A Distinct Slow-Cycling Cancer Stem-like Subpopulation of Pancreatic Adenocarcinoma Cells is maintained in Vivo

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
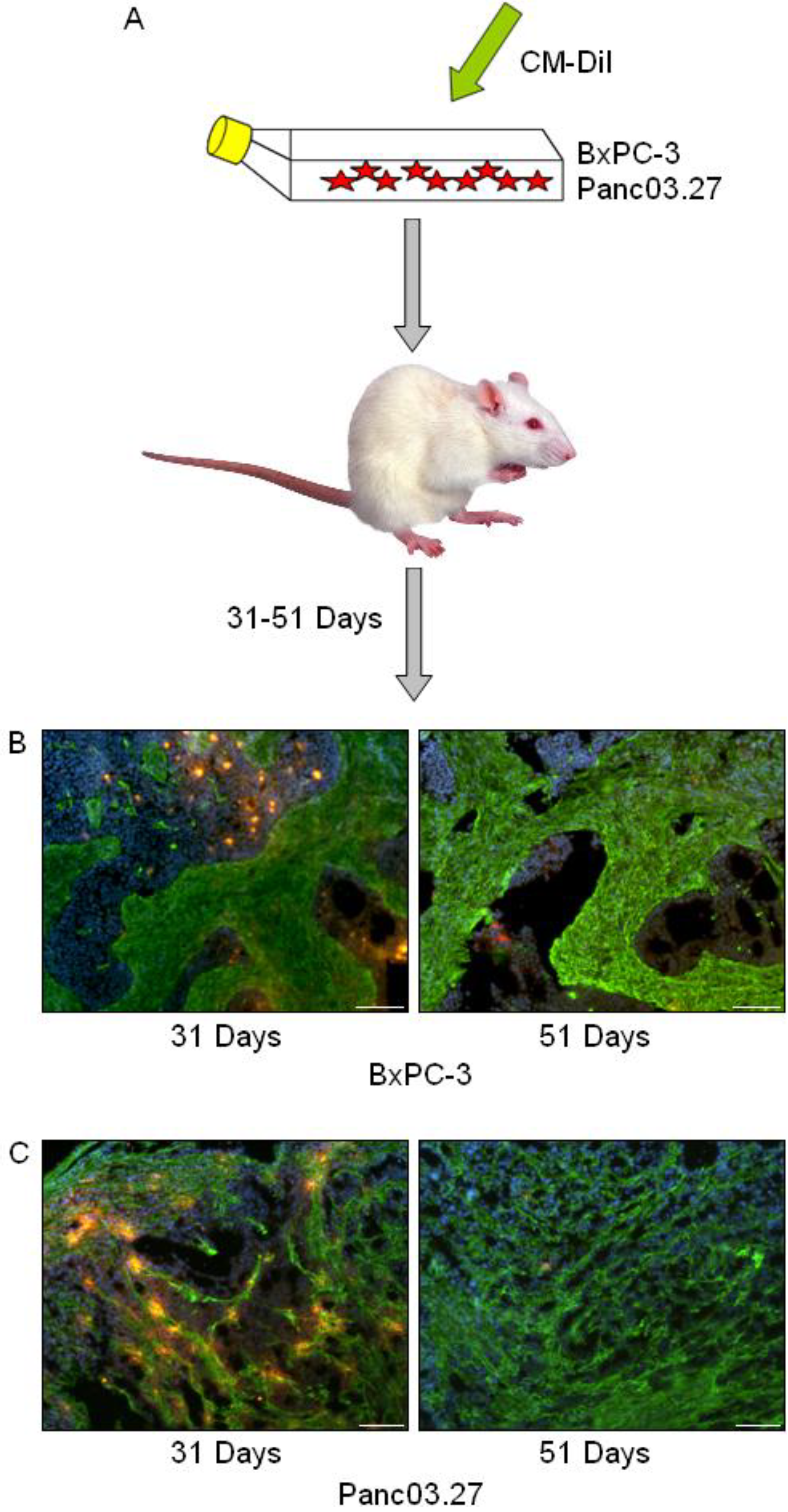
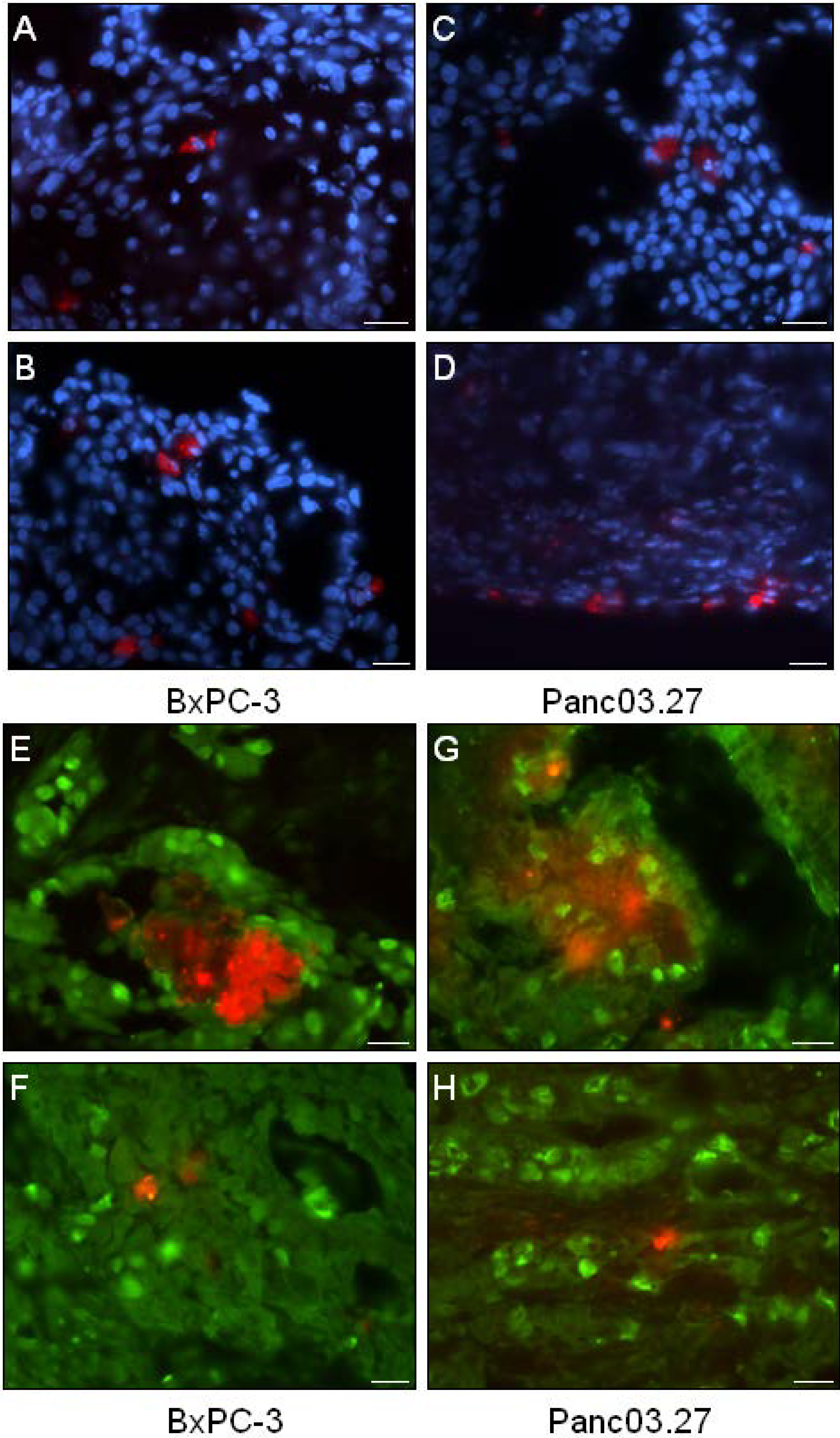
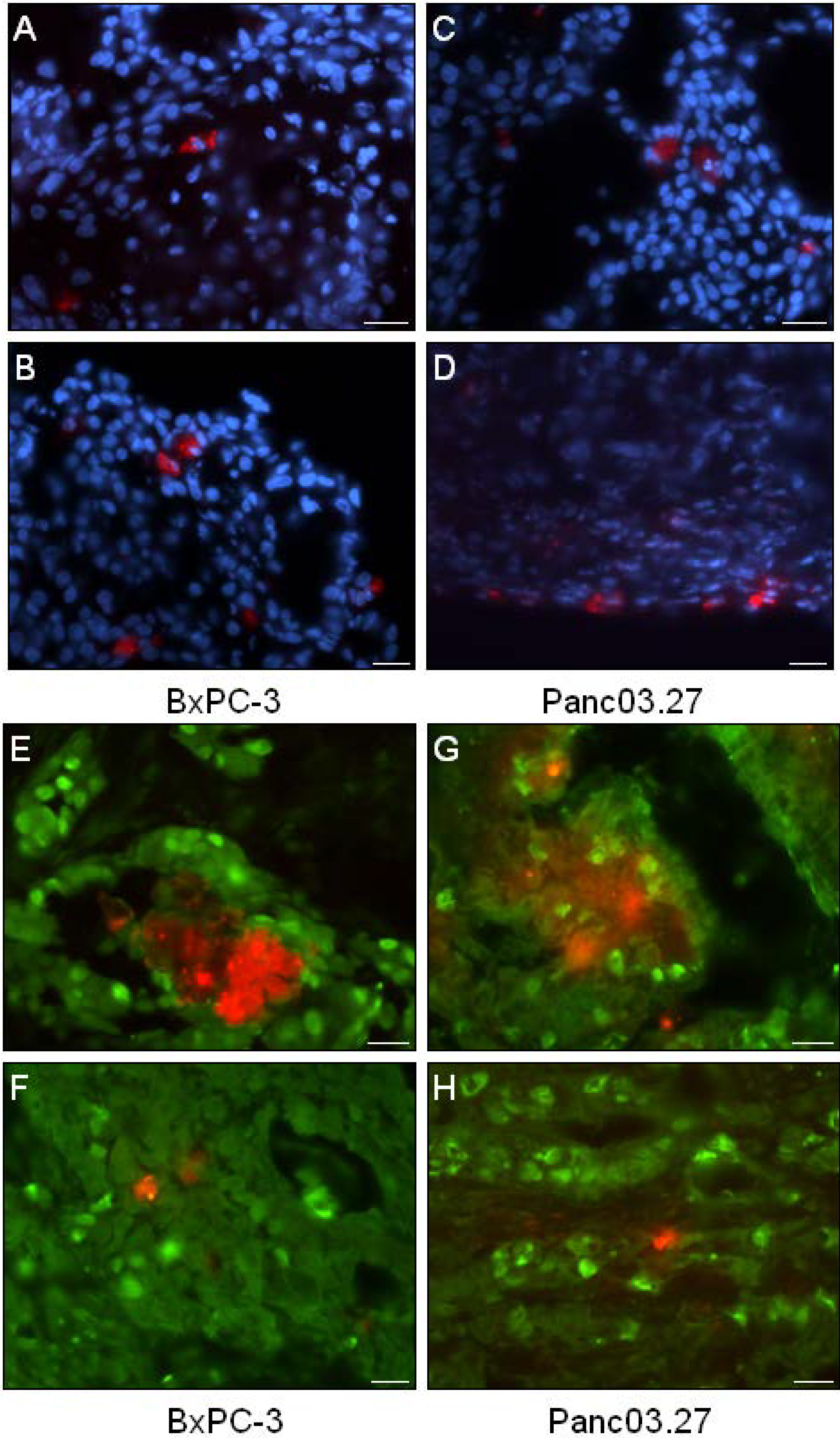
2.1. In Vivo DiI+/SCC Identification

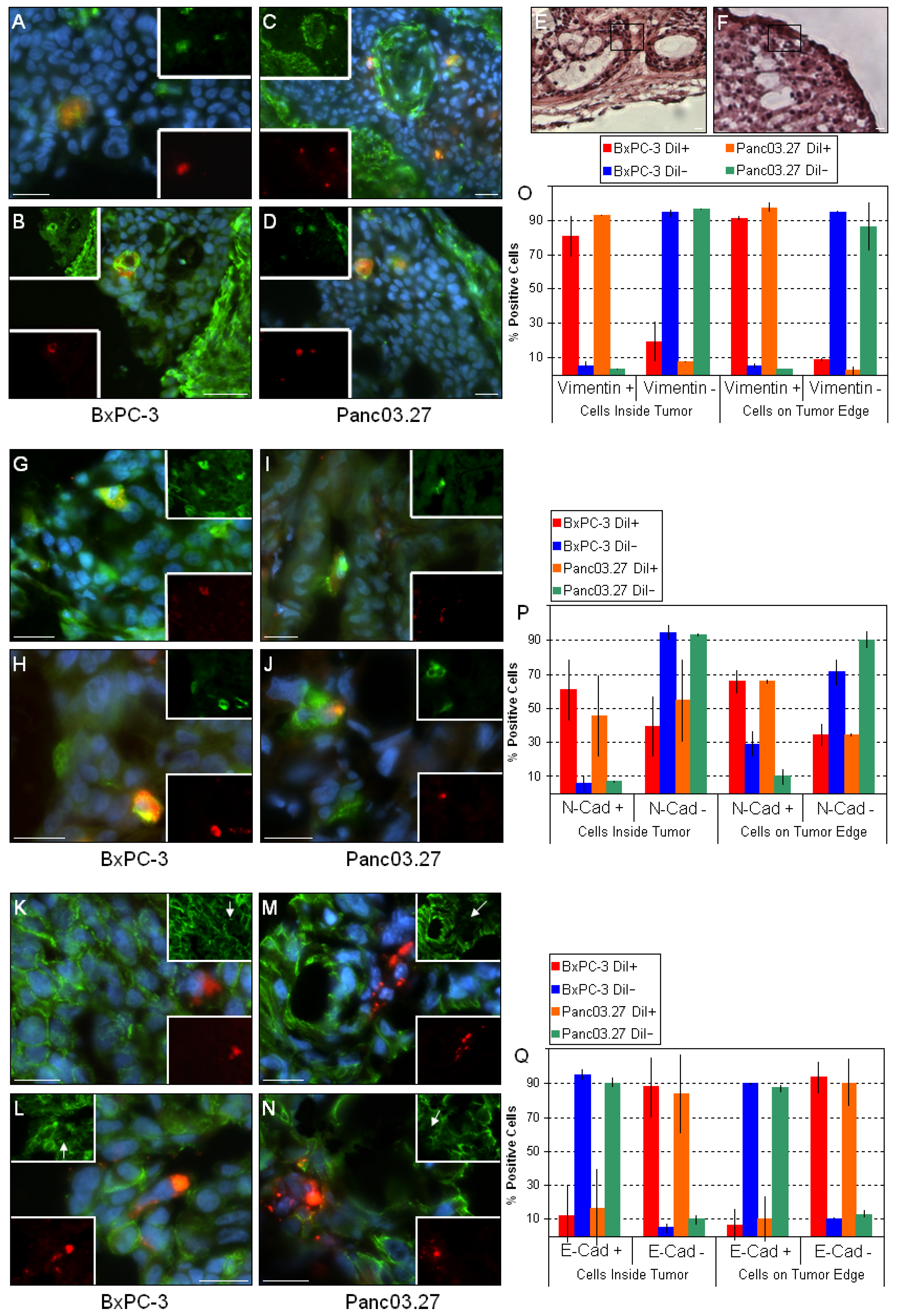
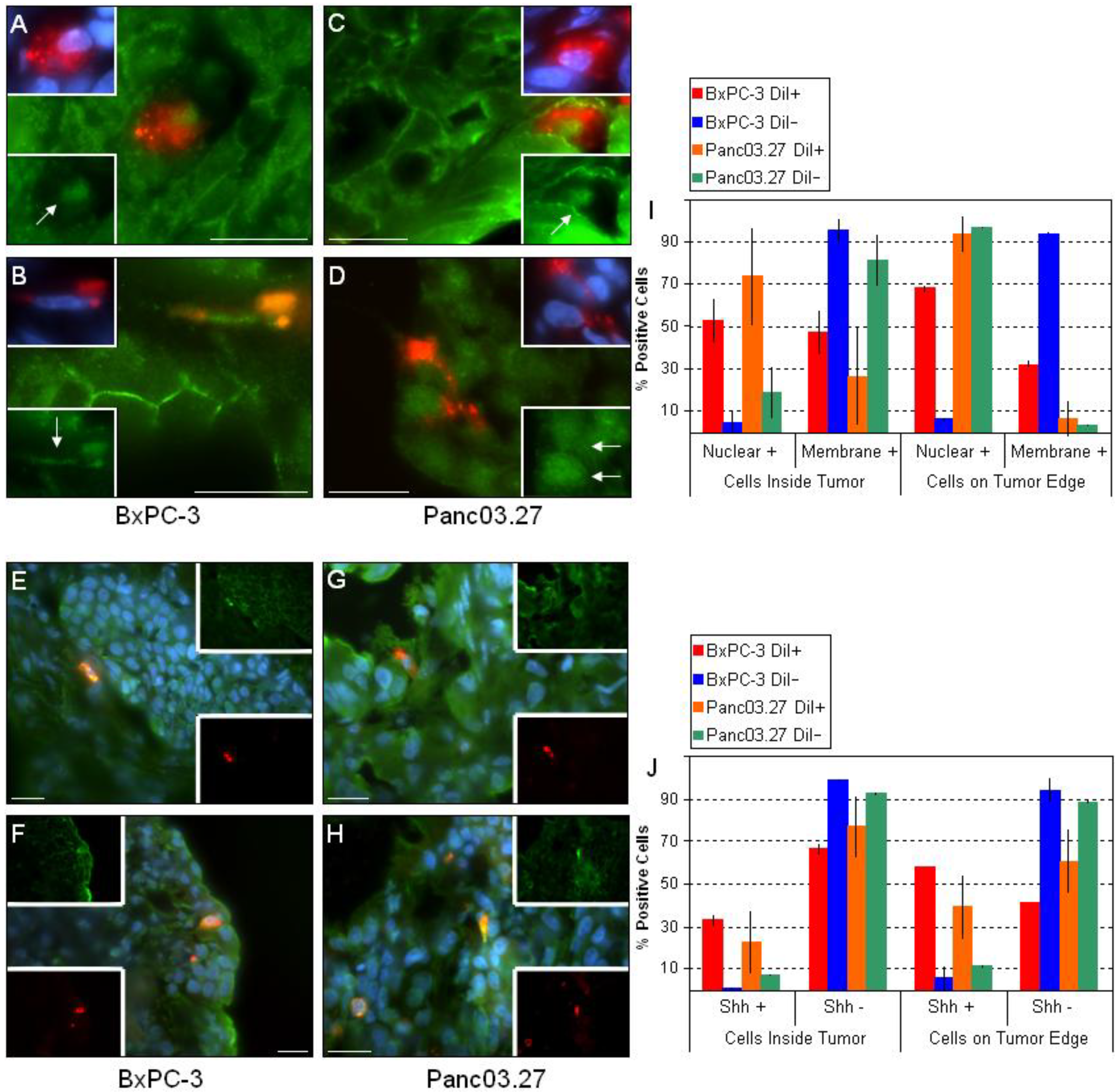
2.2. DiI+/SCC Form a Distinct Subpopulation In Vivo
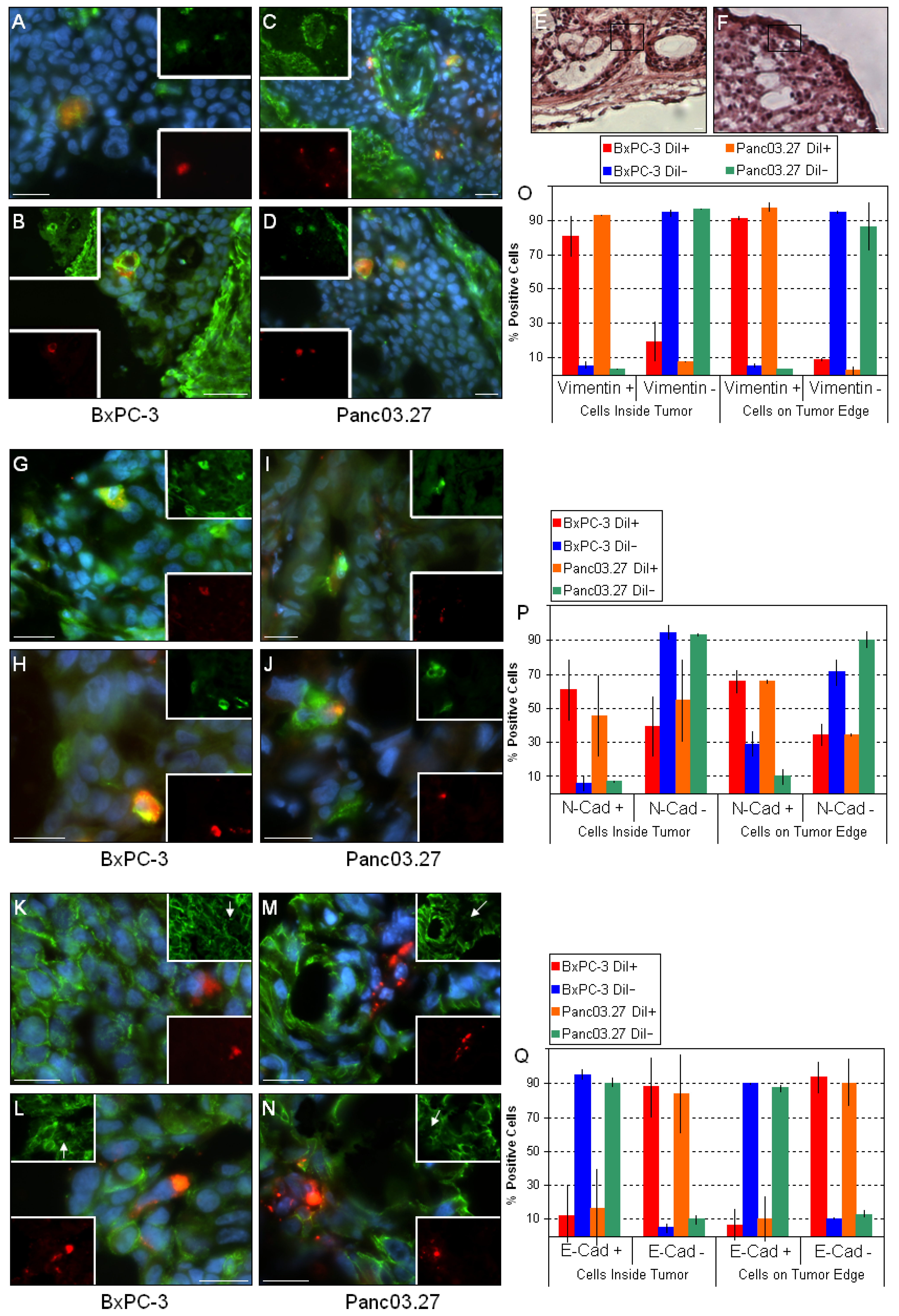
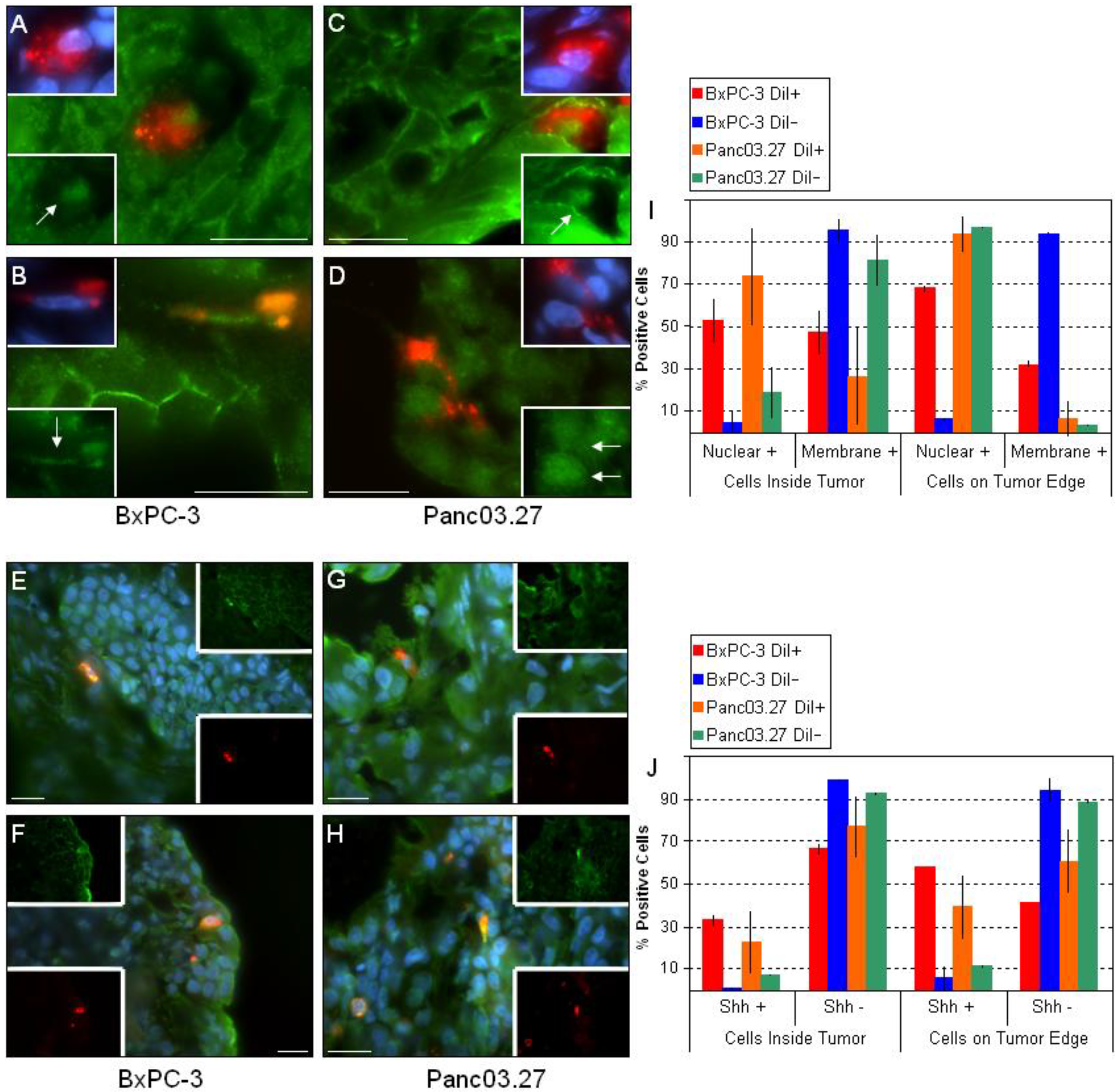
3. Experimental Section
3.1. Cells and Culture Conditions
3.2. Animals
3.3. Tumor Establishment/ In Vivo DiI+/SCC
3.4. Immunohistochemistry/immunofluorescence (IHC/IF)
4. Conclusions
Acknowledgements
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; Hong, S.M.; Fu, B.; Lin, M.T.; Calhoun, E.S.; Kamiyama, M.; Walter, K.; Nikolskaya, T.; Nikolsky, Y.; Hartigan, J.; Smith, D.R.; Hidalgo, M.; Leach, S.D.; Klein, A.P.; Jaffee, E.M.; Goggins, M.; Maitra, A.; Iacobuzio-Donahue, C.; Eshleman, J.R.; Kern, S.E.; Hruban, R.H.; Karchin, R.; Papadopoulos, N.; Parmigiani, G.; Vogelstein, B.; Velculescu, V.E.; Kinzler, K.W. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef]
- Lee, C.J.; Dosch, J.; Simeone, D.M. Pancreatic cancer stem cells. J. Clin. Oncol. 2008, 26, 2806–2812. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; ito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De, M.R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Zabierowski, S.E.; Herlyn, M. Melanoma stem cells: The dark seed of melanoma. J. Clin. Oncol. 2008, 26, 2890–2894. [Google Scholar] [CrossRef]
- Huang, P.; Wang, C.Y.; Gou, S.M.; Wu, H.S.; Liu, T.; Xiong, J.X. Isolation and biological analysis of tumor stem cells from pancreatic adenocarcinoma. World J. Gastroenterol. 2008, 14, 3903–3907. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Krishnamurthy, K.; Wang, G.; Rokhfeld, D.; Bieberich, E. Deoxycholate promotes survival of breast cancer cells by reducing the level of pro-apoptotic ceramide. Breast Cancer Res. 2008, 10, R106. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; Schott, A.; Hayes, D.; Birnbaum, D.; Wicha, M.S.; Dontu, G. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef]
- Lie, D.C.; Colamarino, S.A.; Song, H.J.; Desire, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Dearie, A.R.; Gage, F.H. Wnt signalling regulates adult hippocampal neurogenesis. Nature 2005, 437, 1370–1375. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-Del, C.C.; Yajnik, V.; Antoniu, B.; McMahon, M.; Warshaw, A.L.; Hebrok, M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef]
