
Immunometabolism of Innate Immune Cells in Gastrointestinal Cancer

Simple Summary
Abstract

1. Introduction
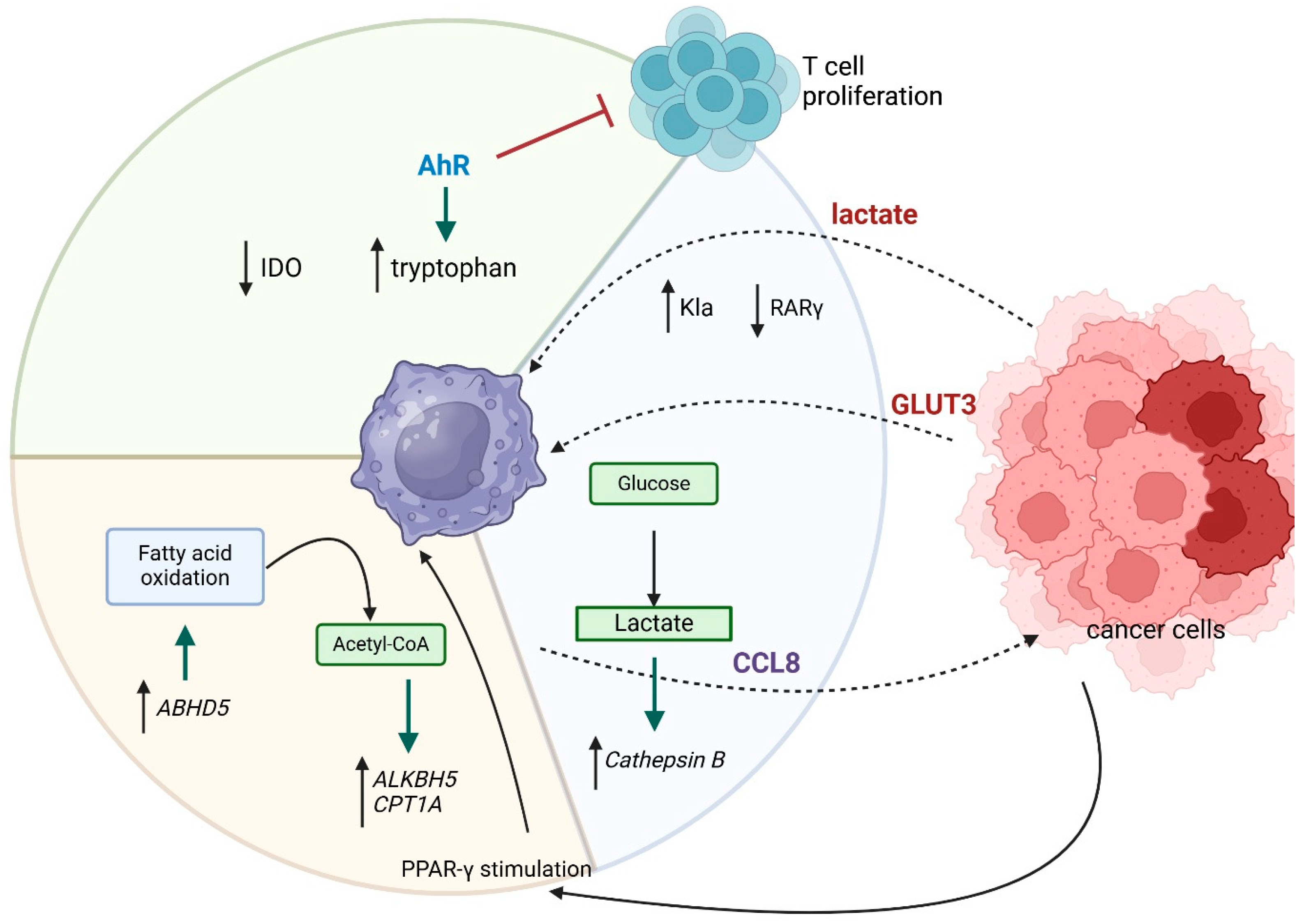
2. Tumor-Associated Macrophages (TAMs)
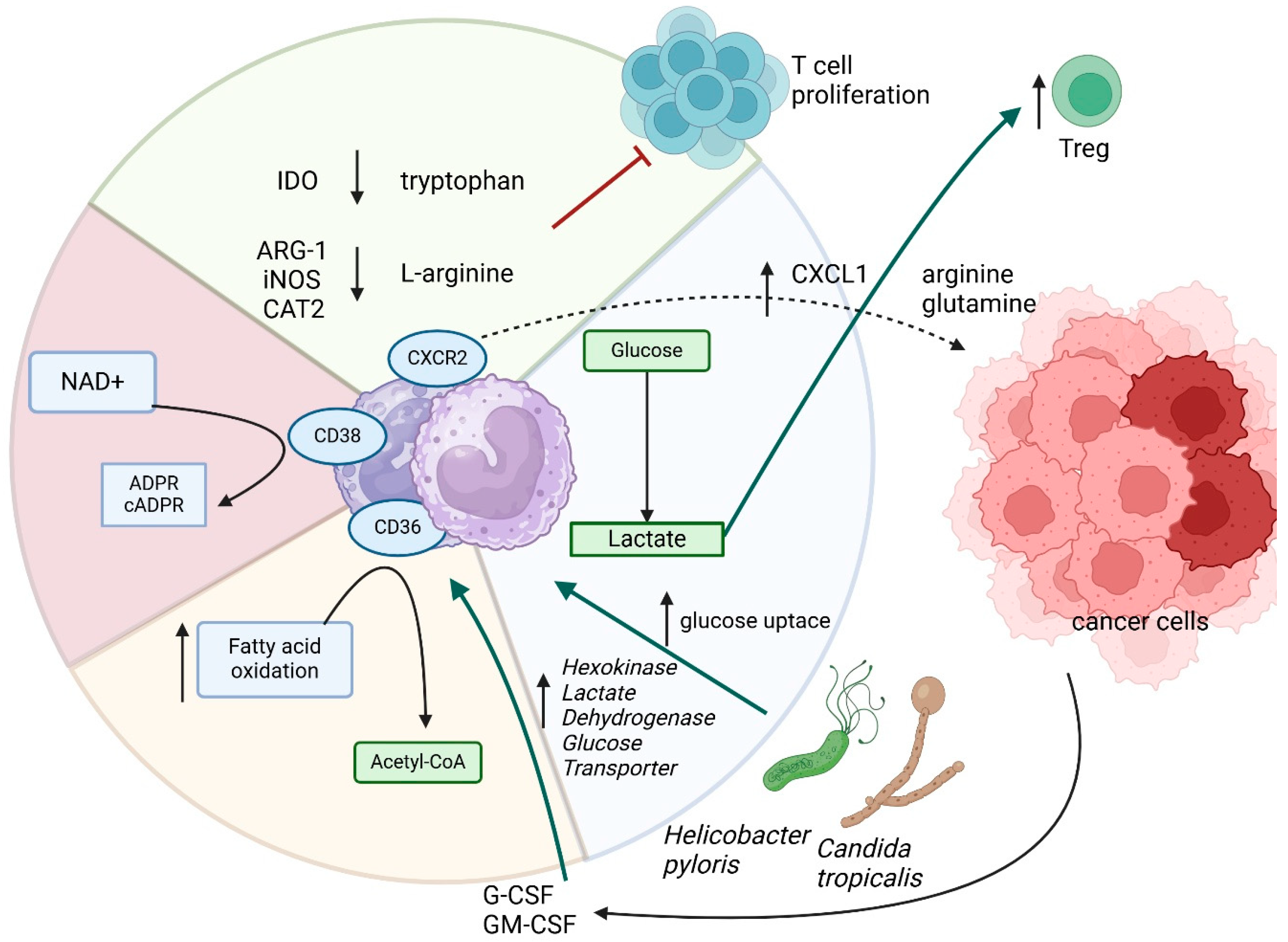
3. Neutrophils
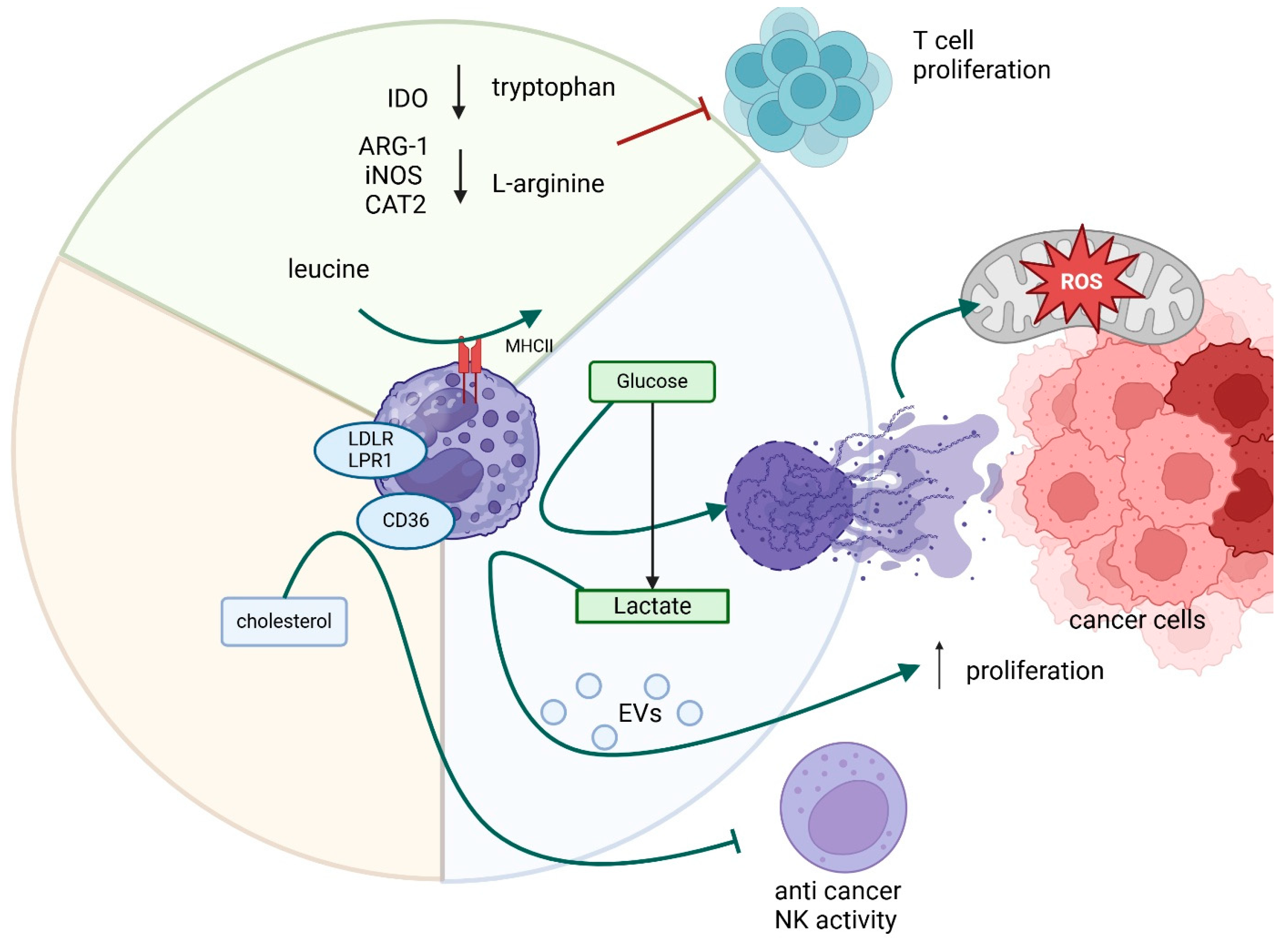
4. Myeloid-Derived Suppressor Cells (MDSCs)
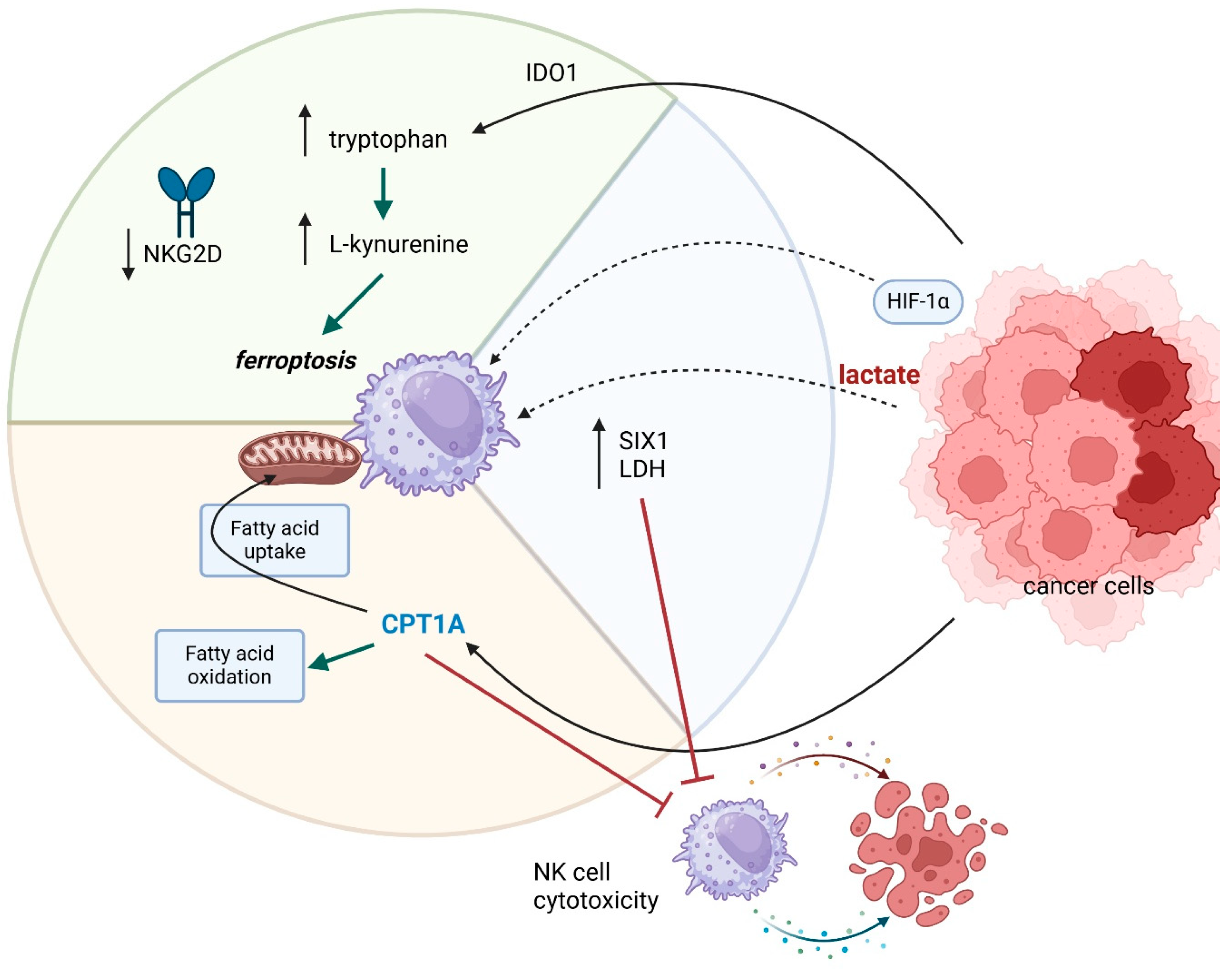
5. NK Cells
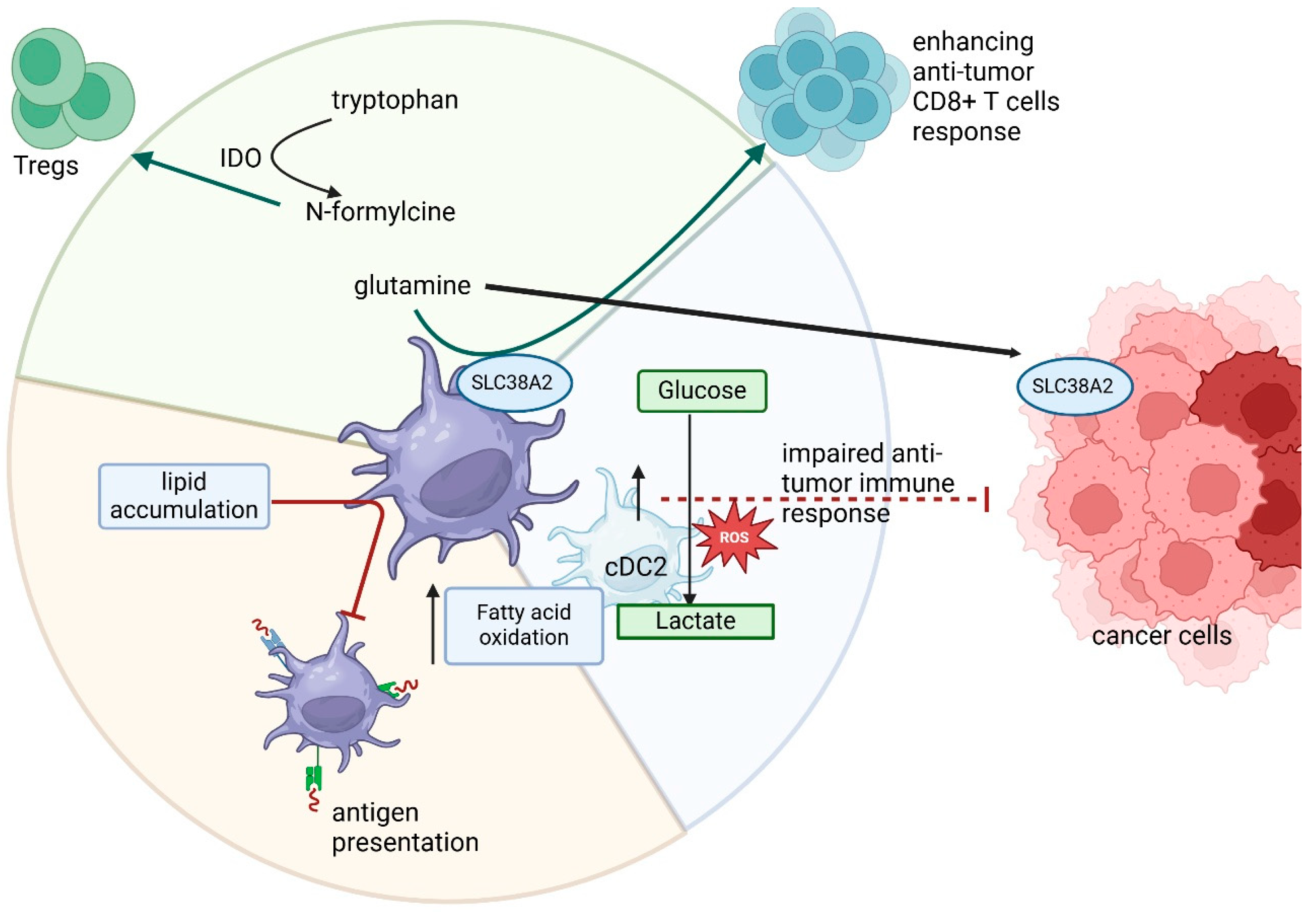
6. Dendritic Cells
7. Immunometabolism-Associated Therapy Approaches
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Álvarez-Carrasco, P.; Maldonado-Bernal, C. The Innate Defenders: A Review of Natural Killer Cell Immunotherapies in Cancer. Front. Immunol. 2024, 15, 1482807. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A.J. Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Basheeruddin, M.; Qausain, S.; Basheeruddin, M.; Qausain, S. Hypoxia-Inducible Factor 1-Alpha (HIF-1α): An Essential Regulator in Cellular Metabolic Control. Cureus 2024, 16, e63852. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Tian, X.; Bai, X.; Han, Y.; Ye, Y.; Peng, M.; Cui, H.; Li, K.; Han, Y. PPAR γ Changing ALDH1A3 Content to Regulate Lipid Metabolism and Inhibit Lung Cancer Cell Growth. Mol. Genet. Genomics 2025, 300, 41. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The Peroxisome Proliferator-Activated Receptor-Gamma Is a Negative Regulator of Macrophage Activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, K.A.; Bianchi-Smiraglia, A. The Aryl Hydrocarbon Receptor as a Tumor Modulator: Mechanisms to Therapy. Front. Oncol. 2024, 14, 1375905. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M.; Cascio, A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. Int. J. Mol. Sci. 2021, 22, 603. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Jumaniyazova, E.; Lokhonina, A.; Dzhalilova, D.; Miroshnichenko, E.; Kosyreva, A.; Fatkhudinov, T. The Role of Macrophages in Various Types of Tumors and the Possibility of Their Use as Targets for Antitumor Therapy. Cancers 2025, 17, 342. [Google Scholar] [CrossRef]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in Tumor-Associated Macrophage (TAM)-Targeted Therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef]
- Jeong, H.; Kim, S.; Hong, B.J.; Lee, C.J.; Kim, Y.E.; Bok, S.; Oh, J.M.; Gwak, S.H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019, 79, 795–806. [Google Scholar] [CrossRef]
- Zhong, Z.; Yang, K.; Li, Y.; Zhou, S.; Yao, H.; Zhao, Y.; Huang, Y.; Zou, J.; Li, Y.; Li, J.; et al. Tumor-Associated Macrophages Drive Glycolysis through the IL-8/STAT3/GLUT3 Signaling Pathway in Pancreatic Cancer Progression. Cancer Lett. 2024, 588, 216784. [Google Scholar] [CrossRef]
- Yu, D.M.; Zhao, J.; Lee, E.E.; Kim, D.; Mahapatra, R.; Rose, E.K.; Zhou, Z.; Hosler, C.; El Kurdi, A.; Choe, J.Y.; et al. GLUT3 Promotes Macrophage Signaling and Function via RAS-Mediated Endocytosis in Atopic Dermatitis and Wound Healing. J. Clin. Investig. 2023, 133, e170706. [Google Scholar] [CrossRef]
- He, Z.; Chen, D.; Wu, J.; Sui, C.; Deng, X.; Zhang, P.; Chen, Z.; Liu, D.; Yu, J.; Shi, J.; et al. Yes Associated Protein 1 Promotes Resistance to 5-Fluorouracil in Gastric Cancer by Regulating GLUT3-Dependent Glycometabolism Reprogramming of Tumor-Associated Macrophages. Arch. Biochem. Biophys. 2021, 702, 108838. [Google Scholar] [CrossRef]
