Review of Adoptive Cellular Therapies for the Treatment of Sarcoma


Simple Summary
Abstract
1. Introduction
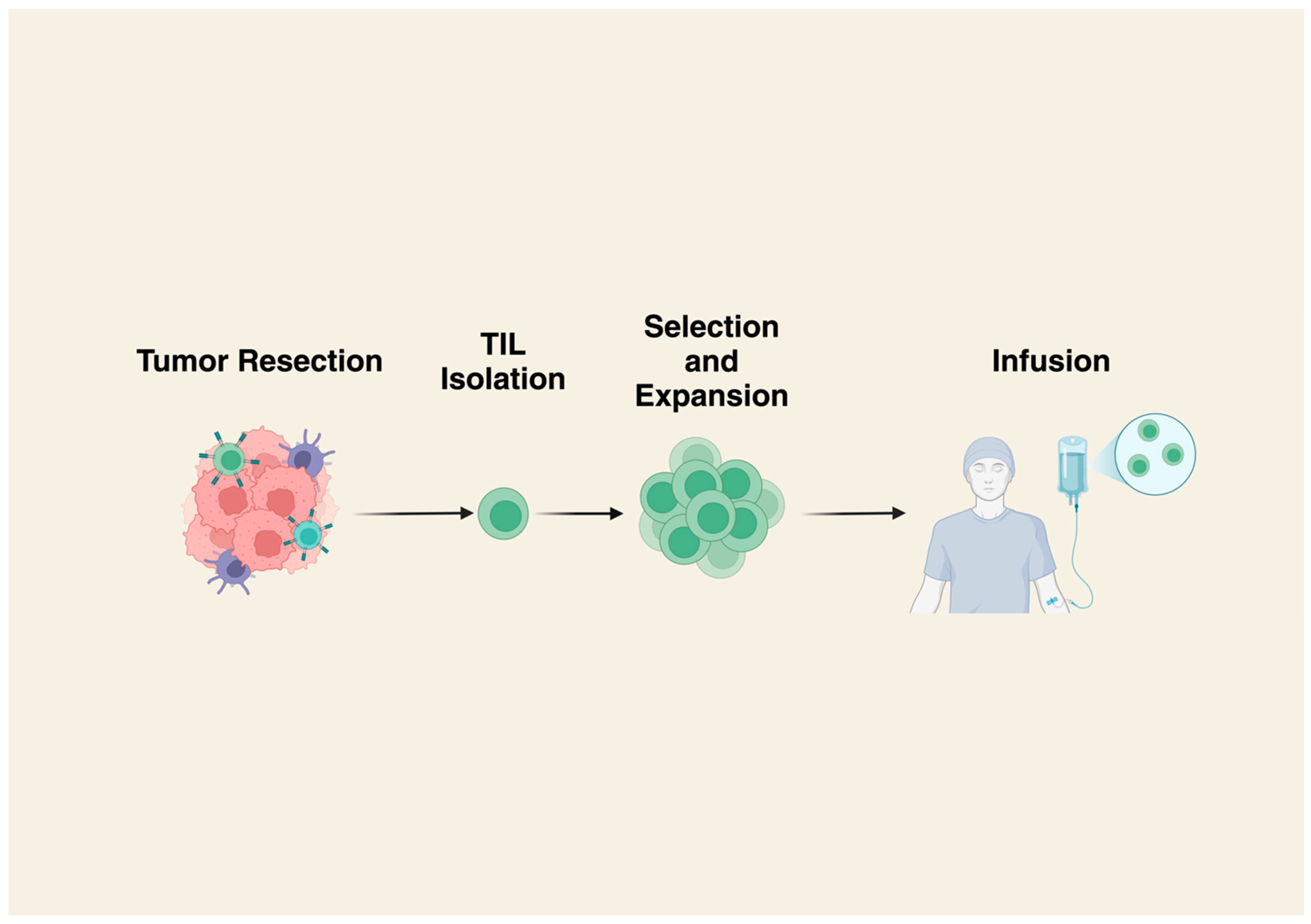
2. Tumor Infiltrating Lymphocytes (TILs)
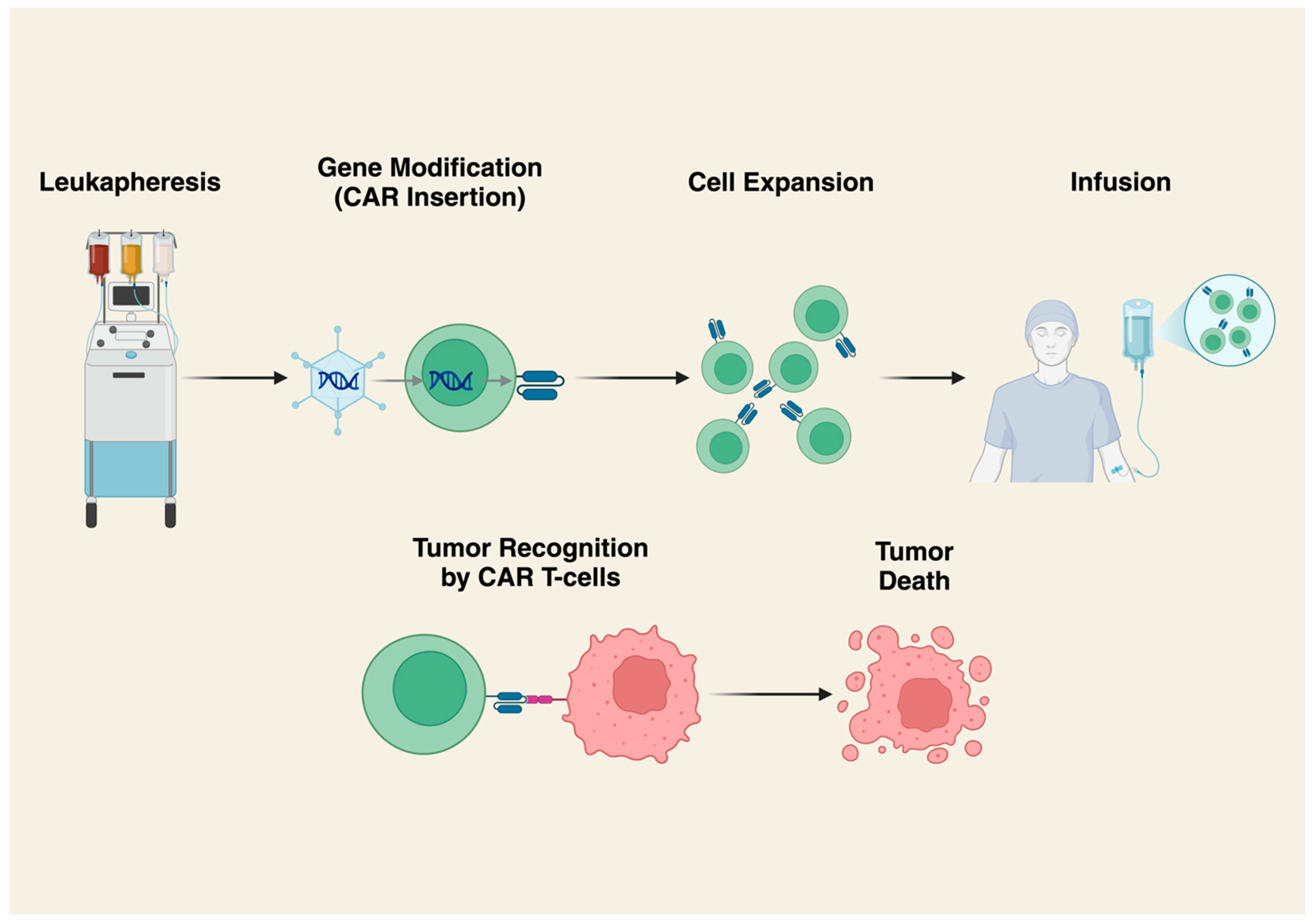
3. Chimeric Antigen Receptor (CAR) T-Cells
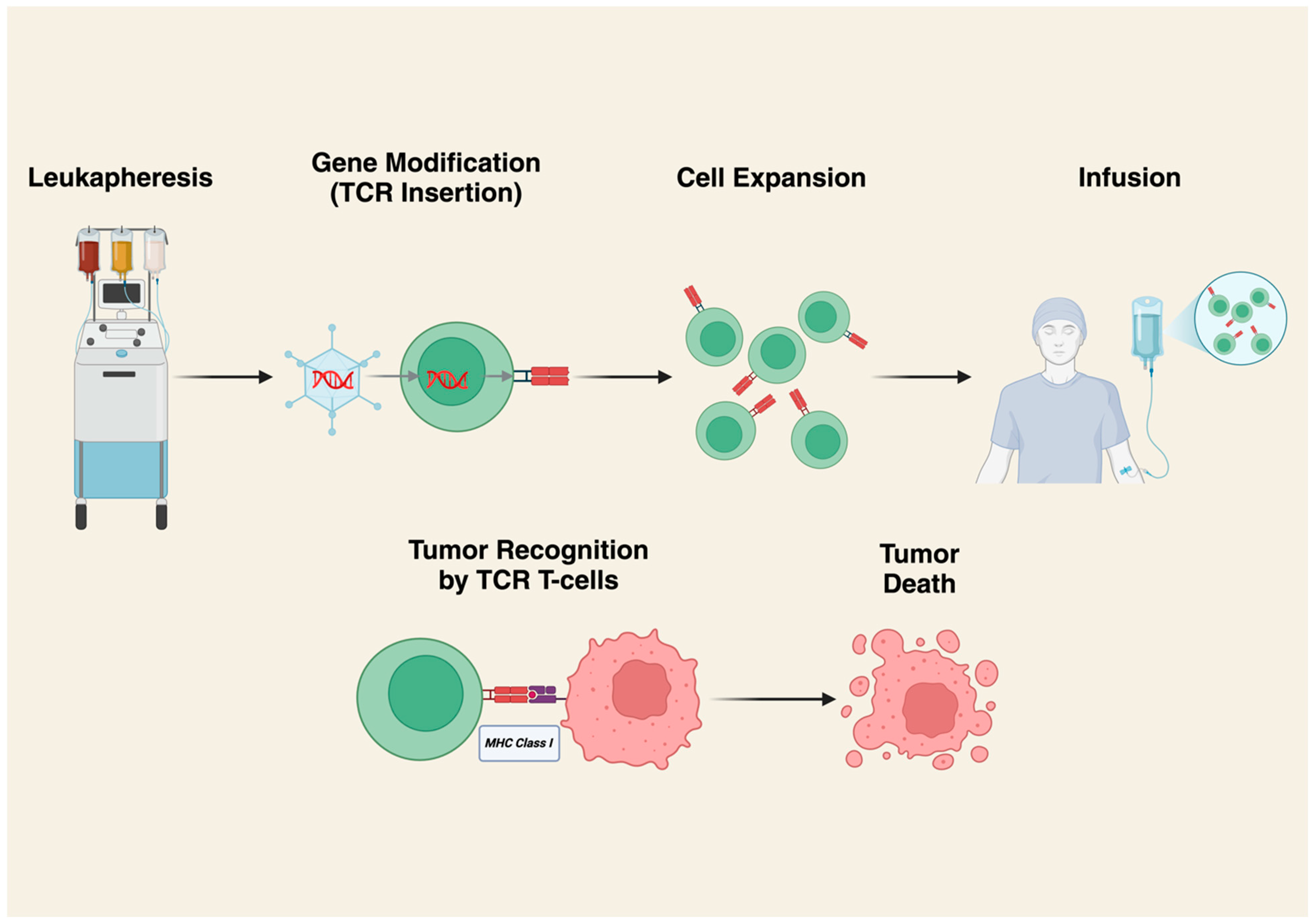
4. T-Cell Receptor (TCR) Gene-Modified T-Cell Therapy
5. Immune Microenvironment in Sarcomas
6. Challenges and Limitations
6.1. Safety Concerns and Adverse Events
6.2. Predictive Markers of Success
6.3. Limitations
6.4. Scalability and Manufacturing
7. Future Directions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACT | Adoptive Cellular Therapy |
| Afami-cel | Afamitresgene Autoleucel |
| CAR | Chimeric Antigen Receptor |
| CR | Complete Response |
| CRS | Cytokine Release Syndrome |
| CXCR2 | C-X-C Motif Chemokine Receptor 2 |
| DoR | Duration of Response |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HLA | Human Leukocyte Antigen |
| ICANS | Immune Effector Cell-Associated Neurotoxicity Syndrome |
| iCasp9 | Inducible Caspase-9 |
| ICI | Immune Checkpoint Inhibitor |
| Lete-cel | Letetresgene Autoleucel |
| LPA | Long Peptide Antigen |
| MAGE-A4 | Melanoma-Associated Antigen A4 |
| MDSC | Myeloid-Derived Suppressor Cell |
| MHC | Major Histocompatibility Complex |
| MRCLS | Myxoid/Round Cell Liposarcoma |
| NY-ESO-1 | New York Esophageal Squamous Cell Carcinoma-1 |
| ORR | Overall Response Rate |
| OS | Overall Survival |
| OTOT | On-Target/Off-Tumor Toxicity |
| PD | Progressive Disease |
| PFS | Progression-Free Survival |
| PR | Partial Response |
| SD | Stable Disease |
| SS | Synovial Sarcoma |
| STS | Soft-Tissue Sarcoma |
| TAM | Tumor-Associated Macrophage |
| TCR | T-Cell Receptor |
| TIL | Tumor-Infiltrating Lymphocyte |
| TLS | Tertiary Lymphoid Structure |
| TME | Tumor Microenvironment |
References
- Sbaraglia, M.; Bellan, E.; Dei Tos, A.P. The 2020 WHO Classification of Soft Tissue Tumours: News and perspectives. Pathologica 2021, 113, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Mathoulin-Pelissier, S.; Cesne, A.L.; Terrier, P.; Bonvalot, S.; Collin, F.; Michels, J.-J.; Blay, J.-Y.; Coindre, J.-M.; Bui, B. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer 2011, 117, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Lochner, J.; Menge, F.; Vassos, N.; Hohenberger, P.; Kasper, B. Prognosis of Patients with Metastatic Soft Tissue Sarcoma: Advances in Recent Years. Oncol. Res. Treat. 2020, 43, 613–619. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Ravi, V.; Subramaniam, A.; Zheng, J.; Amini, B.; Trinh, V.A.; Joseph, J.; Mennel, R.G.; Bishop, A.J.; Sturgis, E.M.; Goepfert, R.P.; et al. Clinical activity of checkpoint inhibitors in angiosarcoma: A retrospective cohort study. Cancer 2022, 128, 3383–3391. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.P.; Sharon, E.; O’Sullivan-Coyne, G.; Moore, N.; Foster, J.C.; Hu, J.S.; Van Tine, B.A.; Conley, A.P.; Read, W.L.; Riedel, R.F.; et al. Atezolizumab for advanced alveolar soft part sarcoma. N. Engl. J. Med. 2023, 389, 911–921. [Google Scholar] [CrossRef]