- Li, X.; Deng, W.; Lobo-Ruppert, S.M.; Ruppert, J.M. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by beta-catenin. Oncogene 2007, 26, 4489–4498. [Google Scholar] [CrossRef]
- Malanchi, I.; Huelsken, J. Cancer stem cells: Never Wnt away from the niche. Curr. Opin. Oncol. 2009, 21, 41–46. [Google Scholar] [CrossRef]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef]
- Le, N.H.; Franken, P.; Fodde, R. Tumour-stroma interactions in colorectal cancer: Converging on beta-catenin activation and cancer stemness. Br. J. Cancer 2008, 98, 1886–1893. [Google Scholar] [CrossRef]
- Rasola, A.; Fassetta, M.; De, B.F.; D'Alessandro, L.; Gramaglia, D.; Di Renzo, M.F.; Comoglio, P.M. A positive feedback loop between hepatocyte growth factor receptor and beta-catenin sustains colorectal cancer cell invasive growth. Oncogene 2007, 26, 1078–1087. [Google Scholar] [CrossRef]
- Yang, L.; Lin, C.; Liu, Z.R. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell 2006, 127, 139–155. [Google Scholar] [CrossRef]
- Dembinski, J.L.; Spaeth, E.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One 2009, 4, e4992. [Google Scholar] [CrossRef]
- Hall, B.; Dembinski, J.; Sasser, A.K.; Studeny, M.; Andreeff, M.; Marini, F. Mesenchymal stem cells in cancer: Tumor-associated fibroblasts and cell-based delivery vehicles. Int. J. Hematol. 2007, 86, 8–16. [Google Scholar] [CrossRef]
- Vermeulen, L.; Melo, De Sousa E.; van der, H.M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; Sprick, M.R.; Kemper, K.; Richel, D.J.; Stassi, G.; Medema, J.P. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Liu, L.K.; Jiang, X.Y.; Zhou, X.X.; Wang, D.M.; Song, X.L.; Jiang, H.B. Upregulation of vimentin and aberrant expression of E-cadherin/beta-catenin complex in oral squamous cell carcinomas: Correlation with the clinicopathological features and patient outcome. Mod. Pathol. 2010, 23, 213–224. [Google Scholar] [CrossRef]