- Shi, Q.; Shen, Q.; Liu, Y.; Shi, Y.; Huang, W.; Wang, X.; Li, Z.; Chai, Y.; Wang, H.; Hu, X.; et al. Increased Glucose Metabolism in TAMs Fuels O-GlcNAcylation of Lysosomal Cathepsin B to Promote Cancer Metastasis and Chemoresistance. Cancer Cell 2022, 40, 1207–1222.e10. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, M.H.; Valli-Mohammed, M.A.; Al-Khayal, K.; Al Shkieh, A.; Zubaidi, A.; Ahmad, R.; Al-Saleh, K.; Al-Obeed, O.; McKerrow, J. Cathepsin B Expression in Colorectal Cancer in a Middle East Population: Potential Value as a Tumor Biomarker for Late Disease Stages. Oncol. Rep. 2017, 37, 3175–3180. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Sun, Z.; Hong, H.; Yang, Y.; Chen, J.; Gao, Z.; Gu, J. Novel Hypoxia- and Lactate Metabolism-Related Molecular Subtyping and Prognostic Signature for Colorectal Cancer. J. Transl. Med. 2024, 22, 587. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Yang, Y.; Jiang, F.Q.; Hu, G.; Wan, S.; Yan, W.Y.; He, X.S.; Xiao, F.; Yang, X.M.; Guo, X.; et al. Histone Lactylation Inhibits RARγ Expression in Macrophages to Promote Colorectal Tumorigenesis through Activation of TRAF6-IL-6-STAT3 Signaling. Cell Rep. 2024, 43, 113688. [Google Scholar] [CrossRef]
- Gao, X.; Zhou, S.; Qin, Z.; Li, D.; Zhu, Y.; Ma, D. Upregulation of HMGB1 in Tumor-Associated Macrophages Induced by Tumor Cell-Derived Lactate Further Promotes Colorectal Cancer Progression. J. Transl. Med. 2023, 21, 53. [Google Scholar] [CrossRef]
- Zhou, H.; Yao, J.; Zhong, Z.; Wei, H.; He, Y.; Li, W.; Hu, K. Lactate-Induced CCL8 in Tumor-Associated Macrophages Accelerates the Progression of Colorectal Cancer through the CCL8/CCR5/MTORC1 Axis. Cancers 2023, 15, 5795. [Google Scholar] [CrossRef]
- Zhang, C.; Cheng, W.; Yang, T.; Fang, H.; Zhang, R. Lactate Secreted by Esophageal Cancer Cells Induces M2 Macrophage Polarization via the AKT/ERK Pathway. Thorac. Cancer 2023, 14, 2139–2148. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, Y.; Zhang, Y.; Bian, Y.; Zeng, Y.; Yao, X.; Wan, J.; Chen, X.; Li, J.; et al. S100A4 Enhances Protumor Macrophage Polarization by Control of PPAR-γ-Dependent Induction of Fatty Acid Oxidation. J. Immunother. Cancer 2021, 9, e002548. [Google Scholar] [CrossRef]
- Sun, M.; Yue, Y.; Wang, X.; Feng, H.; Qin, Y.; Chen, M.; Wang, Y.; Yan, S. ALKBH5-Mediated Upregulation of CPT1A Promotes Macrophage Fatty Acid Metabolism and M2 Macrophage Polarization, Facilitating Malignant Progression of Colorectal Cancer. Exp. Cell Res. 2024, 437, 113994. [Google Scholar] [CrossRef]
- Xiong, J.; He, J.; Zhu, J.; Pan, J.; Liao, W.; Ye, H.; Wang, H.; Song, Y.; Du, Y.; Cui, B.; et al. Lactylation-Driven METTL3-Mediated RNA M6A Modification Promotes Immunosuppression of Tumor-Infiltrating Myeloid Cells. Mol. Cell 2022, 82, 1660–1677.e10. [Google Scholar] [CrossRef]
- Miao, H.; Ou, J.; Peng, Y.; Zhang, X.; Chen, Y.; Hao, L.; Xie, G.; Wang, Z.; Pang, X.; Ruan, Z.; et al. Macrophage ABHD5 Promotes Colorectal Cancer Growth by Suppressing Spermidine Production by SRM. Nat. Commun. 2016, 7, 11716. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose Triglyceride Lipase-Mediated Lipolysis of Cellular Fat Stores Is Activated by CGI-58 and Defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, Q.; Zhao, J.; Xie, H.; Pu, Z. The M6A Methyltransferase METTL3 Modifies Kcnk6 Promoting on Inflammation Associated Carcinogenesis Is Essential for Colon Homeostasis and Defense System through Histone Lactylation Dependent YTHDF2 Binding. Int. Rev. Immunol. 2025, 44, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hezaveh, K.; Shinde, R.S.; Klötgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-Derived Microbial Metabolites Activate the Aryl Hydrocarbon Receptor in Tumor-Associated Macrophages to Suppress Anti-Tumor Immunity. Immunity 2022, 55, 324–340.e8. [Google Scholar] [CrossRef]
- Lee, R.; Li, J.; Li, J.; Wu, C.J.; Jiang, S.; Hsu, W.H.; Chakravarti, D.; Chen, P.; Labella, K.A.; Li, J.; et al. Synthetic Essentiality of Tryptophan 2,3-Dioxygenase 2 in APC-Mutated Colorectal Cancer. Cancer Discov. 2022, 12, 1702–1717. [Google Scholar] [CrossRef]
- Liu, X.; Yan, G.; Xu, B.; Yu, H.; An, Y.; Sun, M. Evaluating the Role of IDO1 Macrophages in Immunotherapy Using ScRNA-Seq and Bulk-Seq in Colorectal Cancer. Front. Immunol. 2022, 13, 1006501. [Google Scholar] [CrossRef]
- Zhong, X.; He, X.; Wang, Y.; Hu, Z.; Huang, H.; Zhao, S.; Wei, P.; Li, D. Warburg Effect in Colorectal Cancer: The Emerging Roles in Tumor Microenvironment and Therapeutic Implications. J. Hematol. Oncol. 2022, 15, 160. [Google Scholar] [CrossRef]
