in clinical trials and is still used in association with chemotherapeutic drugs in some countries (Germany, Italy, Japan, [133]). Therefore, regarding the development of new tools/potential drugs targeting HSFs, it seems important that compounds do not cross the blood-brain barrier in order to minimize neurodegenerative risks and that they take into account HSF3 multiple post-translational modifications, as well as the formation of heterocomplexes between them. Ideally, rather than to simply activate or inhibit a given HSF, it would be more relevant/innovative to target a specific regulatory status, allowing a restrictive effect on designated signaling pathways.


{kind=link}
{kind=link}
| HSF1 | HSF2 | HSF3 | HSF4 | |
|---|---|---|---|---|
| Species Conservation | Human, Mouse, Chicken | Human, Mouse, Chicken | Chicken, Mouse | Human, Mouse, Chicken |
| Oligomerization | NC: Monomer HS: Trimer | HS: Dimer-heterotrimers HSF1/HSF2 Differentiation: Dimer-heterotrimers HSF1/HSF2 Development: homotrimers/heterotrimers HSF1/HSF2 | NC and HS: Dimer | NC and HS: Trimer |
| Target genes in stress responses | Hsp genes (+ non-classical stress genes) | Hsp genes (+ non-classical stress genes) Proteasome subunits | Mainly Hsp genes in chicken Non classical stress genes in mice | Not heat-responsive Repression of hsp genes in ectopic expression experiments |
| Developmental role of HSF | Oogenesis, maternal factor, development and maintenance of germ, ciliated and immune cells | Brain and gametogenesis development | ND | Development and maintenance of sensory organs |
| Adult tissue localization | Ubiquitous | Ubiquitous | Ubiquitous | Tissue specific (heart, brain, skeletal muscle, and pancreas) |
NC: Normal conditions; HS: Heat shock; ND: Not determined.
| HSF | HSF Positive/Negative Regulators of interest in cancer | Effect on HSFs |
|---|---|---|
| HSF1 | HRGβ | Increase HSF1 expression => activation of LDH-A (glycolytic enzyme) and the formation of the complex HSF1/MTA1 (prometastasic) |
| HSF1 | TC1 (Wnt Signaling) | Increases HSF1 expression in gastro-intestinal cancer => tumor aggressiveness |
| HSF1 | GSK3 | Inhibits HSF1 activity-GSK3 is inhibited in breast cancer |
| HSF2 | OncomiR18 | Inhibits HSF2 expression in spermatogenesis |
| HSF4 | DUSP26 | Inhibits HSF4 activation induced by MAPK |
| HSF3 | c-Myb | Induces HSF3 expression |
Acknowledgements
We are very grateful to Ryma Abane, Anne Le Mouël, and Federico Miozzo (UMR7216), and Jessica Gobbo (U866) for helpful comments on the manuscript. Our works are supported by Agence Nationale pour la Recherche (Programme “Neurosciences, Neurologie and Psychiatrie”), ATC Inserm, Association pour la Recherche sur le Cancer (ARC, 3609 and 3997), Institut de Recherche sur les Boissons (IREB), Institut Nationale contre le Cancer (INCa), “Ligue Nationale Contre le Cancer” and its committees in “Nièvre” and “Sâone et Loire” and the ≪Conseil Régional de Bourgogne≫. AdT has an INCa financing. CG's team has the “Label” from the “Ligue Nationale contre le Cancer”.
References
- Murray, J.I.; Whitfield, M.L.; Trinklein, N.D.; Myers, R.M; Brown, P.O.; Botstein, D. Diverse and specific gene expression responses to stresses in cultured human cells. Mol. Biol. Cell 2004, 15, 2361–2374. [Google Scholar]
- Trinklein, N.D.; Chen, W.C.; Kingston, R.E.; Myers, R.M. Transcriptional regulation and binding of heat shock factor 1 and heat shock factor 2 to 32 human heat shock genes during thermal stress and differentiation. Cell Stress Chaperones 2004, 9, 21–28. [Google Scholar]
- Trinklein, N.D.; Murray, J.I.; Hartman, S.J.; Botstein, D.; Myers, R.M. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol. Biol. Cell 2004, 15, 1254–1261. [Google Scholar]
- Fujimoto, M.; Oshima, K.; Shinkawa, T.; Wang, B.B.; Inouye, S.; Hayashida, N.; Takii, R.; Nakai, A. Analysis of HSF4 binding regions reveals its necessity for gene regulation during development and heat shock response in mouse lenses. J. Biol. Chem. 2008, 283, 29961–29970. [Google Scholar]