- Bryne, M.; Boysen, M.; Alfsen, C.G.; Abeler, V.M.; Sudbo, J.; Nesland, J.M.; Kristensen, G.B.; Piffko, J.; Bankfalvi, A. The invasive front of carcinomas. The most important area for tumour prognosis? Anticancer Res. 1998, 18, 4757–4764. [Google Scholar]
- Ganepola, G.A.; Mazziotta, R.M.; Weeresinghe, D.; Corner, G.A.; Parish, C.J.; Chang, D.H.; Tebbutt, N.C.; Murone, C.; Ahmed, N.; Augenlicht, L.H.; Mariadason, J.M. Gene expression profiling of primary and metastatic colon cancers identifies a reduced proliferative rate in metastatic tumors. Clin. Exp. Metastasis 2010, 27, 1–9. [Google Scholar] [CrossRef]
- De, W.O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447. [Google Scholar] [CrossRef]
- Mueller, M.T.; Hermann, P.C.; Witthauer, J.; Rubio-Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D'Haese, J.G.; Schoenberg, M.H.; Berger, F.; Jauch, K.W.; Hidalgo, M.; Heeschen, C. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef]
- Gaspar, C.; Fodde, R. APC dosage effects in tumorigenesis and stem cell differentiation. Int. J. Dev. Biol. 2004, 48, 377–386. [Google Scholar] [CrossRef]
- Spaeth, E.; Klopp, A.; Dembinski, J.; Andreeff, M.; Marini, F. Inflammation and tumor microenvironments: Defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008, 15, 730–738. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef]
- Hall, B.; Dembinski, J.; Sasser, A.K.; Studeny, M.; Andreeff, M.; Marini, F. Mesenchymal stem cells in cancer: Tumor-associated fibroblasts and cell-based delivery vehicles. Int. J. Hematol. 2007, 86, 8–16. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; Campbell, L.L.; Polyak, K.; Brisken, C.; Yang, J.; Weinberg, R.A. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dembinski, J.L.; Krauss, S. A Distinct Slow-Cycling Cancer Stem-like Subpopulation of Pancreatic Adenocarcinoma Cells is maintained in Vivo. Cancers 2010, 2, 2011-2025. https://doi.org/10.3390/cancers2042011
Dembinski JL, Krauss S. A Distinct Slow-Cycling Cancer Stem-like Subpopulation of Pancreatic Adenocarcinoma Cells is maintained in Vivo. Cancers. 2010; 2(4):2011-2025. https://doi.org/10.3390/cancers2042011
Chicago/Turabian StyleDembinski, Jennifer L., and Stefan Krauss. 2010. "A Distinct Slow-Cycling Cancer Stem-like Subpopulation of Pancreatic Adenocarcinoma Cells is maintained in Vivo" Cancers 2, no. 4: 2011-2025. https://doi.org/10.3390/cancers2042011
APA StyleDembinski, J. L., & Krauss, S. (2010). A Distinct Slow-Cycling Cancer Stem-like Subpopulation of Pancreatic Adenocarcinoma Cells is maintained in Vivo. Cancers, 2(4), 2011-2025. https://doi.org/10.3390/cancers2042011