- Shen, X.; Chen, Y.; Tang, Y.; Lu, P.; Liu, M.; Mao, T.; Weng, Y.; Yu, F.; Liu, Y.; Tang, Y.; et al. Targeting Pancreatic Cancer Glutamine Dependency Confers Vulnerability to GPX4-Dependent Ferroptosis. Cell Rep. Med. 2025, 6, 101928. [Google Scholar] [CrossRef]
- Siemińska, I.; Poljańska, E.; Baran, J. Granulocytes and Cells of Granulocyte Origin-The Relevant Players in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 3801. [Google Scholar] [CrossRef]
- Grenader, T.; Nash, S.; Adams, R.; Kaplan, R.; Fisher, D.; Maughan, T.; Bridgewater, J. Derived Neutrophil Lymphocyte Ratio Is Predictive of Survival from Intermittent Therapy in Advanced Colorectal Cancer: A Post Hoc Analysis of the MRC COIN Study. Br. J. Cancer 2016, 114, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.J.; McNamara, M.G.; Šeruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocaña, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2014, 106, dju124. [Google Scholar] [CrossRef] [PubMed]
- Thind, M.K.; Uhlig, H.H.; Glogauer, M.; Palaniyar, N.; Bourdon, C.; Gwela, A.; Lancioni, C.L.; Berkley, J.A.; Bandsma, R.H.J.; Farooqui, A. A Metabolic Perspective of the Neutrophil Life Cycle: New Avenues in Immunometabolism. Front. Immunol. 2024, 14, 1334205. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, H.O.; Roy, E.; Comerci, A.J.; Van Der Windt, D.J.; Zhang, H.; Huang, H.; Loughran, P.; Shiva, S.; Geller, D.A.; Bartlett, D.L.; et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019, 79, 5626–5639. [Google Scholar] [CrossRef]
- Jiang, Z.Z.; Peng, Z.P.; Liu, X.C.; Guo, H.F.; Zhou, M.M.; Jiang, D.; Ning, W.R.; Huang, Y.F.; Zheng, L.; Wu, Y. Neutrophil Extracellular Traps Induce Tumor Metastasis through Dual Effects on Cancer and Endothelial Cells. Oncoimmunology 2022, 11, 2052418. [Google Scholar] [CrossRef]
- Rodríguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.M.B.; López-Villegas, E.O.; Sánchez-García, F.J. Metabolic Requirements for Neutrophil Extracellular Traps Formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Guo, Y.; Ye, L.; Li, D.; Hu, A.; Cai, S.; Yuan, B.; Jin, S.; Zhou, Y.; et al. Therapeutic Targeting of SPIB/SPI1-Facilitated Interplay of Cancer Cells and Neutrophils Inhibits Aerobic Glycolysis and Cancer Progression. Clin. Transl. Med. 2021, 11, e588. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Dai, Y.; Tang, X.; Yin, T.; Wang, C.; Wang, T.; Dong, L.; Shi, M.; Qin, J.; et al. Single-Cell RNA-Seq Analysis Reveals BHLHE40-Driven pro-Tumour Neutrophils with Hyperactivated Glycolysis in Pancreatic Tumour Microenvironment. Gut 2023, 72, 958–971. [Google Scholar] [CrossRef]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L.; et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef]
- Niu, N.; Shen, X.; Zhang, L.; Chen, Y.; Lu, P.; Yang, W.; Liu, M.; Shi, J.; Xu, D.; Tang, Y.; et al. Tumor Cell-Intrinsic SETD2 Deficiency Reprograms Neutrophils to Foster Immune Escape in Pancreatic Tumorigenesis. Adv. Sci. 2023, 10, e2202937. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, J.; Yang, X.; Nan, F.; Zhang, T.; Ji, S.; Rao, D.; Feng, H.; Gao, K.; Gu, X.; et al. Neutrophil Profiling Illuminates Anti-Tumor Antigen-Presenting Potency. Cell 2024, 187, 1422–1439.e24. [Google Scholar] [CrossRef] [PubMed]
- Canè, S.; Barouni, R.M.; Fabbi, M.; Cuozzo, J.; Fracasso, G.; Adamo, A.; Ugel, S.; Trovato, R.; De Sanctis, F.; Giacca, M.; et al. Neutralization of NET-Associated Human ARG1 Enhances Cancer Immunotherapy. Sci. Transl. Med. 2023, 15, eabq6221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Lu, Y.; Sanchez, D.J.; Li, J.; Wang, L.; Meng, X.; Chen, J.; Kien, T.T.; Zhong, M.; et al. Region-Specific CD16+ Neutrophils Promote Colorectal Cancer Progression by Inhibiting Natural Killer Cells. Adv. Sci. 2024, 11, 2403414. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef]
