
Beyond Adaptive Immunity: Trained Innate Immune Responses as a Novel Frontier in Hepatocellular Carcinoma Therapy


Simple Summary
Abstract
1. Introduction
2. Mechanisms of Trained Immunity and Innate Immune Responses in HCC
2.1. Trained Innate Immunity: Definition and Core Mechanisms
2.2. Innate Immune Landscape in HCC: Baseline Immune Dysfunction and Tolerance
3. Impact of Trained Immunity on the HCC Immune Microenvironment
4. Clinical Implications
4.1. Trained Immunity-Based Therapeutic Strategies in HCC
4.2. Immune Modulation of the HCC Microenvironment by Trained Immunity
5. Resistance Mechanisms and Immune Evasion in the Context of Trained Immunity
6. Future Directions Section
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | Antigen-Presenting Cell. |
| BCG | Bacillus Calmette–Guérin |
| CAR | Chimeric Antigen Receptor |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated protein 4 |
| DC | Dendritic Cell |
| HBV | Hepatitis B Virus |
| HCC | Hepatocellular Carcinoma |
| HCV | Hepatitis C Virus |
| HLA | Human Leukocyte Antigen |
| ICI | Immune Checkpoint Inhibitor |
| IFN-γ | Interferon gamma |
| IL | Interleukin (e.g., IL-1β, IL-6, IL-10, IL-12, IL-15, IL-18) |
| iNOS | Inducible Nitric Oxide Synthase |
| LPS | Lipopolysaccharide |
| MDSC | Myeloid-Derived Suppressor Cell |
| MHC | Major Histocompatibility Complex |
| mTOR | Mammalian Target Of Rapamycin |
| NASH | Non-Alcoholic Steatohepatitis |
| NK | Natural Killer (cells) |
| NKG2A | Natural Killer Group 2A |
| NLRP3 | NOD-Like Receptor Protein 3 |
| NOD2 | Nucleotide-binding Oligomerization Domain-containing protein 2 |
| PD-1 | Programmed cell Death protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| ROS | Reactive Oxygen Species |
| TAM | Tumor-Associated Macrophage |
| TAN | Tumor-Associated Neutrophil |
| TGF-β | Transforming Growth Factor beta |
| TLR | Toll-Like Receptor |
| TME | Tumor Microenvironment |
References
- Yu, J.; Li, M.; Ren, B.; Cheng, L.; Wang, X.; Ma, Z.; Yong, W.P.; Chen, X.; Wang, L.; Goh, B.C. Unleashing the efficacy of immune checkpoint inhibitors for advanced hepatocellular carcinoma: Factors, strategies, and ongoing trials. Front. Pharmacol. 2023, 14, 1261575. [Google Scholar] [CrossRef]
- Sui, Y.; Berzofsky, J.A. Trained immunity inducers in cancer immunotherapy. Front. Immunol. 2024, 15, 1427443. [Google Scholar] [CrossRef] [PubMed]
- Kotsari, M.; Dimopoulou, V.; Koskinas, J.; Armakolas, A. Immune System and Hepatocellular Carcinoma (HCC): New Insights into HCC Progression. Int. J. Mol. Sci. 2023, 24, 11471. [Google Scholar] [CrossRef] [PubMed]
- Ficht, X.; Iannacone, M. Immune surveillance of the liver by T cells. Sci. Immunol. 2020, 5, eaba2351. [Google Scholar] [CrossRef]
- Arneth, B. Trained innate immunity. Immunol. Res. 2021, 69, 1–7. [Google Scholar] [CrossRef]
- Daman, A.; Cheong, J.-G.; Berneking, L.; Josefowicz, S. The potency of hematopoietic stem cell reprogramming for changing immune tone. Immunol. Rev. 2024, 323, 197–208. [Google Scholar] [CrossRef]
- Netea, M.; Joosten, L.; Meer, J. Hypothesis: Stimulation of trained immunity as adjunctive immunotherapy in cancer. J. Leukoc. Biol. 2017, 102, 1323–1332. [Google Scholar] [CrossRef]
- Hu, L.; Wang, Z.; Liao, Y.; Jiang, X.; Lian, H.; Lin, Z. Tumor-infiltrating T-Lymphocyte immunity-related immune tolerance and anti–programmed cell death protein 1/ligand of programmed cell death protein 1 therapy for advanced hepatocellular carcinoma. Oncol. Transl. Med. 2024, 10, 162–170. [Google Scholar] [CrossRef]
- Vetvicka, V.; Sima, P.; Vannucci, L. Trained Immunity as an Adaptive Branch of Innate Immunity. Int. J. Mol. Sci. 2021, 22, 10684. [Google Scholar] [CrossRef]
- Tercan, H.; Riksen, N.; Joosten, L.; Netea, M.; Bekkering, S. Trained Immunity: Long-Term Adaptation in Innate Immune Responses. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 55–61. [Google Scholar] [CrossRef]