- Eulo, V.; Van Tine, B.A. Immune checkpoint inhibitor resistance in soft tissue sarcoma. Cancer Drug Resist. 2022, 5, 328–338. [Google Scholar] [CrossRef]
- Granhøj, J.S.; Witness Præst Jensen, A.; Presti, M.; Met, Ö.; Svane, I.M.; Donia, M. Tumor-infiltrating lymphocytes for adoptive cell therapy: Recent advances, challenges, and future directions. Expert Opin. Biol. Ther. 2022, 22, 627–641. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 1985, 313, 1485–1492. [Google Scholar] [CrossRef]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Hölmich, L.R.; Hendel, H.W.; Met, Ö.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef]
- Rohaan, M.W.; Borch, T.H.; van den Berg, J.H.; Met, Ö.; Kessels, R.; Geukes Foppen, M.H.; Stoltenborg Granhøj, J.; Nuijen, B.; Nijenhuis, C.; Jedema, I.; et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2022, 387, 2113–2125. [Google Scholar] [CrossRef]
- Ko, A.; Coward, V.S.; Gokgoz, N.; Dickson, B.C.; Tsoi, K.; Wunder, J.S.; Andrulis, I.L. Investigating the Potential of Isolating and Expanding Tumour-Infiltrating Lymphocytes from Adult Sarcoma. Cancers 2022, 14, 548. [Google Scholar] [CrossRef] [PubMed]
- Mullinax, J.E.; Hall, M.; Beatty, M.; Weber, A.M.; Sannasardo, Z.; Svrdlin, T.; Hensel, J.; Bui, M.; Richards, A.; Gonzalez, R.J.; et al. Expanded Tumor-infiltrating Lymphocytes From Soft Tissue Sarcoma Have Tumor-specific Function. J. Immunother. 2021, 44, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Monberg, T.; Sundvold, V.; Albieri, B.; Hovgaard, D.; Petersen, M.M.; Krarup-Hansen, A.; Met, Ö.; Camilio, K.; Clancy, T.; et al. LTX-315 and adoptive cell therapy using tumor-infiltrating lymphocytes generate tumor specific T cells in patients with metastatic soft tissue sarcoma. Oncoimmunology 2024, 13, 2290900. [Google Scholar] [CrossRef]
- Spicer, J.; Marabelle, A.; Baurain, J.-F.; Jebsen, N.L.; Jøssang, D.E.; Awada, A.; Kristeleit, R.; Loirat, D.; Lazaridis, G.; Jungels, C.; et al. Safety, Antitumor Activity, and T-cell Responses in a Dose-Ranging Phase I Trial of the Oncolytic Peptide LTX-315 in Patients with Solid Tumors. Clin. Cancer Res. 2021, 27, 2755–2763. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009, 21, 215–223. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Manara, M.C.; Hattinger, C.M.; Benini, S.; Perdichizzi, S.; Pasello, M.; Bacci, G.; Zanella, L.; Bertoni, F.; Picci, P.; et al. Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing’s sarcoma. Eur. J. Cancer 2005, 41, 1349–1361. [Google Scholar] [CrossRef]
- Ebb, D.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef]