- Wang, X.; Hu, L.P.; Qin, W.T.; Yang, Q.; Chen, D.Y.; Li, Q.; Zhou, K.X.; Huang, P.Q.; Xu, C.J.; Li, J.; et al. Identification of a Subset of Immunosuppressive P2RX1-Negative Neutrophils in Pancreatic Cancer Liver Metastasis. Nat. Commun. 2021, 12, 174. [Google Scholar] [CrossRef]
- Xu, W.; Liu, J.; Liu, Q.; Xu, J.; Zhou, L.; Liang, Z.; Huang, H.; Huang, B.; Xiao, G.G.; Guo, J. NFE2-Driven Neutrophil Polarization Promotes Pancreatic Cancer Liver Metastasis Progression. Cell Rep. 2025, 44, 115226. [Google Scholar] [CrossRef]
- Chen, L.; Dai, P.; Liu, L.; Chen, Y.; Lu, Y.; Zheng, L.; Wang, H.; Yuan, Q.; Li, X. The Lipid-Metabolism Enzyme ECI2 Reduces Neutrophil Extracellular Traps Formation for Colorectal Cancer Suppression. Nat. Commun. 2024, 15, 7184. [Google Scholar] [CrossRef] [PubMed]
- Sieminska, I.; Baran, J. Myeloid-Derived Suppressor Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 1526. [Google Scholar] [CrossRef]
- Ma, P.; Beatty, P.L.; McKolanis, J.; Brand, R.; Schoen, R.E.; Finn, O.J. Circulating Myeloid Derived Suppressor Cells (MDSC) That Accumulate in Premalignancy Share Phenotypic and Functional Characteristics with MDSC in Cancer. Front. Immunol. 2019, 10, 1401. [Google Scholar] [CrossRef]
- Siemińska, I.; Węglarczyk, K.; Walczak, M.; Czerwińska, A.; Pach, R.; Rubinkiewicz, M.; Szczepanik, A.; Siedlar, M.; Baran, J. Mo-MDSCs Are Pivotal Players in Colorectal Cancer and May Be Associated with Tumor Recurrence after Surgery. Transl. Oncol. 2022, 17, 101346. [Google Scholar] [CrossRef]
- Zhang, Z.; Zheng, Y.; Chen, Y.; Yin, Y.; Chen, Y.; Chen, Q.; Hou, Y.; Shen, S.; Lv, M.; Wang, T. Gut Fungi Enhances Immunosuppressive Function of Myeloid-Derived Suppressor Cells by Activating PKM2-Dependent Glycolysis to Promote Colorectal Tumorigenesis. Exp. Hematol. Oncol. 2022, 11, 88. [Google Scholar] [CrossRef]
- Ding, L.; Sheriff, S.; Sontz, R.A.; Merchant, J.L. Schlafen4+-MDSC in Helicobacter-Induced Gastric Metaplasia Reveals Role for GTPases. Front. Immunol. 2023, 14, 1139391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, A.; Bi, Y.; Wang, Y.; Liu, G. Metabolic Regulation of Myeloid-Derived Suppressor Cell Function in Cancer. Cells 2020, 9, 1011. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Kang, Y.; Wang, L.; Huff, S.; Tang, R.; Hui, H.; Agrawal, K.; Gonzalez, G.M.; Wang, Y.; Patel, S.P.; et al. ALKBH5 Regulates Anti–PD-1 Therapy Response by Modulating Lactate and Suppressive Immune Cell Accumulation in Tumor Microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 20159–20170. [Google Scholar] [CrossRef]
- Cimen Bozkus, C.; Elzey, B.D.; Crist, S.A.; Ellies, L.G.; Ratliff, T.L. Expression of Cationic Amino Acid Transporter 2 Is Required for Myeloid-Derived Suppressor Cell-Mediated Control of T Cell Immunity. J. Immunol. 2015, 195, 5237–5250. [Google Scholar] [CrossRef]
- Won, W.J.; Deshane, J.S.; Leavenworth, J.W.; Oliva, C.R.; Griguer, C.E. Metabolic and Functional Reprogramming of Myeloid-Derived Suppressor Cells and Their Therapeutic Control in Glioblastoma. Cell Stress 2019, 3, 47. [Google Scholar] [CrossRef]
- OuYang, L.Y.; Wu, X.J.; Ye, S.B.; Zhang, R.X.; Li, Z.L.; Liao, W.; Pan, Z.Z.; Zheng, L.M.; Zhang, X.S.; Wang, Z.; et al. Tumor-Induced Myeloid-Derived Suppressor Cells Promote Tumor Progression through Oxidative Metabolism in Human Colorectal Cancer. J. Transl. Med. 2015, 13, 47. [Google Scholar] [CrossRef]
- Zhou, Q.; Peng, Y.; Ji, F.; Chen, H.; Kang, W.; Chan, L.S.; Gou, H.; Lin, Y.; Huang, P.; Chen, D.; et al. Targeting of SLC25A22 Boosts the Immunotherapeutic Response in KRAS-Mutant Colorectal Cancer. Nat. Commun. 2023, 14, 4677. [Google Scholar] [CrossRef]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous Lipid Uptake Induces Metabolic and Functional Reprogramming of Tumor-Associated Myeloid-Derived Suppressor Cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-Type Oxidized LDL Receptor-1 Distinguishes Population of Human Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef]