- Netea, M.; Joosten, L.; Latz, E.; Mills, K.; Natoli, G.; Stunnenberg, H.; O’Neill, L.; Xavier, R. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Fanucchi, S.; Domínguez-Andrés, J.; Joosten, L.A.B.; Netea, M.G.; Mhlanga, M.M. The Intersection of Epigenetics and Metabolism in Trained Immunity. Immunity 2021, 54, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.; Carvalho, A.; La Rocca, C.; Palma, C.; Rodrigues, F.; Silvestre, R.; Kleinnijenhuis, J.; Lachmandas, E.; Gonçalves, L.; Belinha, A.; et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep. 2016, 17, 2562–2571. [Google Scholar] [CrossRef] [PubMed]
- Moorlag, S.; Khan, N.; Novakovic, B.; Kaufmann, E.; Jansen, T.; van Crevel, R.; Divangahi, M.; Netea, M.G. β-Glucan Induces Protective Trained Immunity against Mycobacterium tuberculosis Infection: A Key Role for IL-1. Cell Rep. 2020, 31, 107634. [Google Scholar] [CrossRef]
- Lérias, J.R.; de Sousa, E.; Paraschoudi, G.; Martins, J.; Condeço, C.; Figueiredo, N.; Carvalho, C.; Dodoo, E.; Maia, A.; Castillo-Martin, M.; et al. Trained Immunity for Personalized Cancer Immunotherapy: Current Knowledge and Future Opportunities. Front. Microbiol. 2019, 10, 2924. [Google Scholar] [CrossRef]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Mata-Martínez, P.; Bergón-Gutiérrez, M.; Del Fresno, C. Dectin-1 Signaling Update: New Perspectives for Trained Immunity. Front. Immunol. 2022, 13, 812148. [Google Scholar] [CrossRef]
- Mourits, V.P.; Arts, R.J.W.; Novakovic, B.; Matzaraki, V.; de Bree, L.C.J.; Koeken, V.; Moorlag, S.; van Puffelen, J.H.; Groh, L.; van der Heijden, C.; et al. The role of Toll-like receptor 10 in modulation of trained immunity. Immunology 2020, 159, 289–297. [Google Scholar] [CrossRef]
- Weinberg, A.; Johnson, M.; Crotteau, M.; Ghosh, D.; Vu, T.; Levin, M.J. Trained Immunity Generated by the Recombinant Zoster Vaccine. Res. Sq. 2024. [Google Scholar] [CrossRef]
- Tao, J.; Zhang, J.; Ling, Y.; McCall, C.; Liu, T. Mitochondrial Sirtuin 4 Resolves Immune Tolerance in Monocytes by Rebalancing Glycolysis and Glucose Oxidation Homeostasis. Front. Immunol. 2018, 9, 419. [Google Scholar] [CrossRef]
- De Graaf, D.M.; Teufel, L.U.; van de Veerdonk, F.L.; Joosten, L.A.B.; Netea, M.G.; Dinarello, C.A.; Arts, R.J.W. IL-38 prevents induction of trained immunity by inhibition of mTOR signaling. J. Leukoc. Biol. 2021, 110, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xing, Z. Ligustilide counteracts carcinogenesis and hepatocellular carcinoma cell-evoked macrophage M2 polarization by regulating yes-associated protein-mediated interleukin-6 secretion. Exp. Biol. Med. 2021, 246, 1928–1937. [Google Scholar] [CrossRef]
- Shibata, M.; Nakajima, T.; Mimura, K.; Shimura, T.; Kono, K.; Takenoshita, S. MDSC (myeloid-derived suppressor cells) is an important immunosuppressing factor and functionally related with VEGF and IL-17 in patients with gastrointestinal cancer. Ann. Oncol. 2019, 30, xi4. [Google Scholar] [CrossRef]
- Li, H.-J.; Zhai, N.; Wang, Z.; Song, H.; Yang, Y.; Cui, A.; Li, T.; Wang, G.; Niu, J.; Crispe, I.; et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut 2017, 67, 2035–2044. [Google Scholar] [CrossRef]
- Xue, J.S.; Ding, Z.N.; Meng, G.X.; Yan, L.J.; Liu, H.; Li, H.C.; Yao, S.Y.; Tian, B.W.; Dong, Z.R.; Chen, Z.Q.; et al. The Prognostic Value of Natural Killer Cells and Their Receptors/Ligands in Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. Front. Immunol. 2022, 13, 872353. [Google Scholar] [CrossRef]
- Gao, J.; Duan, Z.; Zhang, L.; Huang, X.; Long, L.; Tu, J.; Liang, H.; Zhang, Y.; Shen, T.; Lu, F. Failure recovery of circulating NKG2D+CD56dimNK cells in HBV-associated hepatocellular carcinoma after hepatectomy predicts early recurrence. Oncoimmunology 2015, 5, e1048061. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Mitroulis, I.; Germanidis, G. Tumor-Associated Neutrophils in Hepatocellular Carcinoma Pathogenesis, Prognosis, and Therapy. Cancers 2021, 13, 2899. [Google Scholar] [CrossRef]
- Leslie, J.; Mackey, J.B.G.; Jamieson, T.; Ramon-Gil, E.; Drake, T.M.; Fercoq, F.; Clark, W.; Gilroy, K.; Hedley, A.; Nixon, C.; et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut 2022, 71, 2093–2106. [Google Scholar] [CrossRef]
- Dudek, M.; Tacke, F. Immature neutrophils bring anti-PD-1 therapy in NASH-HCC to maturity. Gut 2022, 71, 1937–1938. [Google Scholar] [CrossRef]