- Reed, D.R.; Janeway, K.A.; Minard, C.G.; Hall, D.; Crompton, B.D.; Lazar, A.J.; Wang, W.-L.; Voss, S.D.; Militano, O.; Gorlick, R.G.; et al. PEPN1924, a phase 2 study of trastuzumab deruxtecan (DS-8201a, T-DXd) in adolescents and young adults with recurrent HER2+ osteosarcoma: A children’s oncology group pediatric early-phase clinical Trial Network study. JCO 2023, 41, 11527. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Navai, S.; DeRenzo, C.; Joseph, S.K.; Sanber, K.; Wu, M.; Gad, A.Z.; Janeway, K.A.; Campbell, M.; Mullikin, D.; et al. Autologous HER2-specific CAR T cells after lymphodepletion for advanced sarcoma: A phase 1 trial. Nat. Cancer 2024, 5, 880–894. [Google Scholar] [CrossRef]
- Cao, J.W.; Lake, J.; Impastato, R.; Chow, L.; Perez, L.; Chubb, L.; Kurihara, J.; Verneris, M.R.; Dow, S. Targeting osteosarcoma with canine B7-H3 CAR T cells and impact of CXCR2 Co-expression on functional activity. Cancer Immunol. Immunother. 2024, 73, 77. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Q.; Chen, W.; Shan, B.; Ding, Y.; Zhang, G.; Cao, N.; Liu, L.; Zhang, Y. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PLoS ONE 2013, 8, e70689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qin, D.; Shou, A.C.; Liu, Y.; Wang, Y.; Zhou, L. Exploring CAR-T Cell Therapy Side Effects: Mechanisms and Management Strategies. J. Clin. Med. 2023, 12, 6124. [Google Scholar] [CrossRef]
- Xiao, W.; Xu, L.; Wang, J.; Yu, K.; Xu, B.; Que, Y.; Zhao, J.; Pan, Q.; Gao, C.; Zhou, P.; et al. FGFR4-specific CAR-T cells with inducible caspase-9 suicide gene as an approach to treat rhabdomyosarcoma. Cancer Gene Ther. 2024, 31, 1571–1584. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Araujo, D.M.; Abdul Razak, A.R.; Agulnik, M.; Attia, S.; Blay, J.-Y.; Carrasco Garcia, I.; Charlson, J.A.; Choy, E.; Demetri, G.D.; et al. Afamitresgene autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1): An international, open-label, phase 2 trial. Lancet 2024, 403, 1460–1471. [Google Scholar] [CrossRef]
- Sanderson, J.P.; Crowley, D.J.; Wiedermann, G.E.; Quinn, L.L.; Crossland, K.L.; Tunbridge, H.M.; Cornforth, T.V.; Barnes, C.S.; Ahmed, T.; Howe, K.; et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology 2020, 9, 1682381. [Google Scholar] [CrossRef]
- Rytlewski, J.; Milhem, M.M.; Monga, V. Turning “Cold” tumors “Hot”: Immunotherapies in sarcoma. Ann. Transl. Med. 2021, 9, 1039. [Google Scholar] [CrossRef] [PubMed]
- Pollack, S.M.; He, Q.; Yearley, J.H.; Emerson, R.; Vignali, M.; Zhang, Y.; Redman, M.W.; Baker, K.K.; Cooper, S.; Donahue, B.; et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 2017, 123, 3291–3304. [Google Scholar] [CrossRef] [PubMed]
- Iura, K.; Maekawa, A.; Kohashi, K.; Ishii, T.; Bekki, H.; Otsuka, H.; Yamada, Y.; Yamamoto, H.; Harimaya, K.; Iwamoto, Y.; et al. Cancer-testis antigen expression in synovial sarcoma: NY-ESO-1, PRAME, MAGEA4, and MAGEA1. Hum. Pathol. 2017, 61, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Daudi, S.; Eng, K.H.; Mhawech-Fauceglia, P.; Morrison, C.; Miliotto, A.; Beck, A.; Matsuzaki, J.; Tsuji, T.; Groman, A.; Gnjatic, S.; et al. Expression and immune responses to MAGE antigens predict survival in epithelial ovarian cancer. PLoS ONE 2014, 9, e104099. [Google Scholar] [CrossRef]