- Abane, R.; Mezger, V. Roles of heat shock factors in gametogenesis and development. FEBS J. 2010, 277, 4150–4172. [Google Scholar]
- Lecomte, S.; Desmots, F.; Le Masson, F.; Le Goff, P.; Michel, D.; Christians, E.S.; Le Drean, Y. Roles of heat shock factor 1 and 2 in response to proteasome inhibition: Consequence on p53 stability. Oncogene 2010, 29, 4216–4224. [Google Scholar]
- Westerheide, S.D.; Morimoto, R.I. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J. Biol. Chem. 2005, 280, 33097–33100. [Google Scholar]
- Ohtsuka, H.; Azuma, K.; Murakami, H.; Aiba, H. hsf1 (+) extends chronological lifespan through Ecl1 family genes in fission yeast. Mol. Genet. Genomics 2010, 285, 67–77. [Google Scholar]
- Fujimoto, M.; Takaki, E.; Hayashi, T.; Kitaura, Y.; Tanaka, Y.; Inouye, S.; Nakai, A. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 2005, 280, 34908–34916. [Google Scholar]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar]
- Stephanou, A.; Latchman, D.S. Transcriptional modulation of heat-shock protein gene expression. Biochem. Res. Int. 2010, 2011, 238601. [Google Scholar]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar]
- Akerfelt, M.; Trouillet, D.; Mezger, V.; Sistonen, L. Heat shock factors at a crossroad between stress and development. Ann. NY Acad. Sci. 2007, 1113, 15–27. [Google Scholar]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar]
- Anckar, J.; Sistonen, L. Heat shock factor 1 as a coordinator of stress and developmental pathways. Adv. Exp. Med. Biol. 2007, 594, 78–88. [Google Scholar]
- Bjork, J.K.; Sistonen, L. Regulation of the mem- bers of the mammalian heat shock factor family. FEBS J. 2010, 277, 4126–4139. [Google Scholar]
- Fujimoto, M.; Nakai, A. The heat shock factor family and adaptation to proteotoxic stress. FEBS J. 2010, 277, 4112–4125. [Google Scholar]
- Wu, C. Heat shock transcription factors: structure and regulation. Annu. Rev. Cell Dev. Biol. 1995, 11, 441–469. [Google Scholar]
- Rabindran, S.K.; Haroun, R.I.; Clos, J.; Wisniewski, J.; Wu, C. Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science 1993, 259, 230–234. [Google Scholar]
- Kawazoe, Y.; Nakai, A.; Tanabe, M.; Nagata, K. Proteasome inhibition leads to the activation of all members of the heat-shock-factor family. Eur. J. Biochem. 1998, 255, 356–362. [Google Scholar]
- Takemori, Y.; Enoki, Y.; Yamamoto, N.; Fukai, Y.; Adachi, K.; Sakurai, H. Mutational analysis of human heat-shock transcription factor 1 reveals a regulatory role for oligomerization in DNA-binding specificity. Biochem. J. 2009, 424, 253–261. [Google Scholar]
- Sakurai, H.; Enoki, Y. Novel aspects of heat shock factors: DNA recognition, chromatin modulation and gene expression. FEBS J. 2010, 277, 4140–4149. [Google Scholar]
- He, H.; Soncin, F.; Grammatikakis, N.; Li, Y.; Siganou, A.; Gong, J.; Brown, S.A.; Kingston, R.E.; Calderwood, S.K. Elevated expression of heat shock factor (HSF) 2A stimulates HSF1-induced transcription during stress. J. Biol. Chem. 2003, 278, 35465–35475. [Google Scholar]
- Loison, F.; Debure, L.; Nizard, P.; le Goff, P.; Michel, D.; le Drean, Y. Up-regulation of the clusterin gene after proteotoxic stress: implication of HSF1-HSF2 heterocomplexes. Biochem. J. 2006, 395, 223–231. [Google Scholar]
- Ostling, P.; Bjork, J.K.; Roos-Mattjus, P.; Mezger, V.; Sistonen, L. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J. Biol. Chem. 2007, 282, 7077–7086. [Google Scholar]
- Sandqvist, A.; Bjork, J.K.; Akerfelt, M.; Chitikova, Z.; Grichine, A.; Vourc'h, C.; Jolly, C.; Salminen, T.A.; Nymalm, Y.; Sistonen, L. Heterotrimerization of heat-shock factors 1 and 2 provides a transcriptional switch in response to distinct stimuli. Mol. Biol. Cell 2009, 20, 1340–1347. [Google Scholar]
- Ahlskog, J.K.; Bjork, J.K.; Elsing, A.N.; Aspelin, C.; Kallio, M.; Roos-Mattjus, P.; Sistonen, L. Anaphase-promoting complex/cyclosome participates in the acute response to protein-damaging stress. Mol. Cell. Biol. 2010, 30, 5608–5620. [Google Scholar]