- Yu, S.J.; Greten, T.F. Deciphering and Reversing Immunosuppressive Cells in the Treatment of Hepatocellular Carcinoma. J. Liver Cancer 2020, 20, 1–16. [Google Scholar] [CrossRef]
- Pinter, M.; Pinato, D.J.; Ramadori, P.; Heikenwalder, M. NASH and Hepatocellular Carcinoma: Immunology and Immunotherapy. Clin. Cancer Res. 2023, 29, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, Y.; Lin, D.; Lei, L.; Mei, Y.; Jin, Z.; Gong, H.; Zhu, Y.; Hu, B.; Zhang, Y.; et al. NCR(-) group 3 innate lymphoid cells orchestrate IL-23/IL-17 axis to promote hepatocellular carcinoma development. EBioMedicine 2019, 41, 333–344. [Google Scholar] [CrossRef] [PubMed]
- He, L.-H.; Zhang, X.; Sun, K.; Bai, X. Commentary on “The tumour microenvironment shapes innate lymphoid cells in patients with hepatocellular carcinoma”. Hepatobiliary Surg. Nutr. 2022, 11, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Xing, Y.; Wang, Q.; Xiao, W.; Lu, Y.; Hu, X.; Gao, Z.; Xu, M.; Yan, J.; Cao, R.; et al. Improved efficacy of therapeutic vaccination with dendritic cells pulsed with tumor cell lysate against hepatocellular carcinoma by introduction of 2 tandem repeats of microbial HSP70 peptide epitope 407-426 and OK-432. Int. Immunopharmacol. 2011, 11, 2200–2207. [Google Scholar] [CrossRef]
- Lichtenegger, F.; Mueller, K.; Otte, B.; Beck, B.; Hiddemann, W.; Schendel, D.; Subklewe, M. CD86 and IL-12p70 Are Key Players for T Helper 1 Polarization and Natural Killer Cell Activation by Toll-Like Receptor-Induced Dendritic Cells. PLoS ONE 2012, 7, e44266. [Google Scholar] [CrossRef]
- Han, S.; Bao, X.; Zou, Y.; Wang, L.; Li, Y.; Yang, L.; Liao, A.; Zhang, X.; Jiang, X.; Liang, D.; et al. d-lactate modulates M2 tumor-associated macrophages and remodels immunosuppressive tumor microenvironment for hepatocellular carcinoma. Sci. Adv. 2023, 9, eadg2697. [Google Scholar] [CrossRef]
- Du, K.; Li, Y.; Liu, J.; Chen, W.; Wei, Z.; Luo, Y.; Liu, H.; Qi, Y.; Wang, F.; Sui, J. A bispecific antibody targeting GPC3 and CD47 induced enhanced antitumor efficacy against dual antigen-expressing HCC. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 1572–1584. [Google Scholar] [CrossRef]
- Kirchhammer, N.; Trefny, M.; Natoli, M.; Brücher, D.; Smith, S.; Werner, F.; Koch, V.; Schreiner, D.; Bartoszek, E.; Buchi, M.; et al. NK cells with tissue-resident traits shape response to immunotherapy by inducing adaptive antitumor immunity. Sci. Transl. Med. 2022, 14, eabm9043. [Google Scholar] [CrossRef]
- Jiang, Z.-Z.; Peng, Z.-P.; Liu, X.-C.; Guo, H.-F.; Zhou, M.-M.; Jiang, D.; Ning, W.-R.; Huang, Y.-F.; Zheng, L.; Wu, Y. Neutrophil extracellular traps induce tumor metastasis through dual effects on cancer and endothelial cells. Oncoimmunology 2022, 11, 2052418. [Google Scholar] [CrossRef]
- Martin, K.; Schreiner, J.; Zippelius, A. Modulation of APC Function and Anti-Tumor Immunity by Anti-Cancer Drugs. Front. Immunol. 2015, 6, 501. [Google Scholar] [CrossRef]
- Sojoodi, M.; Barrett, S.; Erstad, D.; Jordan, V.; Gale, E.; Salloum, S.; Wang, Y.; Lanuti, M.; Zukerberg, L.; Caravan, P.; et al. Abstract LB278: IL-12 immunotherapy prevents hepatocellular carcinoma in a murine NAFLD induced cirrhosis model. Cancer Res. 2023, 83, LB278. [Google Scholar] [CrossRef]
- Oncology Live® Staff. Superagonists Pull IL-15 Into Focus in Oncology Care. Oncol. Live 2022, 23, 82. [Google Scholar]
- Sajid, M.; Liu, L.; Sun, C. The Dynamic Role of NK Cells in Liver Cancers: Role in HCC and HBV Associated HCC and Its Therapeutic Implications. Front. Immunol. 2022, 13, 887186. [Google Scholar] [CrossRef]
- Lee, M.; Park, C.-S.; Lee, Y.-R.; Im, S.A.; Song, S.; Lee, C. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch. Pharmacal Res. 2014, 37, 1234–1240. [Google Scholar] [CrossRef]
- Michel, K.; Fulton, R.; Leonardo, S.; Gorden, K.; Graff, J.; Danielson, M. Abstract 1703: A novel tumor vaccine platform: Direct conjugation of antigens to the β glucan PAMP Imprime PGG enhances antigen presentation and T cell priming. Cancer Res. 2017, 77, 1703. [Google Scholar] [CrossRef]