- Taye, A.; El-Sheikh, A.A.K. Lectin-like Oxidized Low-Density Lipoprotein Receptor 1 Pathways. Eur. J. Clin. Investig. 2013, 43, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, C.; Han, F.; Tang, J.; Deng, M.; Niu, Y.; Lai, M.; Zhang, H. Long Noncoding RNA MIR4435-2HG Suppresses Colorectal Cancer Initiation and Progression by Reprogramming Neutrophils. Cancer Immunol. Res. 2022, 10, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Dominguez, G.A.; Hashimoto, A.; Lin, E.W.; Chiu, C.; Sasser, K.; Lee, J.W.; Beatty, G.L.; Gabrilovich, D.I.; Rustgi, A.K. CD38+ M-MDSC Expansion Characterizes a Subset of Advanced Colorectal Cancer Patients. JCI Insight 2018, 3, e97022. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Waldron, T.J.; Eruslanov, E.; Kim, S.B.; Lee, J.S.; O’Brien, S.; Hicks, P.D.; Basu, D.; Singhal, S.; Malavasi, F.; et al. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res. 2015, 75, 4074. [Google Scholar] [CrossRef]
- Kang, J.; Lee, D.; Lee, K.J.; Yoon, J.E.; Kwon, J.H.; Seo, Y.; Kim, J.; Chang, S.Y.; Park, J.; Kang, E.A.; et al. Tumor-Suppressive Effect of Metformin via the Regulation of M2 Macrophages and Myeloid-Derived Suppressor Cells in the Tumor Microenvironment of Colorectal Cancer. Cancers 2022, 14, 2881. [Google Scholar] [CrossRef]
- Juarez, D.; Fruman, D.A. Targeting the Mevalonate Pathway in Cancer. Trends Cancer 2021, 7, 525. [Google Scholar] [CrossRef]
- Fionda, C.; Scarno, G.; Stabile, H.; Molfetta, R.; Di Censo, C.; Gismondi, A.; Paolini, R.; Sozzani, S.; Santoni, A.; Sciumè, G. NK Cells and Other Cytotoxic Innate Lymphocytes in Colorectal Cancer Progression and Metastasis. Int. J. Mol. Sci. 2022, 23, 7859. [Google Scholar] [CrossRef]
- Han, B.; He, J.; Chen, Q.; Yuan, M.; Zeng, X.; Li, Y.; Zeng, Y.; He, M.; Zhou, Q.; Feng, D.; et al. ELFN1-AS1 Promotes GDF15-Mediated Immune Escape of Colorectal Cancer from NK Cells by Facilitating GCN5 and SND1 Association. Discov. Oncol. 2023, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, C.; D’Antonio, L.; Fieni, C.; Ciummo, S.L.; Di Carlo, E. Colorectal Cancer-Associated Immune Exhaustion Involves T and B Lymphocytes and Conventional NK Cells and Correlates with a Shorter Overall Survival. Front. Immunol. 2021, 12, 778329. [Google Scholar] [CrossRef] [PubMed]
- Tumino, N.; Nava Lauson, C.B.; Tiberti, S.; Besi, F.; Martini, S.; Fiore, P.F.; Scodamaglia, F.; Mingari, M.C.; Moretta, L.; Manzo, T.; et al. The Tumor Microenvironment Drives NK Cell Metabolic Dysfunction Leading to Impaired Antitumor Activity. Int. J. Cancer 2023, 152, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, A.; Nelius, E.; Fan, Z.; Khatchatourova, E.; Alvarado-Diaz, A.; He, J.; Krzywinska, E.; Sobecki, M.; Nagarajan, S.; Kerdiles, Y.; et al. Resting Natural Killer Cell Homeostasis Relies on Tryptophan/NAD+ Metabolism and HIF-1α. EMBO Rep. 2023, 24, e56156. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef]
- Ge, W.; Meng, L.; Cao, S.; Hou, C.; Zhu, X.; Huang, D.; Li, Q.; Peng, Y.; Jiang, K. The SIX1/LDHA Axis Promotes Lactate Accumulation and Leads to NK Cell Dysfunction in Pancreatic Cancer. J. Immunol. Res. 2023, 2023, 6891636. [Google Scholar] [CrossRef]
- Zaiatz Bittencourt, V.; Jones, F.; Tosetto, M.; Doherty, G.A.; Ryan, E.J. Dysregulation of Metabolic Pathways in Circulating Natural Killer Cells Isolated from Inflammatory Bowel Disease Patients. J. Crohns Colitis 2021, 15, 1316–1325. [Google Scholar] [CrossRef]
- Sheppard, S.; Srpan, K.; Lin, W.; Lee, M.; Delconte, R.B.; Owyong, M.; Carmeliet, P.; Davis, D.M.; Xavier, J.B.; Hsu, K.C.; et al. Fatty Acid Oxidation Fuels Natural Killer Cell Responses against Infection and Cancer. Proc. Natl. Acad. Sci. USA 2024, 121, e2319254121. [Google Scholar] [CrossRef]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.S.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty Acid Carbon Is Essential for DNTP Synthesis in Endothelial Cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef]