- Sullivan, E.K.; Weirich, C.S.; Guyon, J.R.; Sif, S.; Kingston, R.E. Transcriptional activation domains of human heat shock factor 1 recruit human SWI/SNF. Mol. Cell. Biol. 2001, 21, 5826–5837. [Google Scholar]
- Tu, N.; Hu, Y.; Mivechi, N.F. Heat shock transcription factor (Hsf)-4b recruits Brg1 during the G1 phase of the cell cycle and regulates the expression of heat shock proteins. J. Cell. Biochem. 2006, 98, 1528–1542. [Google Scholar]
- Thomson, S.; Hollis, A.; Hazzalin, C.A.; Mahadevan, L.C. Distinct stimulus-specific histone modifications at hsp70 chromatin targeted by the transcription factor heat shock factor-1. Mol. Cell. 2004, 15, 585–594. [Google Scholar]
- Jolly, C.; Metz, A.; Govin, J.; Vigneron, M.; Turner, B.M.; Khochbin, S.; Vourc'h, C. Stress-induced transcription of satellite III repeats. J. Cell Biol. 2004, 164, 25–33. [Google Scholar]
- Valgardsdottir, R.; Chiodi, I.; Giordano, M.; Cobianchi, F.; Riva, S.; Biamonti, G. Structural and functional characterization of noncoding repetitive RNAs transcribed in stressed human cells. Mol. Biol. Cell 2005, 16, 2597–2604. [Google Scholar]
- Eymery, A.; Souchier, C.; Vourc'h, C.; Jolly, C. Heat shock factor 1 binds to and transcribes satellite II and III sequences at several pericentromeric regions in heat-shocked cells. Exp. Cell Res. 2010, 316, 1845–1855. [Google Scholar]
- Xing, H.; Wilkerson, D.C.; Mayhew, C.N.; Lubert, E.J.; Skaggs, H.S.; Goodson, M.L.; Hong, Y.; Park-Sarge, O.K.; Sarge, K.D. Mechanism of hsp70i gene bookmarking. Science 2005, 307, 421–423. [Google Scholar]
- Christians, E.; Davis, A.A.; Thomas, S.D.; Benjamin, I.J. Maternal effect of Hsf1 on reproductive success. Nature 2000, 407, 693–694. [Google Scholar]
- Metchat, A; Akerfelt, M.; Bierkamp, C; Delsinne, V.; Sistonen, L.; Alexandre, H.; Christians, E.S. Mammalian heat shock factor 1 is essential for oocyte meiosis and directly regulates Hsp90alpha expression. J. Biol. Chem. 2009, 284, 9521–9528. [Google Scholar]
- Bierkamp, C.; Luxey, M.; Metchat, A.; Audouard, C.; Dumollard, R.; Christians, E. Lack of maternal Heat Shock Factor 1 results in multiple cellular and developmental defects, including mitochondrial damage and altered redox homeostasis, and leads to reduced survival of mammalian oocytes and embryos. Dev. Biol. 2010, 339, 338–353. [Google Scholar]
- Santos, S.D.; Saraiva, M.J. Enlarged ventricles, astrogliosis and neurodegeneration in heat shock factor 1 null mouse brain. Neuroscience 2004, 126, 657–663. [Google Scholar]
- Homma, S.; Jin, X.; Wang, G.; Tu, N.; Min, J.; Yanasak, N.; Mivechi, N.F. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in HSF1-deficient mice. J. Neurosci. 2007, 27, 7974–7986. [Google Scholar]
- Uchida, S.; Har, K.; Kobayashi, A.; Fujimoto, M.; Otsuki, K.; Yamagata, H.; Hobara, T.; Abe, N.; Higuchi, F.; Shibata, T.; Hasegawa, S.; Kida, S.; Nakai, A.; Watanabe, Y. Impaired hippocampal spinogenesis and neurogenesis and altered affective behavior in mice lacking heat shock factor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 1681–1686. [Google Scholar]
- Nakai, A.; Suzuki, M.; Tanabe, M. Arrest of spermatogenesis in mice expressing an active heat shock transcription factor 1. EMBO J. 2000, 19, 1545–1554. [Google Scholar]
- Izu, H.; Inouye, S.; Fujimoto, M; Shiraishi, K; Naito, K.; Nakai, A. Heat shock transcription factor 1 is involved in quality-control mechanisms in male germ cells. Biol. Reprod. 2004, 70, 18–24. [Google Scholar]
- Wang, G; Ying, Z.; Jin, X.; Tu, N.; Zhang, Y.; Phillips, M.; Moskophidis, D.; Mivechi, N.F. Essential requirement for both HSF1 and HSF2 transcriptional activity in spermatogenesis and male fertility. Genesis 2004, 38, 66–80. [Google Scholar]
- Takaki, E.; Fujimoto, M.; Sugahara, K.; Nakahari, T.; Yonemura, S.; Tanaka, Y.; Hayashida, N.; Inouye, S.; Takemoto, T.; Yamashita, H.; Nakai, A. Maintenance of olfactory neurogenesis requires HSF1, a major heat shock transcription factor in mice. J. Biol. Chem. 2006, 281, 4931–4937. [Google Scholar]