- Chen, K.; Wu, Z.; Zang, M.; Wang, C.; Wang, Y.; Wang, D.; Ma, Y.; Qu, C. Immunization with glypican-3 nanovaccine containing TLR7 agonist prevents the development of carcinogen-induced precancerous hepatic lesions to cancer in a murine model. Am. J. Transl. Res. 2018, 10, 1736–1749. [Google Scholar]
- Buffen, K.; Oosting, M.; Quintin, J.; Ng, A.; Kleinnijenhuis, J.; Kumar, V.; Van De Vosse, E.; Wijmenga, C.; Van Crevel, R.; Oosterwijk, E.; et al. Autophagy Controls BCG-Induced Trained Immunity and the Response to Intravesical BCG Therapy for Bladder Cancer. PLoS Pathog. 2014, 10, e1004485. [Google Scholar] [CrossRef]
- Singh, A.; Netea, M.; Bishai, W. BCG turns 100: Its nontraditional uses against viruses, cancer, and immunologic diseases. J. Clin. Investig. 2021, 131, e148291. [Google Scholar] [CrossRef]
- Vaziri, F.; Setayesh, T.; Hu, Y.; Ravindran, R.; Wei, D.; Wan, Y.Y. BCG as an Innovative Option for HCC Treatment: Repurposing and Mechanistic Insights. Adv. Sci. 2024, 11, e2308242. [Google Scholar] [CrossRef]
- Vuscan, P.; Kischkel, B.; Hatzioannou, A.; Markaki, E.; Sarlea, A.; Tintoré, M.; Cune, J.; Verginis, P.; De Lecea, C.; Chavakis, T.; et al. Potent induction of trained immunity by Saccharomyces cerevisiae β-glucans. Front. Immunol. 2024, 15, 1323333. [Google Scholar] [CrossRef]
- Camilli, G.; Tabouret, G.; Quintin, J. The Complexity of Fungal β-Glucan in Health and Disease: Effects on the Mononuclear Phagocyte System. Front. Immunol. 2018, 9, 673. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Pan, J.; Xiang, W.; You, Z.; Zhang, Y.; Wang, J.; Zhang, A. β-glucan: A potent adjuvant in immunotherapy for digestive tract tumors. Front. Immunol. 2024, 15, 1424261. [Google Scholar] [CrossRef] [PubMed]
- Cheung, I.Y.; Mauguen, A.; Modak, S.; Ragupathi, G.; Basu, E.M.; Roberts, S.S.; Kushner, B.H.; Cheung, N.K. Effect of Oral β-Glucan on Antibody Response to Ganglioside Vaccine in Patients With High-Risk Neuroblastoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2023, 9, 242–250. [Google Scholar] [CrossRef]
- Thwe, P.M.; Fritz, D.I.; Snyder, J.P.; Smith, P.R.; Curtis, K.D.; O’Donnell, A.; Galasso, N.A.; Sepaniac, L.A.; Adamik, B.J.; Hoyt, L.R.; et al. Syk-dependent glycolytic reprogramming in dendritic cells regulates IL-1β production to β-glucan ligands in a TLR-independent manner. J. Leukoc. Biol. 2019, 106, 1325–1335. [Google Scholar] [CrossRef]
- Tran, J.; Wong, P.; Fan, C.; Russler-Germain, D.; Cubitt, C.; Marin, N.; Schappe, T.; Berrien-Elliott, M.; Wang, T.; Foltz, J.; et al. Combined IL-12, IL-15 and IL-18 activation induces epigenetic changes in human NK cells during the memory-like state transition. J. Immunol. 2023, 210, 160.12. [Google Scholar] [CrossRef]
- Lui, G.; Minnar, C.; Soon-Shiong, P.; Schlom, J.; Gameiro, S. Exploiting an Interleukin-15 Heterodimeric Agonist (N803) for Effective Immunotherapy of Solid Malignancies. Cells 2023, 12, 1611. [Google Scholar] [CrossRef]
- Chen, W.; Bamford, R.; Edmondson, E.; Waldmann, T. IL-15 and anti-PD-1 augment the efficacy of agonistic intratumoral anti-CD40 in a mouse model with multiple TRAMP-C2 tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 2082–2093. [Google Scholar] [CrossRef]
- Zhou, Z.; Lin, L.; An, Y.; Zhan, M.; Chen, Y.; Cai, M.; Zhu, X.; Lu, L.; Zhu, K. The Combination Immunotherapy of TLR9 Agonist and OX40 Agonist via Intratumoural Injection for Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 529–543. [Google Scholar] [CrossRef]
- Karapetyan, L.; Luke, J.; Davar, D. Toll-Like Receptor 9 Agonists in Cancer. OncoTargets Ther. 2020, 13, 10039–10060. [Google Scholar] [CrossRef]
- Naour, J.L.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 2020, 9, 1777624. [Google Scholar] [CrossRef]
- Yu, T.; Girard, M.; Watts, T. Abstract A012: STING agonists drive intrinsic type I interferon responses in monocytes for optimal anti-tumor immunity. Cancer Immunol. Res. 2023, 11, A012. [Google Scholar] [CrossRef]
- Jekle, A.; Thatikonda, S.; Jaisinghani, R.; Ren, S.; Kinkade, A.; Stevens, S.; Stoycheva, A.; Rajwanshi, V.; Williams, C.; Deval, J.; et al. Tumor Regression upon Intratumoral and Subcutaneous Dosing of the STING Agonist ALG-031048 in Mouse Efficacy Models. Int. J. Mol. Sci. 2023, 24, 16274. [Google Scholar] [CrossRef] [PubMed]