- Hong, D.S.; Van Tine, B.A.; Biswas, S.; McAlpine, C.; Johnson, M.L.; Olszanski, A.J.; Clarke, J.M.; Araujo, D.; Blumenschein, G.R.; Kebriaei, P.; et al. Autologous T cell therapy for MAGE-A4+ solid cancers in HLA-A*02+ patients: A phase 1 trial. Nat. Med. 2023, 29, 104–114. [Google Scholar] [CrossRef]
- Endo, M.; de Graaff, M.A.; Ingram, D.R.; Lim, S.; Lev, D.C.; Briaire-de Bruijn, I.H.; Somaiah, N.; Bovée, J.V.M.G.; Lazar, A.J.; Nielsen, T.O. NY-ESO-1 (CTAG1B) expression in mesenchymal tumors. Mod. Pathol. 2015, 28, 587–595. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Lai, J.-P.; Rosenberg, A.Z.; Miettinen, M.M.; Lee, C.-C.R. NY-ESO-1 expression in sarcomas: A diagnostic marker and immunotherapy target. Oncoimmunology 2012, 1, 1409–1410. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 c259T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef]
- Gyurdieva, A.; Zajic, S.; Chang, Y.-F.; Houseman, E.A.; Zhong, S.; Kim, J.; Nathenson, M.; Faitg, T.; Woessner, M.; Turner, D.C.; et al. Biomarker correlates with response to NY-ESO-1 TCR T cells in patients with synovial sarcoma. Nat. Commun. 2022, 13, 5296. [Google Scholar] [CrossRef]
- Oike, N.; Kawashima, H.; Ogose, A.; Hotta, T.; Hatano, H.; Ariizumi, T.; Sasaki, T.; Yamagishi, T.; Umezu, H.; Endo, N. Prognostic impact of the tumor immune microenvironment in synovial sarcoma. Cancer Sci. 2018, 109, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Furness, A.J.S.; Thistlethwaite, F.; Burgess, M.A.; Riedel, R.F.; Haanen, J.; Noujaim, J.; Chalmers, A.W.; Pousa, A.L.; Chugh, R.; et al. Lete-cel in patients with synovial sarcoma or myxoid/round cell liposarcoma: Planned interim analysis of the pivotal IGNYTE-ESO trial. JCO 2024, 42, 2500. [Google Scholar] [CrossRef]
- Adaptimmune’s Lete-cel Achieves Primary Endpoint in Pivotal Trial :: Adaptimmune (ADAP). Available online: https://www.adaptimmune.com/investors-and-media/news-center/press-releases/detail/277/adaptimmunes-lete-cel-achieves-primary-endpoint-in-pivotal (accessed on 12 January 2025).
- D’Angelo, S.P.; Druta, M.; Van Tine, B.A.; Liebner, D.; Schuetze, S.M.; Tap, W.D.; Preston, J.; Goodison, S.; D’Souza, J.W.; Kapoor, G.S.; et al. Letetresgene autoleucel in advanced/metastatic myxoid/round cell liposarcoma. J. Clin. Oncol. 2025, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Weng, D.; Liu, J.; Han, Z.; Ou, Y.; Xu, B.; Peng, R.; Que, Y.; Wen, X.; Yang, J.; et al. Phase 1 clinical trial to assess safety and efficacy of NY-ESO-1-specific TCR T cells in HLA-A∗02:01 patients with advanced soft tissue sarcoma. Cell Rep. Med. 2023, 4, 101133. [Google Scholar] [CrossRef]
- Ishihara, M.; Nishida, Y.; Kitano, S.; Kawai, A.; Muraoka, D.; Momose, F.; Harada, N.; Miyahara, Y.; Seo, N.; Hattori, H.; et al. A phase 1 trial of NY-ESO-1-specific TCR-engineered T-cell therapy combined with a lymph node-targeting nanoparticulate peptide vaccine for the treatment of advanced soft tissue sarcoma. Int. J. Cancer 2023, 152, 2554–2566. [Google Scholar] [CrossRef]