- Inouye, S.; Izu, H.; Takaki, E.; Suzuki, H.; Shirai, M.; Yokota, Y.; Ichikawa, H.; Fujimoto, M.; Nakai, A. Impaired IgG production in mice deficient for heat shock transcription factor 1. J. Biol. Chem. 2004, 279, 38701–38709. [Google Scholar]
- Zheng, H.; Li, Z. Cutting edge: cross-presentation of cell-associated antigens to MHC class I molecule is regulated by a major transcription factor for heat shock proteins. J. Immunol. 2004, 173, 5929–5933. [Google Scholar]
- Kallio, M.; Chang, Y.; Manuel, M.; Alastalo, T.P; Rallu, M.; Gitton, Y.; Pirkkala, L.; Loones, M.T.; Paslaru, L.; Larney, S.; Hiard, S.; Morange, M.; Sistonen, L.; Mezger, V. Brain abnormalities, defective meiotic chromosome synapsis and female subfertility in HSF2 null mice. EMBO J. 2002, 21, 2591–2601. [Google Scholar]
- Wang, G.; Zhang, J.; Moskophidis, D.; Mivechi, N.F. Targeted disruption of the heat shock transcription factor (HSF)-2 gene results in increased embryonic lethality, neuronal defects, and reduced spermatogenesis. Genesis 2003, 36, 48–61. [Google Scholar]
- Chang, Y.; Ostling, P.; Akerfelt, M.; Trouillet, D.; Rallu, M.; Gitton, Y.; El Fatimy, R.; Fardeau, V.; Le Crom, S.; Morange, M.; Sistonen, L.; Mezger, V. Role of heat-shock factor 2 in cerebral cortex formation and as a regulator of p35 expression. Genes Dev. 2006, 20, 836–847. [Google Scholar]
- Alastalo, T.P.; Lonnstrom, M.; Leppa, S.; Kaarniranta, K.; Pelto-Huikko, M.; Sistonen, L.; Parvinen, M. Stage-specific expression and cellular localization of the heat shock factor 2 isoforms in the rat seminiferous epithelium. Exp. Cell Res. 1998, 240, 16–27. [Google Scholar]
- Bjork, J.K.; Sandqvist, A.; Elsing, A.N.; Kotaja, N.; Sistonen, L. miR-18, a member of Oncomir-1, targets heat shock transcription factor 2 in spermatogenesis. Development 2010, 137, 3177–3184. [Google Scholar]
- Akerfelt, M.; Henriksson, E.; Laiho, A.; Vihervaara, A.; Rautoma, K.; Kotaja, N.; Sistonen, L. Promoter ChIP-chip analysis in mouse testis reveals Y chromosome occupancy by HSF2. Proc. Natl. Acad. Sci. USA 2008, 105, 11224–11229. [Google Scholar]
- Fujimoto, M.; Hayashida, N.; Katoh, T.; Oshima, K.; Shinkawa, T.; Prakasam, R.; Tan, K.; Inouye, S.; Takii, R.; Nakai, A. A novel mouse HSF3 has the potential to activate nonclassical heat-shock genes during heat shock. Mol. Biol. Cell 2010, 21, 106–116. [Google Scholar]
- Tanabe, M.; Nakai, A.; Kawazoe, Y.; Nagata, K. Different thresholds in the responses of two heat shock transcription factors, HSF1 and HSF3. J. Biol. Chem. 1997, 272, 15389–15395. [Google Scholar]
- Nakai, A.; Tanabe, M; Kawazoe, Y.; Inazawa, J.; Morimoto, R.I.; Nagata, K. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol. Cell. Biol. 1997, 17, 469–481. [Google Scholar]
- Somasundaram, T.; Bhat, S.P. Canonical heat shock element in the alpha B-crystallin gene shows tissue-specific and developmentally controlled interactions with heat shock factor. J. Biol. Chem. 2000, 275, 17154–17159. [Google Scholar]
- Nakai, A. Heat shock transcription factors and sensory placode development. BMB Rep. 2009, 42, 631–635. [Google Scholar]
- Fujimoto, M.; Izu, H.; Seki, K.; Fukuda, K.; Nishida, T.; Yamada, S.; Kato, K.; Yonemura, S.; Inouye, S.; Nakai, A. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J. 2004, 23, 4297–4306. [Google Scholar]
- Takaki, E.; Fujimoto, M.; Nakahari, T.; Yonemura, S.; Miyata, Y.; Hayashida, N.; Yamamoto, K.; Vallee, R.B.; Mikuriya, T.; Sugahara, K.; Yamashita, H.; Inouye, S.; Nakai, A. Heat shock transcription factor 1 is required for maintenance of ciliary beating in mice. J. Biol. Chem. 2007, 282, 37285–37292. [Google Scholar]
- Shi, X.; Cui, B.; Wang, Z.; Weng, L.; Xu, Z.; Ma, J.; Xu, G.; Kong, X.; Hu, L. Removal of Hsf4 leads to cataract development in mice through down-regulation of gamma S-crystallin and Bfsp expression. BMC Mol. Biol. 2009, 10, 10. [Google Scholar]