- Kapate, N.; Dunne, M.; Gottlieb, A.; Mukherji, M.; Suja, V.; Prakash, S.; Park, K.; Kumbhojkar, N.; Guerriero, J.; Mitragotri, S. Polymer Backpack-loaded Tissue Infiltrating Monocytes for Treating Cancer. Adv. Healthc. Mater. 2024, 14, e2304144. [Google Scholar] [CrossRef]
- Chow, S.; Dorigo, O. Monocytes—A Promising New TRAIL in Ovarian Cancer Cell Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 29, 299–301. [Google Scholar] [CrossRef]
- Facciorusso, A.; Tartaglia, N.; Villani, R.; Serviddio, G.; Ramai, D.; Mohan, B.P.; Chandan, S.; Abd El Aziz, M.A.; Evangelista, J.; Cotsoglou, C.; et al. Lenvatinib versus sorafenib as first-line therapy of advanced hepatocellular carcinoma: A systematic review and meta-analysis. Am. J. Transl. Res. 2021, 13, 2379–2387. [Google Scholar]
- Contreras, L.; Rodríguez-Gil, A.; Muntané, J.; de la Cruz, J. Sorafenib-associated translation reprogramming in hepatocellular carcinoma cells. RNA Biol. 2025, 22, 1–11. [Google Scholar] [CrossRef]
- Napoletano, C.; Ruscito, I.; Bellati, F.; Zizzari, I.; Rahimi, H.; Gasparri, M.; Antonilli, M.; Panici, P.; Rughetti, A.; Nuti, M. Bevacizumab-Based Chemotherapy Triggers Immunological Effects in Responding Multi-Treated Recurrent Ovarian Cancer Patients by Favoring the Recruitment of Effector T Cell Subsets. J. Clin. Med. 2019, 8, 380. [Google Scholar] [CrossRef]
- Lee, J.; Lozano-Ruiz, B.; Yang, F.; Fan, D.D.; Shen, L.; González-Navajas, J. The Multifaceted Role of Th1, Th9, and Th17 Cells in Immune Checkpoint Inhibition Therapy. Front. Immunol. 2021, 12, 625667. [Google Scholar] [CrossRef]
- Fathi, F.; Saidi, R.; Banafshe, H.; Arbabi, M.; Lotfinia, M.; Motedayyen, H. Changes in immune profile affect disease progression in hepatocellular carcinoma. Int. J. Immunopathol. Pharmacol. 2022, 36, 03946320221078476. [Google Scholar] [CrossRef]
- Haber, P.; Castet, F.; Torres-Martín, M.; Andreu-Oller, C.; Puigvehí, M.; Maeda, M.; Radu, P.; Dufour, J.; Verslype, C.; Czauderna, C.; et al. Molecular markers of response to anti-PD1 therapy in advanced hepatocellular carcinoma. Gastroenterology 2022, 164, 72–88.e18. [Google Scholar] [CrossRef]
- Chew, V.; Lai, L.; Pan, L.; Lim, C.; Li, J.; Ong, R.; Chua, C.; Leong, J.; Lim, K.; Toh, H.; et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc. Natl. Acad. Sci. USA 2017, 114, E5900–E5909. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, C.; Sun, Q.; Wang, Y.; Yu, W.; Wei, F.; Ren, X. Trained Immunity of IL-12-, IL-15-, and IL-18-Induced CD3+CD56+ NKT-Like Cells. J. Oncol. 2022, 2022, 8724933. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-J.; Tsou, P.-Y.; Chou, S.; Lee, M.-N.; Chen, S.-C. Abstract 6352: Combination treatment with recombinant murine IL-12 and anti-PD-1 antibody enhanced anti-tumor efficacy through the activation of cytotoxic T cell response. Cancer Res. 2023, 83, 6352. [Google Scholar] [CrossRef]
- Ghosh, C.C.; Cournoyer, L.; Liu, Y.; Ballarin, A.; Layman, I.B.; LaPorte, J.; Morrissey, M.; Fraser, K.; Perati, S.; Cox, B.F.; et al. Subcutaneous checkpoint inhibition is equivalent to systemic delivery when combined with nelitolimod delivered via pressure-enabled drug delivery for depletion of intrahepatic myeloid-derived suppressor cells and control of liver metastases. J. Immunother. Cancer 2024, 12, e008837. [Google Scholar] [CrossRef]
- Choi, G.; Na, H.; Kuen, D.S.; Kim, B.S.; Chung, Y. Autocrine TGF-β1 Maintains the Stability of Foxp3(+) Regulatory T Cells via IL-12Rβ2 Downregulation. Biomolecules 2020, 10, 819. [Google Scholar] [CrossRef]
- Sarkar, O.; Donninger, H.; Rayyan, N.A.; Chew, L.; Stamp, B.; Zhang, X.; Whitt, A.; Li, C.; Hall, M.; Mitchell, R.; et al. Tumor-educated monocytes suppress T cells via adenosine and depletion of adenosine in the tumor microenvironment with adenosine deaminase enzyme promotes response to immunotherapy. bioRxiv 2022. [Google Scholar] [CrossRef]
- Humbert, M.; Guery, L.; Brighouse, D.; Lemeille, S.; Hugues, S. Intratumoral CpG-B Promotes Antitumoral Neutrophil, cDC, and T-cell Cooperation without Reprograming Tolerogenic pDC. Cancer Res. 2018, 78, 3280–3292. [Google Scholar] [CrossRef]