- Muraoka, D.; Harada, N.; Hayashi, T.; Tahara, Y.; Momose, F.; Sawada, S.; Mukai, S.; Akiyoshi, K.; Shiku, H. Nanogel-based immunologically stealth vaccine targets macrophages in the medulla of lymph node and induces potent antitumor immunity. ACS Nano 2014, 8, 9209–9218. [Google Scholar] [CrossRef]
- Kageyama, S.; Wada, H.; Muro, K.; Niwa, Y.; Ueda, S.; Miyata, H.; Takiguchi, S.; Sugino, S.H.; Miyahara, Y.; Ikeda, H.; et al. Dose-dependent effects of NY-ESO-1 protein vaccine complexed with cholesteryl pullulan (CHP-NY-ESO-1) on immune responses and survival benefits of esophageal cancer patients. J. Transl. Med. 2013, 11, 246. [Google Scholar] [CrossRef]
- Hong, D.S.; Clarke, J.M.; Asch, A.; Charlson, J.; Johanns, T.M.; Calvo, E.; Boni, V.; de Miguel, M.J.; Garcia, V.M.; Lawrence, D.P.; et al. 540P Safety and efficacy from the SURPASS trial with ADP-A2M4CD8, a SPEAR T-cell therapy incorporating a CD8α co-receptor and an affinity optimized TCR targeting MAGE-A4. Ann. Oncol. 2021, 32, S604–S605. [Google Scholar] [CrossRef]
- Albarrán, V.; Villamayor, M.L.; Pozas, J.; Chamorro, J.; Rosero, D.I.; San Román, M.; Guerrero, P.; Pérez de Aguado, P.; Calvo, J.C.; García de Quevedo, C.; et al. Current landscape of immunotherapy for advanced sarcoma. Cancers 2023, 15, 2287. [Google Scholar] [CrossRef] [PubMed]
- Recine, F.; Vanni, S.; Bongiovanni, A.; Fausti, V.; Mercatali, L.; Miserocchi, G.; Liverani, C.; Pieri, F.; Casadei, R.; Cavaliere, D.; et al. Clinical and translational implications of immunotherapy in sarcomas. Front. Immunol. 2024, 15, 1378398. [Google Scholar] [CrossRef]
- Wood, G.E.; Meyer, C.; Petitprez, F.; D’Angelo, S.P. Immunotherapy in sarcoma: Current data and promising strategies. Am. Soc. Clin. Oncol. Educ. Book 2024, 44, e432234. [Google Scholar] [CrossRef]
- Shimizu, K.; Iyoda, T.; Okada, M.; Yamasaki, S.; Fujii, S.-I. Immune suppression and reversal of the suppressive tumor microenvironment. Int. Immunol. 2018, 30, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef]
- Fujiwara, T.; Healey, J.; Ogura, K.; Yoshida, A.; Kondo, H.; Hata, T.; Kure, M.; Tazawa, H.; Nakata, E.; Kunisada, T.; et al. Role of Tumor-Associated Macrophages in Sarcomas. Cancers 2021, 13, 1086. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.-M.; Marie, B.; Larousserie, F.; Entz-Werlé, N.; de Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. Oncoimmunology 2017, 6, e1331193. [Google Scholar]
- Dumars, C.; Ngyuen, J.-M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.-F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343–78354. [Google Scholar] [CrossRef]
- Fridman, W.H.; Sibéril, S.; Pupier, G.; Soussan, S.; Sautès-Fridman, C. Activation of B cells in Tertiary Lymphoid Structures in cancer: Anti-tumor or anti-self? Semin. Immunol. 2023, 65, 101703. [Google Scholar] [CrossRef]
- Sorbye, S.W.; Kilvaer, T.; Valkov, A.; Donnem, T.; Smeland, E.; Al-Shibli, K.; Bremnes, R.M.; Busund, L.-T. Prognostic impact of lymphocytes in soft tissue sarcomas. PLoS ONE 2011, 6, e14611. [Google Scholar] [CrossRef]