- Akerfelt, M.; Vihervaara, A.; Laiho, A.; Conter, A.; Christians, E.S.; Sistonen, L.; Henriksson, E. Heat shock transcription factor 1 localizes to sex chromatin during meiotic repression. J. Biol. Chem. 2010, 285, 34469–34476. [Google Scholar]
- Meng, L.; Gabai, V.L.; Sherman, M.Y. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 2010, 29, 5204–5213. [Google Scholar]
- Elenbaas, B.; Spirio, L.; Koerner, F.; Fleming, M.D.; Zimonjic, D.B.; Donaher, J.L.; Popescu, N.C.; Hahn, W.C.; Weinberg, R.A. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001, 15, 50–65. [Google Scholar]
- O'Callaghan-Sunol, C.; Sherman, M.Y. Heat shock transcription factor (HSF1) plays a critical role in cell migration via maintaining MAP kinase signaling. Cell Cycle 2006, 5, 1431–1437. [Google Scholar]
- Gallo, G.J.; Prentice, H.; Kingston, R.E. Heat shock factor is required for growth at normal temperatures in the fission yeast Schizosaccharomyces pombe. Mol. Cell. Biol. 1993, 13, 749–761. [Google Scholar]
- Hahn, J.S.; Hu, Z.; Thiele, D.J.; Iyer, V.R. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 2004, 24, 5249–5256. [Google Scholar]
- Xiao, X.; Zuo, X.; Davis, A.A.; McMillan, D.R.; Curry, B.B.; Richardson, J.A.; Benjamin, I.J. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999, 18, 5943–5952. [Google Scholar]
- Whitesell, L.; Lindquist, S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Exp. Opin. Ther. Targets 2009, 13, 469–478. [Google Scholar]
- Tang, D.; Khaleque, M.A.; Jones, E.L.; Theriault, J.R.; Li, C.; Wong, W.H.; Stevenson, M.A.; Calderwood, S.K. Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones 2005, 10, 46–58. [Google Scholar]
- Hoang, A.T.; Huang, J.; Rudra-Ganguly, N.; Zheng, J.; Powell, W.C.; Rabindran, S.K.; Wu, C.; Roy-Burman, P. A novel association between the human heat shock transcription factor 1 (HSF1) and prostate adenocarcinoma. Am. J. Pathol. 2000, 156, 857–864. [Google Scholar]
- Kurahashi, T.; Miyake, H.; Hara, I.; Fujisawa, M. Expression of major heat shock proteins in prostate cancer: correlation with clinicopathological outcomes in patients undergoing radical prostatectomy. J. Urol. 2007, 177, 757–761. [Google Scholar]
- Wang, Y.; Theriault, J.R.; He, H.; Gong, J.; Calderwood, S.K. Expression of a dominant negative heat shock factor-1 construct inhibits aneuploidy in prostate carcinoma cells. J. Biol. Chem. 2004, 279, 32651–32659. [Google Scholar]
- Khaleque, M.A.; Bharti, A.; Sawyer, D.; Gong, J.; Benjamin, I.J.; Stevenson, M.A.; Calderwood, S.K. Induction of heat shock proteins by heregulin beta1 leads to protection from apoptosis and anchorage-independent growth. Oncogene 2005, 24, 6564–6573. [Google Scholar]
- Fanelli, M.A.; Montt-Guevara, M.; Diblasi, A.M.; Gago, F.E.; Tello, O.; Cuello-Carrion, F.D.; Callegari, E.; Bausero, M.A.; Ciocca, D.R. P-cadherin and beta-catenin are useful prognostic markers in breast cancer patients; beta-catenin interacts with heat shock protein Hsp27. Cell Stress Chaperones 2008, 13, 207–220. [Google Scholar]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar]
- Cen, H.; Zheng, S.; Fang, Y.M.; Tang, X.P.; Dong, Q. Induction of HSF1 expression is associated with sporadic colorectal cancer. World J. Gastroenterol. 2004, 10, 3122–3126. [Google Scholar]
- Jacobs, A.T.; Marnett, L.J. HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J. Biol. Chem. 2009, 284, 9176–9183. [Google Scholar]
- Potter, J.D.; Bigler, J.; Fosdick, L.; Bostick, R.M.; Kampman, E.; Chen, C.; Louis, T.A.; Grambsch, P. Colorectal adenomatous and hyperplastic polyps: smoking and N-acetyltransferase 2 polymorphisms. Cancer Epidemiol. Biomarker. Prev. 1999, 8, 69–75. [Google Scholar]
- Tejpar, S.; Li, C.; Yu, C.; Poon, R.; Denys, H.; Sciot, R.; Van Cutsem, E.; Cassiman, J.J.; Alman, B.A. Tcf-3 expression and beta-catenin mediated transcriptional activation in aggressive fibromatosis (desmoid tumour). Br. J. Cancer 2001, 85, 98–101. [Google Scholar]
- Ehrenfried, J.A.; Herron, B.E.; Townsend, C.M., Jr.; Evers, B.M. Heat shock proteins are differentially expressed in human gastrointestinal cancers. Surg. Oncol. 1995, 4, 197–203. [Google Scholar]