- Lee, P.C.; Wu, C.J.; Hung, Y.W.; Lee, C.J.; Chi, C.T.; Lee, I.C.; Yu-Lun, K.; Chou, S.H.; Luo, J.C.; Hou, M.C.; et al. Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor-treated unresectable hepatocellular carcinoma. J. Immunother. Cancer 2022, 10, e004779. [Google Scholar] [CrossRef]
- Qu, S.; Worlikar, T.; Felsted, A.; Ganguly, A.; Beems, M.; Hubbard, R.; Pepple, A.; Kevelin, A.; Garavaglia, H.; Dib, J.; et al. Non-thermal histotripsy tumor ablation promotes abscopal immune responses that enhance cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000200. [Google Scholar] [CrossRef]
- Löffler, M.W.; Nussbaum, B.; Jäger, G.; Jurmeister, P.S.; Budczies, J.; Pereira, P.L.; Clasen, S.; Kowalewski, D.J.; Mühlenbruch, L.; Königsrainer, I.; et al. A Non-interventional Clinical Trial Assessing Immune Responses After Radiofrequency Ablation of Liver Metastases From Colorectal Cancer. Front. Immunol. 2019, 10, 2526. [Google Scholar] [CrossRef]
- Tao, H.; Zhong, X.; Zeng, A.; Song, L. Unveiling the veil of lactate in tumor-associated macrophages: A successful strategy for immunometabolic therapy. Front. Immunol. 2023, 14, 1208870. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, Y.; Qu, Q.; Zhu, J.; Liu, Z.; Ning, W.; Zeng, H.; Zhang, N.; Du, W.; Chen, C.; et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int. J. Clin. Oncol. 2017, 22, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, C.; Wang, B.; Yang, J.; Wang, Y.; Luo, F.; Xu, J.; Zhao, C.; Liu, R.; Chu, Y. The IFN-γ/PD-L1 axis between T cells and tumor microenvironment: Hints for glioma anti-PD-1/PD-L1 therapy. J. Neuroinflamm. 2018, 15, 290. [Google Scholar] [CrossRef]
- Shklovskaya, E.; Rizos, H. MHC Class I Deficiency in Solid Tumors and Therapeutic Strategies to Overcome It. Int. J. Mol. Sci. 2021, 22, 6741. [Google Scholar] [CrossRef]
- Seo, H.; Jeon, I.; Kim, B.-S.; Park, M.; Bae, E.-A.; Song, B.; Koh, C.-H.; Shin, K.-S.; Kim, I.-K.; Choi, K.-Y.; et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat. Commun. 2017, 8, 15776. [Google Scholar] [CrossRef]
- Nicolai, C.J.; Wolf, N.; Chang, I.C.; Kirn, G.; Marcus, A.; Ndubaku, C.O.; McWhirter, S.M.; Raulet, D.H. NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci. Immunol. 2020, 5, eaaz2738. [Google Scholar] [CrossRef]
- Yu, H.; Aravindan, N.; Xu, J.; Natarajan, M. Inter- and intra-cellular mechanism of NF-kB-dependent survival advantage and clonal expansion of radio-resistant cancer cells. Cell. Signal. 2017, 31, 105–111. [Google Scholar] [CrossRef]
- Zhang, F.-B.; Wang, B.; Zhang, W.; Xu, Y.-F.; Zhang, C.; Xue, X. Transcription Factor MAZ Potentiates the Upregulated NEIL3-mediated Aerobic Glycolysis, Thereby Promoting Angiogenesis in Hepatocellular Carcinoma. Curr. Cancer Drug Targets 2024, 24, 1235–1249. [Google Scholar] [CrossRef]
- Hoerning, A.; Koss, K.; Datta, D.; Boneschansker, L.; Jones, C.; Wong, I.; Irimia, D.; Calzadilla, K.; Benítez, F.; Hoyer, P.; et al. Subsets of human CD4+ regulatory T cells express the peripheral homing receptor CXCR3. Eur. J. Immunol. 2011, 41, 2291–2302. [Google Scholar] [CrossRef]
- Scurr, M.; Pembroke, T.; Bloom, A.; Roberts, D.; Thomson, A.; Smart, K.; Bridgeman, H.; Adams, R.; Brewster, A.; Jones, R.; et al. Low-Dose Cyclophosphamide Induces Antitumor T-Cell Responses, which Associate with Survival in Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 6771–6780. [Google Scholar] [CrossRef]
- Wu, J.-M.; Chang, K.-H.; Hsu, F.L.-T. Immunonutrition of perioperative therapy for colorectal cancer. Formos. J. Surg. 2023, 56, 9–11. [Google Scholar]
- Gao, Z.-G.; Jacobson, K. A2B Adenosine Receptor and Cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef] [PubMed]
- Perrot, I.; Michaud, H.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425. [Google Scholar] [CrossRef] [PubMed]
- Wurm, M.; Schaaf, O.; Reutner, K.; Ganesan, R.; Mostböck, S.; Pelster, C.; Böttcher, J.; De Andrade Pereira, B.; Taubert, C.; Alt, I.; et al. A Novel Antagonistic CD73 Antibody for Inhibition of the Immunosuppressive Adenosine Pathway. Mol. Cancer Ther. 2021, 20, 2250–2261. [Google Scholar] [CrossRef]