- Weng, W.; Yu, L.; Li, Z.; Tan, C.; Lv, J.; Lao, I.W.; Hu, W.; Deng, Z.; Liu, Z.; Wang, J.; et al. The immune subtypes and landscape of sarcomas. BMC Immunol. 2022, 23, 46. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Livingston, J.A.A.; Ravi, V.; Lin, H.Y.; Amini, B.; Solis, L.M.; Conley, A.P.; Zarzour, M.A.; Ludwig, J.A.; Ratan, R.; et al. A phase II multi-arm study to test the efficacy of oleclumab and durvalumab in specific sarcoma subtypes. JCO 2022, 40, TPS11594. [Google Scholar] [CrossRef]
- Lin, C.-C. Clinical Development of Colony-Stimulating Factor 1 Receptor (CSF1R) Inhibitors. J. Immunother. Precis. Oncol. 2021, 4, 105–114. [Google Scholar] [CrossRef]
- Movva, S.; Druta, M.; Davis, L.E.; Monga, V.; Milhem, M.M.; Bailey, H.H.; Chugh, R.; Bruns, I.; Allgood, V.E.; Chawla, S.P. Safety and clinical activity of TTI-621 in combination with doxorubicin in patients with unresectable or metastatic high-grade leiomyosarcoma: Results from the low-dose expansion cohort. JCO 2023, 41, 11508. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Targeting macrophages in cancer: From bench to bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Algarra, I.; Garrido, F.; Garcia-Lora, A.M. MHC heterogeneity and response of metastases to immunotherapy. Cancer Metastasis Rev. 2021, 40, 501–517. [Google Scholar] [CrossRef]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef]
- Springuel, L.; Lonez, C.; Alexandre, B.; Van Cutsem, E.; Machiels, J.-P.H.; Van Den Eynde, M.; Prenen, H.; Hendlisz, A.; Shaza, L.; Carrasco, J.; et al. Chimeric Antigen Receptor-T Cells for Targeting Solid Tumors: Current Challenges and Existing Strategies. BioDrugs 2019, 33, 515–537. [Google Scholar] [CrossRef]
- Johnson, L.A.; June, C.H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017, 27, 38–58. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-Cell Therapy for Solid Tumors: Lessons Learned from Lymphoma Treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef]
- Janelle, V.; Delisle, J.-S. T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers 2021, 13, 598. [Google Scholar] [CrossRef]
- Workman, M.J.; Mahe, M.M.; Trisno, S.; Poling, H.M.; Watson, C.L.; Sundaram, N.; Chang, C.-F.; Schiesser, J.; Aubert, P.; Stanley, E.G.; et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat. Med. 2017, 23, 49–59. [Google Scholar] [CrossRef]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, J.; Jiang, H.; Zhou, M. Strategies to Overcome Antigen Heterogeneity in CAR-T Cell Therapy. Cells 2025, 14, 320. [Google Scholar] [CrossRef]
- Moon, E.K.; Ranganathan, R.; Eruslanov, E.; Kim, S.; Newick, K.; O’Brien, S.; Lo, A.; Liu, X.; Zhao, Y.; Albelda, S.M. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin. Cancer Res. 2016, 22, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Joseph Espat, N.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Su, J.; Sun, R.; Sun, Y.; Wang, Y.; Dong, Y.; Shi, B.; Jiang, H.; Li, Z. Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors without Requiring Preconditioned Lymphodepletion. Clin. Cancer Res. 2020, 26, 5494–5505. [Google Scholar] [CrossRef]