- Wang, J.; He, H.; Yu, L.; Xia, H.H.; Lin, M.C.; Gu, Q.; Li, M.; Zou, B.; An, X.; Jiang, B.; Kung, H.F.; Wong, B.C. HSF1 down-regulates XAF1 through transcriptional regulation. J. Biol. Chem. 2006, 281, 2451–2459. [Google Scholar]
- Park, J.; Jung, Y; Kim, J.; Kim, K.Y.; Ahn, S.G.; Song, K.; Lee, I. TC1 (C8orf4) is upregulated by cellular stress and mediates heat shock response. Biochem. Biophys. Res. Commun. 2007, 360, 447–452. [Google Scholar]
- Kim, B.; Koo, H.; Yang, S.; Bang, S.; Jung, Y.; Kim, Y.; Kim, J.; Park, J.; Moon, R.T.; Song, K.; Lee, I. TC1(C8orf4) correlates with Wnt/beta-catenin target genes and aggressive biological behavior in gastric cancer. Clin. Cancer Res. 2006, 12, 3541–3548. [Google Scholar]
- Strik, H.M.; Weller, M.; Frank, B.; Hermisson, M.; Deininger, M.H.; Dichgans, J.; Meyermann, R. Heat shock protein expression in human gliomas. Anticancer Res. 2000, 20, 4457–4462. [Google Scholar]
- Mustafa, D.A.; Sieuwerts, A.M.; Zheng, P.P.; Kros, J.M. Overexpression of colligin 2 in glioma vasculature is associated with overexpression of heat shock factor 2. Gene Regul. Syst. Biol. 2010, 4, 103–107. [Google Scholar]
- Hu, Y.; Mivechi, N.F. Association and regulation of heat shock transcription factor 4b with both extracellular signal-regulated kinase mitogen-activated protein kinase and dual-specificity tyrosine phosphatase DUSP26. Mol. Cell. Biol. 2006, 26, 3282–3294. [Google Scholar]
- Tanikawa, J.; Ichikawa-Iwata, E.; Kanei-Ishii, C.; Nakai, A.; Matsuzawa, S.; Reed, J.C.; Ishii, S. p53 suppresses the c-Myb-induced activation of heat shock transcription factor 3. J. Biol. Chem. 2000, 275, 15578–15585. [Google Scholar]
- Min, J.N.; Huang, L.; Zimonjic, D.B.; Moskophidis, D.; Mivechi, N.F. Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 2007, 26, 5086–5097. [Google Scholar]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar]
- Paul, S. Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: therapeutic approaches. Bioessays 2008, 30, 1172–1184. [Google Scholar]
- Borissenko, L.; Groll, M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem. Rev. 2007, 107, 687–717. [Google Scholar]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Altieri, D.C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003, 22, 8581–8589. [Google Scholar]
- Khaleque, M.A.; Bharti, A.; Gong, J.; Gray, P.J.; Sachdev, V.; Ciocca, D.R.; Stati, A.; Fanelli, M.; Calderwood, S.K. Heat shock factor 1 represses estrogen-dependent transcription through association with MTA1. Oncogene 2008, 27, 1886–1893. [Google Scholar]
- Martin, M.D.; Hilsenbeck, S.G.; Mohsin, S.K.; Hopp, T.A.; Clark, G.M.; Osborne, C.K.; Allred, D.C.; O'Connell, P. Breast tumors that overexpress nuclear metastasis-associated 1 (MTA1) protein have high recurrence risks but enhanced responses to systemic therapies. Breast Cancer Res. Treat. 2006, 95, 7–12. [Google Scholar]
- Hofer, M.D.; Kuefer, R.; Varambally, S.; Li, H.; Ma, J.; Shapiro, G.I.; Gschwend, J.E.; Hautmann, R.E.; Sanda, M.G.; Giehl, K.; Menke, A.; Chinnaiyan, A.M.; Rubin, M.A. The role of metastasis-associated protein 1 in prostate cancer progression. Cancer Res. 2004, 64, 825–829. [Google Scholar]
- Toh, Y.; Oki, E.; Oda, S.; Tokunaga, E.; Ohno, S.; Maehara, Y.; Nicolson, G.L.; Sugimachi, K. Overexpression of the MTA1 gene in gastrointestinal carcinomas: correlation with invasion and metastasis. Int. J. Cancer 1997, 74, 459–463. [Google Scholar]
- Biamonti, G. Nuclear stress bodies: a heterochromatin affair? Nat. Rev. Mol. Cell Biol. 2004, 5, 493–498. [Google Scholar]
- Eymery, A.; Callanan, M.; Vourc'h, C. The secret message of heterochromatin: new insights into the mechanisms and function of centromeric and pericentric repeat sequence transcription. Int. J. Dev. Biol. 2009, 53, 259–268. [Google Scholar]
- King, B.L.; Carcangiu, M.L.; Carter, D.; Kiechle, M.; Pfisterer, J.; Pfleiderer, A.; Kacinski, B.M. Microsatellite instability in ovarian neoplasms. Br. J. Cancer 1995, 72, 376–382. [Google Scholar]