- Lu, Y.; Gu, X.; Chen, L.; Yao, Z.; Song, J.; Niu, X.; Xiang, R.; Cheng, T.; Qin, Z.; Deng, W.; et al. Interferon-γ produced by tumor-infiltrating NK cells and CD4+ T cells downregulates TNFSF15 expression in vascular endothelial cells. Angiogenesis 2014, 17, 529–540. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Y.; Wang, P.; Li, M.; Xiang, T.; Xie, S.; Li, M.; Wang, Y.; Weng, D.; Zhao, J. Targeting PDHK1 by DCA to Restore NK Cell Function in Hepatocellular Carcinoma. Mol. Cancer Ther. 2024, 23, 1731–1742. [Google Scholar] [CrossRef]
- Gregory, S.; Perati, S.; Brown, Z. Alteration in immune function in patients with fatty liver disease. Hepatoma Res. 2022, 8, 31. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Quitt, O.; Luo, S.; Meyer, M.; Xie, Z.; Golsaz-Shirazi, F.; Loffredo-Verde, E.; Festag, J.; Bockmann, J.-H.; Zhao, L.; Stadler, D.; et al. T-cell engager antibodies enable T cells to control HBV infection and to target HBsAg-positive hepatoma in mice. J. Hepatol. 2021, 75, 1058–1071. [Google Scholar] [CrossRef]

{kind=link}
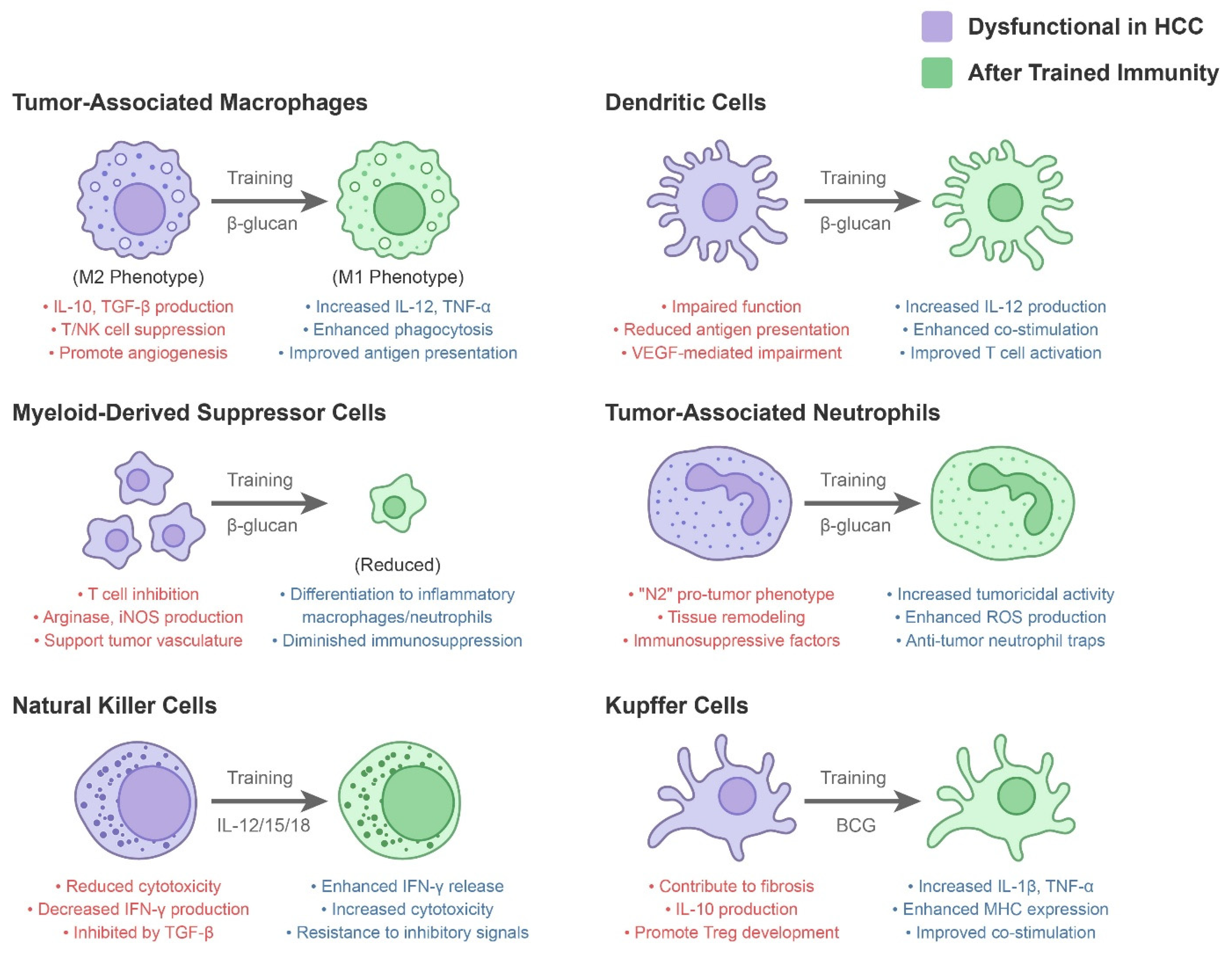
| Innate Immune Component | Dysfunctional Role in HCC Microenvironment | Modulation by Trained Immunity |
|---|---|---|
| Tumor-Associated Macrophages (TAMs) | Often M2-polarized; secrete IL-10, TGF-β, VEGF; suppress T/NK cells; promote angiogenesis and tumor growth [3]. High TAM burden correlates with poor prognosis [3]. | Training (e.g., with BCG or β-glucan) can repolarize macrophages toward an M1 phenotype, increasing IL-12, TNF-α, and antigen presentation [34]. Trained macrophages show enhanced phagocytosis and ROS production against tumor cells, potentially converting an immunosuppressive milieu into an inflammatory one [2]. |
| Myeloid-Derived Suppressor Cells (MDSCs) | Expanded in HCC; inhibit T-cell proliferation via arginase, iNOS, IL-10; support tumor vasculature and metastasis [23]. Present in blood and tumors of HCC patients, especially in advanced disease [23]. | Certain trained immunity strategies aim to differentiate or deplete MDSCs. For example, β-glucan can drive myeloid precursors towards inflammatory macrophages and neutrophils, reducing the immunosuppressive MDSC pool [2]. Trained monocytes may also be resistant to tumor-derived suppressive signals, diminishing MDSC accumulation [2]. |
| Natural Killer (NK) Cells | Key anti-tumor effectors, but often dysfunctional in HCC due to TGF-β and chronic stimulation. Show reduced cytotoxicity and IFN-γ production; some HCCs upregulate HLA-E or shed NKG2D ligands to evade NK attack. High NK infiltration predicts better survival [25,26]. | Cytokine-induced trained NK cells have enhanced IFN-γ release and cytotoxicity upon encountering tumor cells [2]. IL-12/15/18 “memory-like” NK cells or IL-15 superagonist-expanded NK cells can overcome some inhibitory signals and more efficiently lyse HCC cells (even those with low MHC-I) [2]. Trained NK cells are being tested in adaptive cell therapy to improve tumor control in HCC [2]. |
| Neutrophils (TANs) | Often exhibit an “N2” pro-tumor phenotype: secreting proteases, ROS that cause tissue remodeling, and suppressive factors. Neutrophil-to-lymphocyte ratio is a negative prognostic indicator in HCC [28,29]. | Trained immunity (e.g., via β-glucan) can reprogram neutrophil production and function. β-glucan has been shown to induce trained granulopoiesis, yielding neutrophils with increased tumoricidal activity (via ROS and neutrophil extracellular traps) [2]. Trained neutrophils may be less prone to the immature, immunosuppressive phenotype seen in NASH-HCC [28,29]. However, excessive neutrophil activation must be balanced to avoid collateral damage. |
| Dendritic Cells (DCs) | Conventional DCs in HCC are often functionally impaired [30]. High VEGF levels in the HCC microenvironment interfere with DC differentiation and antigen presentation [1]. As a result, DCs in HCC may have reduced capacity to prime anti-tumor T cells [30]. | Trained monocytes give rise to more potent DCs with increased IL-12 production and co-stimulatory molecule expression [2]. In a trained environment, DCs may overcome tumor-induced paralysis, leading to better activation of tumor-specific T cells. Some trained immunity adjuvants (like CpG DNA or LPS analogs) directly activate DCs to mature and migrate into lymph nodes, bridging innate and adaptive responses [2]. |