- Delconte, R.B.; Owyong, M.; Santosa, E.K.; Srpan, K.; Sheppard, S.; McGuire, T.J.; Abbasi, A.; Diaz-Salazar, C.; Chun, J.; Rogatsky, I.; et al. Fasting Reshapes Tissue-Specific Niches to Improve NK Cell-Mediated Anti-Tumor Immunity. Immunity 2024, 57, 1923–1938.e7. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.X.; Xu, X.H.; He, T.; Liu, J.J.; Xie, T.Y.; Tian, W.; Liu, J.Y. L-Kynurenine Induces NK Cell Loss in Gastric Cancer Microenvironment via Promoting Ferroptosis. J. Exp. Clin. Cancer Res. 2023, 42, 52. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Guo, L.; Xing, Z.; Shi, L.; Liang, H.; Li, A.; Kuang, C.; Tao, B.; Yang, Q. IDO1 Can Impair NK Cells Function against Non-Small Cell Lung Cancer by Downregulation of NKG2D Ligand via ADAM10. Pharmacol. Res. 2022, 177, 106132. [Google Scholar] [CrossRef]
- Song, H.; Park, H.; Kim, Y.S.; Kim, K.D.; Lee, H.K.; Cho, D.H.; Yang, J.W.; Hur, D.Y. L-Kynurenine-Induced Apoptosis in Human NK Cells Is Mediated by Reactive Oxygen Species. Int. Immunopharmacol. 2011, 11, 932–938. [Google Scholar] [CrossRef]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 483200. [Google Scholar] [CrossRef]
- Zheng, X.; Qian, Y.; Fu, B.; Jiao, D.; Jiang, Y.; Chen, P.; Shen, Y.; Zhang, H.; Sun, R.; Tian, Z.; et al. Mitochondrial Fragmentation Limits NK Cell-Based Tumor Immunosurveillance. Nat. Immunol. 2019, 20, 1656–1667. [Google Scholar] [CrossRef]
- Lin, J.H.; Huffman, A.P.; Wattenberg, M.M.; Walter, D.M.; Carpenter, E.L.; Feldser, D.M.; Beatty, G.L.; Furth, E.E.; Vonderheide, R.H. Type 1 Conventional Dendritic Cells Are Systemically Dysregulated Early in Pancreatic Carcinogenesis. J. Exp. Med. 2020, 217, e20190673. [Google Scholar] [CrossRef]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e7. [Google Scholar] [CrossRef]
- Alston James, C.; Baer, J.M.; Zou, C.; Panni, U.Y.; Knolhoff, B.L.; Hogg, G.D.; Kingston, N.L.; Kang, L.I.; Lander, V.E.; Luo, J.; et al. Systemic Alterations in Type-2 Conventional Dendritic Cells Leads to Impaired Tumor Immunity in Pancreatic Cancer. Cancer Immunol. Res. 2023, 11, 1055. [Google Scholar] [CrossRef]
- Kießler, M.; Plesca, I.; Sommer, U.; Wehner, R.; Wilczkowski, F.; Müller, L.; Tunger, A.; Lai, X.; Rentsch, A.; Peuker, K.; et al. Tumor-Infiltrating Plasmacytoid Dendritic Cells Are Associated with Survival in Human Colon Cancer. J. Immunother. Cancer 2021, 9, e001813. [Google Scholar] [CrossRef]
- Tjomsland, V.; Sandström, P.; Spångeus, A.; Messmer, D.; Emilsson, J.; Falkmer, U.; Falkmer, S.; Magnusson, K.E.; Borch, K.; Larsson, M. Pancreatic Adenocarcinoma Exerts Systemic Effects on the Peripheral Blood Myeloid and Plasmacytoid Dendritic Cells: An Indicator of Disease Severity? BMC Cancer 2010, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Boyette, L.B.; MacEdo, C.; Hadi, K.; Elinoff, B.D.; Walters, J.T.; Ramaswami, B.; Chalasani, G.; Taboas, J.M.; Lakkis, F.G.; Metes, D.M. Phenotype, Function, and Differentiation Potential of Human Monocyte Subsets. PLoS ONE 2017, 12, e0176460. [Google Scholar] [CrossRef]
- Guo, C.; You, Z.; Shi, H.; Sun, Y.; Du, X.; Palacios, G.; Guy, C.; Yuan, S.; Chapman, N.M.; Lim, S.A.; et al. SLC38A2 and Glutamine Signalling in CDC1s Dictate Anti-Tumour Immunity. Nature 2023, 620, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Elomaa, H.; Härkönen, J.; Väyrynen, S.A.; Ahtiainen, M.; Ogino, S.; Nowak, J.A.; Lau, M.C.; Helminen, O.; Wirta, E.V.; Seppälä, T.T.; et al. Quantitative Multiplexed Analysis of Indoleamine 2,3-Dioxygenase (IDO) and Arginase-1 (ARG1) Expression and Myeloid Cell Infiltration in Colorectal Cancer. Mod. Pathol. 2024, 37, 100450. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid Dendritic Cells from Mouse Tumor-Draining Lymph Nodes Directly Activate Mature Tregs via Indoleamine 2,3-Dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef]