- Lee, J.D.; Unger, E.R.; Gittenger, C.; Lee, D.R.; Hebert, R.; Maize, J.C. Interphase cytogenetic analysis of 1q12 satellite III DNA in melanocytic lesions: increased aneuploidy with malignant histology. Am. J. Dermatopathol. 2001, 23, 176–180. [Google Scholar]
- Flactif, M.; Zandecki, M.; Lai, J.L.; Bernardi, F.; Obein, V.; Bauters, F.; Facon, T. Interphase fluorescence in situ hybridization (FISH) as a powerful tool for the detection of aneuploidy in multiple myeloma. Leukemia 1995, 9, 2109–2114. [Google Scholar]
- Lee, Y.J.; Lee, H.J.; Lee, J.S.; Jeoung, D.; Kang, C.M.; Bae, S.; Lee, S.J.; Kwon, S.H.; Kang, D.; Lee, Y.S. A novel function for HSF1-induced mitotic exit failure and genomic instability through direct interaction between HSF1 and Cdc20. Oncogene 2008, 27, 2999–3009. [Google Scholar]
- Lee, Y.J.; Kim, E.H.; Lee, J.S.; Jeoung, D.; Bae, S.; Kwon, S.H.; Lee, Y.S. HSF1 as a mitotic regulator: Phosphorylation of HSF1 by Plk1 is essential for mitotic progression. Cancer Res. 2008, 68, 7550–7560. [Google Scholar]
- Kavak, E.; Najafov, A.; Ozturk, N.; Seker, T.; Cavusoglu, K.; Aslan, T.; Duru, A.D.; Saygili, T.; Hoxhaj, G.; Hiz, M.C.; Unal, D.O.; Birgül-Iyison, N.; Ozturk, M.; Koman, A. Analysis of the Wnt/B-catenin/TCF4 pathway using SAGE, genome-wide microarray and promoter analysis: Identification of BRI3 and HSF2 as novel targets. Cell Signal. 2010, 22, 1523–1535. [Google Scholar]
- Schutte, J.; Viallet, J.; Nau, M.; Segal, S.; Fedorko, J.; Minna, J. jun-B inhibits and c-fos stimulates the transforming and trans-activating activities of c-jun. Cell 1989, 59, 987–997. [Google Scholar]
- Wilkerson, D.C.; Skaggs, H.S.; Sarge, K.D. HSF2 binds to the Hsp90, Hsp27, and c-Fos promoters constitutively and modulates their expression. Cell Stress Chaperones 2007, 12, 283–290. [Google Scholar]
- Cho, K.; Ishiwata, T.; Uchida, E; Nakazawa, N.; Korc, M.; Naito, Z.; Tajiri, T. Enhanced expression of keratinocyte growth factor and its receptor correlates with venous invasion in pancreatic cancer. Am. J. Pathol. 2007, 170, 1964–1974. [Google Scholar]
- Hishikawa, Y.; Tamaru, N.; Ejima, K.; Hayashi, T.; Koji, T. Expression of keratinocyte growth factor and its receptor in human breast cancer: its inhibitory role in the induction of apoptosis possibly through the overexpression of Bcl-2. Arch. Histol. Cytol. 2004, 67, 455–464. [Google Scholar]
- Shaoul, R.; Eliahu, L.; Sher, I.; Hamlet, Y.; Miselevich, I.; Goldshmidt, O.; Ron, D. Elevated expression of FGF7 protein is common in human gastric diseases. Biochem. Biophys. Res. Commun. 2006, 350, 825–833. [Google Scholar]
- Chikama, T.; Liu, C.Y.; Meij, J.T.; Hayashi, Y.; Wang, I.J.; Yang, L.; Nishida, T.; Kao, W.W. Excess FGF-7 in corneal epithelium causes corneal intraepithelial neoplasia in young mice and epithelium hyperplasia in adult mice. Am. J. Pathol. 2008, 172, 638–649. [Google Scholar]
- Rossi, A.; Ciafre, S.; Balsamo, M.; Pierimarchi, P.; Santoro, M.G. Targeting the heat shock factor 1 by RNA interference: a potent tool to enhance hyperthermochemotherapy efficacy in cervical cancer. Cancer Res. 2006, 66, 7678–7685. [Google Scholar]
- Akagawa, H.; Takano, Y.; Ishii, A.; Mizuno, S.; Izui, R.; Sameshima, T.; Kawamura, N.; Dobashi, K.; Yoshioka, T. Stresgenin B, an inhibitor of heat-induced heat shock protein gene expression, produced by Streptomyces sp. AS-9. J. Antibiot. (Tokyo) 1999, 52, 960–970. [Google Scholar]
- Limtrakul, P.; Khantamat, O.; Pintha, K. Inhibition of P-glycoprotein function and expression by kaempferol and quercetin. J. Chemother. 2005, 17, 86–95. [Google Scholar]
- Jakubowicz-Gil, J.; Paduch, R.; Piersiak, T.; Glowniak, K.; Gawron, A.; Kandefer-Szerszen, M. The effect of quercetin on pro-apoptotic activity of cisplatin in HeLa cells. Biochem. Pharmacol. 2005, 69, 1343–1350. [Google Scholar]
- Shen, F.; Herenyiova, M.; Weber, G. Synergistic down-regulation of signal transduction and cytotoxicity by tiazofurin and quercetin in human ovarian carcinoma cells. Life Sci. 1999, 64, 1869–1876. [Google Scholar]