- Xiong, X.; Xi, J.; Liu, Q.; Wang, C.; Jiang, Z.; Yue, S.-Y.; Shi, L.; Rong, Y. Co-expression of IL-7 and PH20 promote anti-GPC3 CAR-T tumour suppressor activity in vivo and in vitro. Liver Int. 2021, 41, 1033–1043. [Google Scholar] [CrossRef]
- Zhang, L.; Davies, J.S.; Serna, C.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A.; Hinrichs, C.S. Enhanced efficacy and limited systemic cytokine exposure with membrane-anchored interleukin-12 T-cell therapy in murine tumor models. J. Immunother. Cancer 2020, 8, e000210. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, G.; Wan, X. Challenges and new technologies in adoptive cell therapy. J. Hematol. Oncol. 2023, 16, 97. [Google Scholar]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: A phase 1 dose escalation and expansion trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef]
- Qu, C.; Zou, R.; Wang, P.; Zhu, Q.; Kang, L.; Ping, N.; Xia, F.; Liu, H.; Kong, D.; Yu, L.; et al. Decitabine-primed tandem CD19/CD22 CAR-T therapy in relapsed/refractory diffuse large B-cell lymphoma patients. Front. Immunol. 2022, 13, 969660. [Google Scholar] [CrossRef]
- Park, A.K.; Fong, Y.; Kim, S.-I.; Yang, J.; Murad, J.P.; Lu, J.; Jeang, B.; Chang, W.-C.; Chen, N.G.; Thomas, S.H.; et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020, 12, eaaz1863. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liao, Q.; Zhao, C.; Zhu, C.; Feng, M.; Liu, Z.; Jiang, L.; Zhang, L.; Ding, X.; Yuan, M.; et al. Conditioned CAR-T cells by hypoxia-inducible transcription amplification (HiTA) system significantly enhances systemic safety and retains antitumor efficacy. J. Immunother. Cancer 2021, 9, e002755. [Google Scholar] [CrossRef] [PubMed]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. “Final common pathway” of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for neoantigen identification. Front. Immunol. 2019, 10, 1392. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Trial | Cancer Treated * | Adoptive Cellular Product | Status ** |
|---|---|---|---|
| NCT04052334 | Soft-Tissue Sarcoma | TIL | Completed |
| NCT05607095 | Undifferentiated Pleomorphic Sarcoma Dedifferentiated Liposarcoma | TIL | Recruiting |
| NCT03725605 | Soft-Tissue Sarcoma | TIL | Completed |
| NCT03935893 | Solid Tumor | TIL | Recruiting |
| NCT06566092 | Rhabdomyosarcoma Ewing Sarcoma | TIL | Recruiting |
| NCT03449108 | Bone Sarcoma Soft-Tissue Sarcoma | TIL | Active |
| NCT03449108 | Bone Sarcoma Soft-Tissue Sarcoma | TIL | Active |
- To the best of our knowledge, this table contains all active, completed, and recruiting clinical trials using TILs for the treatment of sarcomas accessible on ClinicalTrials.gov. Sarcomas treated in the trials are listed if specified by subtype in the study’s online entry; however, many trials recruited participants with other malignancies (may be found via respective trial numbers).