- Cao, W.; Ramakrishnan, R.; Tuyrin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Min Zhang, L.; Klein-Seetharaman, J.; et al. Oxidized Lipids Block Antigen Cross-Presentation by Dendritic Cells in Cancer Oxidized Lipids and DCs in Cancer. J. Immunol. 2014, 192, 2920. [Google Scholar] [CrossRef]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid Accumulation and Dendritic Cell Dysfunction in Cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef]
- Wang, X.; Qu, Y.; Ji, J.; Liu, H.; Luo, H.; Li, J.; Han, X. Colorectal Cancer Cells Establish Metabolic Reprogramming with Cancer-Associated Fibroblasts (CAFs) through Lactate Shuttle to Enhance Invasion, Migration, and Angiogenesis. Int. Immunopharmacol. 2024, 143, 113470. [Google Scholar] [CrossRef]
- Chang, Y.; Ou, Q.; Zhou, X.; Nie, K.; Zheng, P.; Liu, J.; Chen, L.; Yan, H.; Guo, D.; Zhang, S. Jianpi Jiedu Decoction Suppresses Colorectal Cancer Growth by Inhibiting M2 Polarization of TAMs through the Tryptophan Metabolism-AhR Pathway. Int. Immunopharmacol. 2024, 138, 112610. [Google Scholar] [CrossRef]
- Lee, K.J.; Ko, E.J.; Park, Y.Y.; Park, S.S.; Ju, E.J.; Park, J.; Shin, S.H.; Suh, Y.A.; Hong, S.M.; Park, I.J.; et al. A Novel Nanoparticle-Based Theranostic Agent Targeting LRP-1 Enhances the Efficacy of Neoadjuvant Radiotherapy in Colorectal Cancer. Biomaterials 2020, 255, 120151. [Google Scholar] [CrossRef]
- Tan, Z.; Xiao, L.; Tang, M.; Bai, F.; Li, J.; Li, L.; Shi, F.; Li, N.; Li, Y.; Du, Q.; et al. Targeting CPT1A-Mediated Fatty Acid Oxidation Sensitizes Nasopharyngeal Carcinoma to Radiation Therapy. Theranostics 2018, 8, 2329–2347. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Wang, H.; Xu, Q.; Pan, Y.; Jiang, Z.; Li, S.; Qu, Y.; Hu, Y.; Wu, H.; Wang, X. A Novel CPT1A Covalent Inhibitor Modulates Fatty Acid Oxidation and CPT1A-VDAC1 Axis with Therapeutic Potential for Colorectal Cancer. Redox Biol. 2023, 68, 102959. [Google Scholar] [CrossRef] [PubMed]
- DePeaux, K.; Delgoffe, G.M. Metabolic Barriers to Cancer Immunotherapy. Nat. Rev. Immunol. 2021, 21, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial Stress Induced by Continuous Stimulation under Hypoxia Rapidly Drives T Cell Exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Zhu, L.; Zhu, X.; Wu, Y. Effects of Glucose Metabolism, Lipid Metabolism, and Glutamine Metabolism on Tumor Microenvironment and Clinical Implications. Biomolecules 2022, 12, 580. [Google Scholar] [CrossRef]
- Ning, S.; Hahn, G. Cytotoxicity of Lonidamine Alone and in Combination with Other Drugs against Murine RIF-1 and Human HT1080 Cells in Vitro—PubMed. Cancer Res. 1990, 50, 7867–7870. [Google Scholar]
- Fan, Y.; Pedersen, O. Gut Microbiota in Human Metabolic Health and Disease. Nat. Rev. Microbiol. 2020, 19, 55–71. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.J.; Wang, R.; Gong, F.L.; Shi, Y.H.; Li, S.N.; Chen, P.P.; Yuan, Y.F. Fasting Mimicking Diet Inhibits Tumor-Associated Macrophage Survival and pro-Tumor Function in Hypoxia: Implications for Combination Therapy with Anti-Angiogenic Agent. J. Transl. Med. 2023, 21, 754. [Google Scholar] [CrossRef]
- Luo, M.; Wang, Q.; Sun, Y.; Jiang, Y.; Wang, Q.; Gu, Y.; Hu, Z.; Chen, Q.; Xu, J.; Chen, S.; et al. Fasting-Mimicking Diet Remodels Gut Microbiota and Suppresses Colorectal Cancer Progression. Npj Biofilm. Microbiomes 2024, 10, 53. [Google Scholar] [CrossRef]