- Zanini, C.; Giribaldi, G.; Mandili, G.; Carta, F.; Crescenzio, N.; Bisaro, B.; Doria, A.; Foglia, L.; di Montezemolo, L.C.; Timeus, F.; Turrini, F. Inhibition of heat shock proteins (HSP) expression by quercetin and differential doxorubicin sensitization in neuroblastoma and Ewing's sarcoma cell lines. J. Neurochem. 2007, 103, 1344–1354. [Google Scholar]
- Asea, A.; Ara, G.; Teicher, B.A.; Stevenson, M.A.; Calderwood, S.K. Effects of the flavonoid drug quercetin on the response of human prostate tumours to hyperthermia in vitro and in vivo. Int. J. Hyperther. 2001, 17, 347–356. [Google Scholar]
- Westerheide, S.D.; Kawahara, T.L.; Orton, K.; Morimoto, R.I. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J. Biol. Chem. 2006, 281, 9616–9622. [Google Scholar]
- Phillips, P.A.; Dudeja, V.; McCarroll, J.A.; Borja-Cacho, D.; Dawra, R.K.; Grizzle, W.E.; Vickers, S.M.; Saluja, A.K. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007, 67, 9407–9416. [Google Scholar]
- Neznanov, N.; Gorbachev, A.V.; Neznanova, L.; Komarov, A.P.; Gurova, K.V.; Gasparian, A.V.; Banerjee, A.K.; Almasan, A.; Fairchild, R.L.; Gudkov, A.V. Anti-malaria drug blocks proteotoxic stress response: anti-cancer implications. Cell Cycle 2009, 8, 3960–3970. [Google Scholar]
- Mathew, A.; Mathur, S.K.; Morimoto, R.I. Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 1998, 18, 5091–5098. [Google Scholar]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.P.; Vourc'h, C.; Matthias, P.; Khochbin, S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181. [Google Scholar]
- Conde, R.; Belak, Z.R.; Nair, M.; O'Carroll, R.F.; Ovsenek, N. Modulation of Hsf1 activity by novobiocin and geldanamycin. Biochem. Cell Biol. 2009, 87, 845–851. [Google Scholar]
- Zaarur, N.; Gabai, V.L.; Porco, J.A., Jr.; Calderwood, S.; Sherman, M.Y. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 2006, 66, 1783–1791. [Google Scholar]
- Au, Q.; Zhang, Y.; Barber, J.R.; Ng, S.C.; Zhang, B. Identification of inhibitors of HSF1 functional activity by high-content target-based screening. J. Biomol. Screen. 2009, 14, 1165–1175. [Google Scholar]
- Wang, C.; Dai, Z.; Fan, R.; Deng, Y.; Lv, G.; Lu, G. HSF1 overexpression enhances oncolytic effect of replicative adenovirus. J. Transl. Med. 2010, 8, 44. [Google Scholar]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; van der Heijden, J.W.; Ylstra, B.; Peters, G.J.; Kaspers, G.L.; Dijkmans, B.A.; Scheper, R.J.; Jansen, G. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar]
- Ruckrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar]
- Lu, A.; Ran, R.; Parmentier-Batteur, S.; Nee, A.; Sharp, F.R. Geldanamycin induces heat shock proteins in brain and protects against focal cerebral ischemia. J. Neurochem. 2002, 81, 355–364. [Google Scholar]
- Wang, J.; Gines, S.; MacDonald, M.E.; Gusella, J.F. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci. 2005, 6, 1. [Google Scholar]
- Ciocca, D.R.; Fanelli, M.A.; Cuello-Carrion, F.D.; Castro, G.N. Heat shock proteins in prostate cancer: from tumorigenesis to the clinic. Int. J. Hyperther. 2010, 26, 737–747. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Thonel, A.; Mezger, V.; Garrido, C. Implication of Heat Shock Factors in Tumorigenesis: Therapeutical Potential. Cancers 2011, 3, 1158-1181. https://doi.org/10.3390/cancers3011158
De Thonel A, Mezger V, Garrido C. Implication of Heat Shock Factors in Tumorigenesis: Therapeutical Potential. Cancers. 2011; 3(1):1158-1181. https://doi.org/10.3390/cancers3011158
Chicago/Turabian StyleDe Thonel, Aurelie, Valerie Mezger, and Carmen Garrido. 2011. "Implication of Heat Shock Factors in Tumorigenesis: Therapeutical Potential" Cancers 3, no. 1: 1158-1181. https://doi.org/10.3390/cancers3011158
APA StyleDe Thonel, A., Mezger, V., & Garrido, C. (2011). Implication of Heat Shock Factors in Tumorigenesis: Therapeutical Potential. Cancers, 3(1), 1158-1181. https://doi.org/10.3390/cancers3011158