| Kupffer Cells (Liver Resident Macrophages) | In chronic liver disease, Kupffer cells contribute to fibrosis and can become tolerant to endotoxin (reducing their cytokine output). They form part of the immunosuppressive stroma in HCC, producing IL-10 and promoting Treg development [2]. | BCG or other inducers can potentially train Kupffer cells. Trained Kupffer cells would secrete more pro-inflammatory cytokines (IL-1β, TNF-α) upon sensing tumor antigens or danger signals, thereby activating other immune cells in the liver. In trained mice, liver macrophages have shown increased expression of MHC and co-stimulatory molecules [2,35], suggesting improved capacity to stimulate anti-tumor T cells locally. |
| Mechanism Category | Therapy | Mode of Action and Relevance to Trained Immunity in HCC |
|---|---|---|
| Trained Immunity Induction | BCG Vaccine | Engages pattern recognition receptors (NOD2, TLRs) on monocytes/macrophages, driving them into a trained state. In preclinical HCC models, a single BCG dose significantly reduced tumor burden and outperformed anti-PD-1 therapy [2]. |
| β-Glucan (yeast-derived) | Binds dectin-1 on myeloid cells, triggering Syk–NLRP3 inflammasome signaling and IL-1β release, a key trained immunity mechanism. β-Glucan-trained macrophages and neutrophils have increased tumoricidal activity [2]. Synergized with PD-1 blockade in murine models [2]. | |
| Cytokine-Based Therapy | IL-15 Superagonist (N-803) | Expands and activates NK and CD8^+ T cells in vivo. Being tested in clinical trials for solid tumors, including liver cancer [42]. |
| Adoptive Cell Therapy | Cytokine-Trained NK Cells | IL-12/15/18-trained NK cells exhibit enhanced IFN-γ secretion and cytotoxicity against HCC cells [2]. Early-phase trials for NK cell therapy in HCC are ongoing [43]. |
| Immune Checkpoint Modulation | Anti-NKG2A and Anti-CD47 | Anti-NKG2A prevents HCC cells from engaging NK-cell inhibitory receptors, enhancing NK cytotoxicity [25]. Anti-CD47 removes the “don’t eat me” signal, enabling trained macrophage phagocytosis [34]. |
| Innate Immune Adjuvants | TLR Agonists (CpG, R848) | Activate TLR9 and TLR7/8 pathways in DCs and macrophages, promoting tumor antigen presentation and T-cell recruitment [44]. Clinical trials are evaluating liver-targeted delivery of these agonists. |
| Trained Immunity-Based Vaccines | BCG/β-Glucan + Tumor Antigen | A combination strategy to engage both innate and adaptive immunity. β-Glucan-based vaccines have shown enhanced T-cell responses in preclinical studies [45,46]. |
| Combination Therapy | Checkpoint Inhibitor + Trained Immunity Inducer | Combining β-glucan or BCG with anti-PD-1 therapy increased immune infiltration and tumor control in preclinical models [2]. Atezolizumab + Bevacizumab already demonstrates microenvironment modification. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, C.-H.; Chuang, P.-C.; Liu, Y.-W. Beyond Adaptive Immunity: Trained Innate Immune Responses as a Novel Frontier in Hepatocellular Carcinoma Therapy. Cancers 2025, 17, 1250. https://doi.org/10.3390/cancers17071250
Hsieh C-H, Chuang P-C, Liu Y-W. Beyond Adaptive Immunity: Trained Innate Immune Responses as a Novel Frontier in Hepatocellular Carcinoma Therapy. Cancers. 2025; 17(7):1250. https://doi.org/10.3390/cancers17071250
Chicago/Turabian StyleHsieh, Ching-Hua, Pei-Chin Chuang, and Yueh-Wei Liu. 2025. "Beyond Adaptive Immunity: Trained Innate Immune Responses as a Novel Frontier in Hepatocellular Carcinoma Therapy" Cancers 17, no. 7: 1250. https://doi.org/10.3390/cancers17071250
APA StyleHsieh, C.-H., Chuang, P.-C., & Liu, Y.-W. (2025). Beyond Adaptive Immunity: Trained Innate Immune Responses as a Novel Frontier in Hepatocellular Carcinoma Therapy. Cancers, 17(7), 1250. https://doi.org/10.3390/cancers17071250