- Rizvanović, N.; Nesek Adam, V.; KalajdŽija, M.; Čaušević, S.; Dervišević, S.; Smajić, J. Effects of Preoperative Oral Carbohydrate Loading on Neutrophil/Lymphocyte Ratio and Postoperative Complications Following Colorectal Cancer Surgery: A Randomized Controlled Study. Eur. Surg. Res. 2023, 64, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pei, T.; Liu, C.; Cao, M.; Hu, X.; Yuan, J.; Chen, F.; Guo, B.; Hong, Y.; Liu, J.; et al. Glutamine Metabolic Competition Drives Immunosuppressive Reprogramming of Intratumour GPR109A+myeloid Cells to Promote Liver Cancer Progression. Gut 2024, 74, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Izabela, S.; Edyta, R.-B.; Janusz, J.; Mikołaj, P.; Karolina, B.-S.; Janusz, R.; Jan, D.; Maciej, S.; Jarek, B. The Level of Myeloid Derived-Suppressor Cells in Peripheral Blood of Patients with Prostate Cancerafter Various Types of Therapy. Pol. J. Pathol. 2020, 71, 46–54. [Google Scholar]
- Fu, C.; Fu, Z.; Jiang, C.; Xia, C.; Zhang, Y.; Gu, X.; Zheng, K.; Zhou, D.; Tang, S.; Lyu, S.; et al. CD205+ Polymorphonuclear Myeloid-Derived Suppressor Cells Suppress Antitumor Immunity by Overexpressing GLUT3. Cancer Sci. 2021, 112, 1011–1025. [Google Scholar] [CrossRef]
- Lenart, M.; Rutkowska-Zapała, M.; Siedlar, M. NK-Cell Receptor Modulation in Viral Infections. Clin. Exp. Immunol. 2024, 217, 151–158. [Google Scholar] [CrossRef]
- Littwitz-Salomon, E.; Moreira, D.; Frost, J.N.; Choi, C.; Liou, K.T.; Ahern, D.K.; O’Shaughnessy, S.; Wagner, B.; Biron, C.A.; Drakesmith, H.; et al. Metabolic Requirements of NK Cells during the Acute Response against Retroviral Infection. Nat. Commun. 2021, 12, 5376. [Google Scholar] [CrossRef]
- Schimmer, S.; Sridhar, V.; Satan, Z.; Grebe, A.; Saad, M.; Wagner, B.; Kahlert, N.; Werner, T.; Richter, D.; Dittmer, U.; et al. Iron Improves the Antiviral Activity of NK Cells. Front. Immunol. 2024, 15, 1526197. [Google Scholar] [CrossRef]
- Santosa, E.K.; Kim, H.; Rückert, T.; Le Luduec, J.B.; Abbasi, A.J.; Wingert, C.K.; Peters, L.; Frost, J.N.; Hsu, K.C.; Romagnani, C.; et al. Control of Nutrient Uptake by IRF4 Orchestrates Innate Immune Memory. Nat. Immunol. 2023, 24, 1685–1697. [Google Scholar] [CrossRef]
- Raines, L.N.; Zhao, H.; Wang, Y.; Chen, H.Y.; Gallart-Ayala, H.; Hsueh, P.C.; Cao, W.; Koh, Y.; Alamonte-Loya, A.; Liu, P.S.; et al. PERK Is a Critical Metabolic Hub for Immunosuppressive Function in Macrophages. Nat. Immunol. 2022, 23, 431–445. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Singh, K.; Ritchie, T.M.; Aguiar, J.A.; Fan, I.Y.; Portillo, A.L.; Rojas, E.A.; Vahedi, F.; El-Sayes, A.; Xing, S.; et al. Metabolic Flexibility Determines Human NK Cell Functional Fate in the Tumor Microenvironment. Cell Metab. 2021, 33, 1205–1220.e5. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Potential Target | Potential or Approved Therapeutic | Literature |
|---|---|---|---|
| Macrophages | ABHD5/SRM/spermidine axis | - | [31] |
| IL-8/STAT3/GLUT3 signaling axis | reparixin | [18] | |
| RARγ-dependant TRAF6-IL-6-STAT3 signaling | nordihydroguaiaretic acid (NDGA) | [24] | |
| lactate transport function of MCT1 | AZD3965, an MCT1 (monocarboxylate transporter 1) inhibitor | [108] | |
| AhR expression and M2-type polarization | JianpiJiedu decoction (traditional Chinese medicine) | [109] | |
| Neutrophils | LRP1 | LRP1 inhibitor | [53,110] |
| MDSCs | CD38 | anti-CD38 monoclonal antibody (Daratumumab) | [77] |
| activation of 5′AMP-activated protein kinase (AMPK) MDSCs | metformin | [78] | |
| Dendritic cells | inhibitor of acetylCoA carboxylase | 5(tetradecycloxy)2furoic acid (TOFA) in the research phase | [107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siemińska, I.; Lenart, M. Immunometabolism of Innate Immune Cells in Gastrointestinal Cancer. Cancers 2025, 17, 1467. https://doi.org/10.3390/cancers17091467
Siemińska I, Lenart M. Immunometabolism of Innate Immune Cells in Gastrointestinal Cancer. Cancers. 2025; 17(9):1467. https://doi.org/10.3390/cancers17091467
Chicago/Turabian StyleSiemińska, Izabela, and Marzena Lenart. 2025. "Immunometabolism of Innate Immune Cells in Gastrointestinal Cancer" Cancers 17, no. 9: 1467. https://doi.org/10.3390/cancers17091467
APA StyleSiemińska, I., & Lenart, M. (2025). Immunometabolism of Innate Immune Cells in Gastrointestinal Cancer. Cancers, 17(9), 1467. https://doi.org/10.3390/cancers17091467