- * When abundantly clear, we include in this column the sarcoma subtype that was assessed in the clinical trial. However, many trials investigated solid tumors broadly, including sarcomas. These studies are marked as such in this column. ** This table may not be exhaustive as there are trials listed with statuses of “unknown” and “not yet recruiting” that were excluded.
| Trial | Cancer Treated * | CAR Target | Status ** |
|---|---|---|---|
| NCT04995003 | Sarcoma | HER2 | Recruiting |
| NCT00902044 | Sarcoma | HER2 | Active |
| NCT04511871 | Solid Tumor | HER2 | Active |
| NCT03635632 | Solid Tumor | GD2 | Active |
| NCT03373097 | Solid Tumor | GD2 | Recruiting |
| NCT03721068 | Osteosarcoma | GD2 | Recruiting |
| NCT04539366 | Osteosarcoma | GD2 | Recruiting |
| NCT04483778 | Solid Tumor | B7-H3 | Active |
| NCT06500819 | Solid Tumor | B7-H3 | Recruiting |
| NCT04556669 | Solid Tumor | CD22 | Recruiting |
| NCT06087341 | Sarcoma | NKG2D | Recruiting |
| NCT05312411 | Osteosarcoma | FITC-E2 | Active |
| NCT05103631 | Solid Tumor | GPC3 | Recruiting |
| NCT04377932 | Solid Tumor | GPC3 | Recruiting |
| NCT04715191 | Solid Tumor | GPC3 | Recruiting |
| NCT05120271 | Solid Tumor | GPC3 | Recruiting |
- To the best of our knowledge, this table contains all active, completed, and recruiting clinical trials using CAR T-cells for treatment of sarcomas accessible on ClinicalTrials.gov. Sarcomas treated in the trials are listed if specified by subtype in the study’s online entry, however, many trials recruited participants with other malignancies (may be found via respective trial numbers).
- * When abundantly clear, we include in this column the sarcoma subtype that was assessed in the clinical trial. However, many trials investigated solid tumors broadly, including sarcomas. These studies are marked as such in this column. ** This table may not be exhaustive as there are trials listed with statuses of “unknown” and “not yet recruiting” that were excluded.
| Trial | Cancer Treated * | TCR Target | Status ** |
|---|---|---|---|
| NCT04044768 | Synovial Sarcoma Myxoid/Round Cell Liposarcoma | MAGE-A4 | Recruiting |
| NCT03132922 | Synovial Sarcoma Myxoid Round Cell Liposarcoma | MAGE-A4 | Active |
| NCT05642455 | Synovial Sarcoma Osteosarcoma | MAGE-A4 | Recruiting |
| NCT06703346 (Sub-Study of NCT03967223) | Synovial Sarcoma Myxoid Round Cell Liposarcoma | NY-ESO-1 | Active |
| NCT05993299 (Sub-Study of NCT03967223) | Synovial Sarcoma Myxoid Round Cell Liposarcoma | NY-ESO-1 | Active |
| NCT01343043 | Synovial Sarcoma | NY-ESO-1 | Completed |
| NCT01477021 | Synovial Sarcoma | NY-ESO-1 | Completed |
| NCT06083883 | Synovial Sarcoma Myxoid Round Cell Liposarcoma | NY-ESO-1 | Recruiting |
| NCT02650986 | Synovial Sarcoma | NY-ESO-1 | Active |
| NCT03450122 | Synovial Sarcoma Myxoid Round Cell Liposarcoma | NY-ESO-1 | Completed |
| NCT03967223 | Synovial Sarcoma Myxoid Round Cell Liposarcoma | NY-ESO-1 | Active |
| NCT03250325 | Synovial sarcoma | NY-ESO-1 | Completed |
| NCT05296564 | Synovial Sarcoma Soft-Tissue Sarcoma | NY-ESO-1 | Recruiting |
| NCT02992743 | Myxoid Round Cell Liposarcoma | NY-ESO-1 | Completed |
| NCT04318964 | Soft-Tissue Sarcoma | NY-ESO-1 | Active |
| NCT03462316 | Bone Sarcoma Soft-Tissue Sarcoma | NY-ESO-1 | Active |
| NCT02319824 | Sarcoma | NY-ESO-1 | Completed |
| NCT03450122 | Synovial Sarcoma Myxoid Round Cell Liposarcoma | NY-ESO-1 | Completed |
| NCT02869217 | Synovial Sarcoma | NY-ESO-1 | Active |
| NCT05621668 | Bone Sarcoma Soft-Tissue Sarcoma | Attil12 | Recruiting |
- To the best of our knowledge, this table contains all active, completed, and recruiting clinical trials using TCR gene-modified T-cells for the treatment of sarcomas accessible on ClinicalTrials.gov. The sarcomas treated in the trials are listed if specified by subtype in the study’s online entry; however, many trials recruited participants with other malignancies (may be found via respective trial numbers).
- * When abundantly clear, we include in this column the sarcoma subtype that was assessed in the clinical trial. However, many trials investigated solid tumors broadly, including sarcomas. These studies are marked as such in this column. ** This table may not be exhaustive as there are trials listed with statuses of “unknown” and “not yet recruiting” that were excluded.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fradin, J.J.; Charlson, J.A. Review of Adoptive Cellular Therapies for the Treatment of Sarcoma. Cancers 2025, 17, 1302. https://doi.org/10.3390/cancers17081302
Fradin JJ, Charlson JA. Review of Adoptive Cellular Therapies for the Treatment of Sarcoma. Cancers. 2025; 17(8):1302. https://doi.org/10.3390/cancers17081302
Chicago/Turabian StyleFradin, James J., and John A. Charlson. 2025. "Review of Adoptive Cellular Therapies for the Treatment of Sarcoma" Cancers 17, no. 8: 1302. https://doi.org/10.3390/cancers17081302
APA StyleFradin, J. J., & Charlson, J. A. (2025). Review of Adoptive Cellular Therapies for the Treatment of Sarcoma. Cancers, 17(8), 1302. https://doi.org/10.3390/cancers17081302