
A Network-Based Approach Exploiting Transcriptomics and Interactomics Data for Predicting Drug Repurposing Solutions Across Human Cancers



Simple Summary
Abstract
1. Introduction
- Text-mining-based approaches. They involve extracting new knowledge from existing scientific literature [9].
- Semantic-based approaches. They integrate multiple sources (chemical, pharmacological, biological, biomedical, etc.) to build a semantic network, from which it is possible to predict new drug–disease relationships. While these methods have the advantage of making use of a massive number of databases, it is still a challenge to integrate all of these different sources [9].
- Transcriptional signature-based approaches. Transcriptomics data provide us with a list of over- and under-expressed genes in a biological system, treated with a drug or affected by a disease/condition. Comparing the transcriptional signatures can give us new insights on relationships between drugs and diseases [10].
- Molecular Docking. Here, we try to score the interaction between a small-molecule ligand and a protein to evaluate if a drug can bind to a new target [10].
- Network-based approaches. These methods organize the relationships between biological entities in the form of a network. Nodes can be many kinds of biological entities, like drugs, diseases, or proteins, while edges represent a direct or indirect interaction between two nodes [10]. A well-established algorithm falling in this category is SAveRUNNER [11], broadly applied in several biological contexts from viral infections to complex human diseases [12,13,14,15,16].
2. Materials and Methods
2.1. Data Retrieval
2.1.1. Gene Expression Data
2.1.2. Drug–Target Interactions
2.1.3. The SIGnaling Network Open Resource
2.2. Preprocessing
2.2.1. Differentially Expressed Genes Analysis
2.2.2. Disease Genes
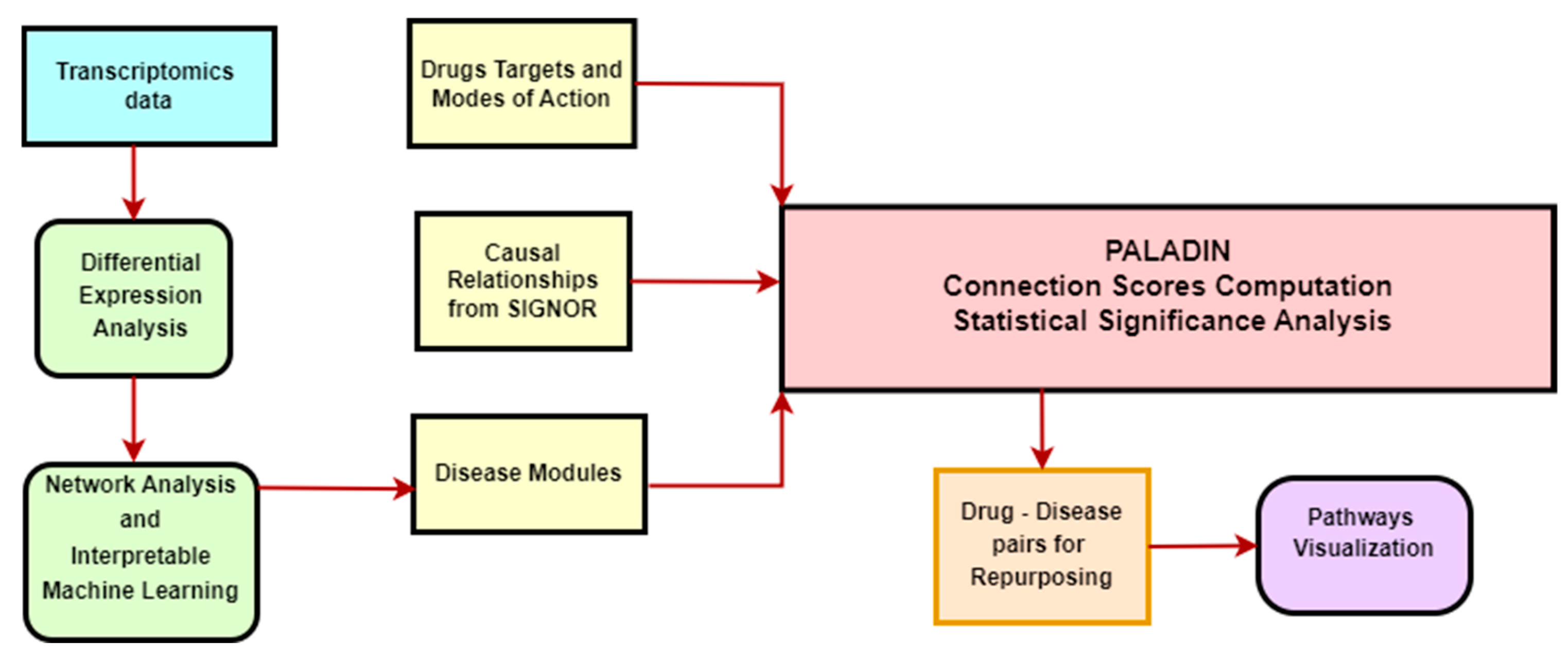
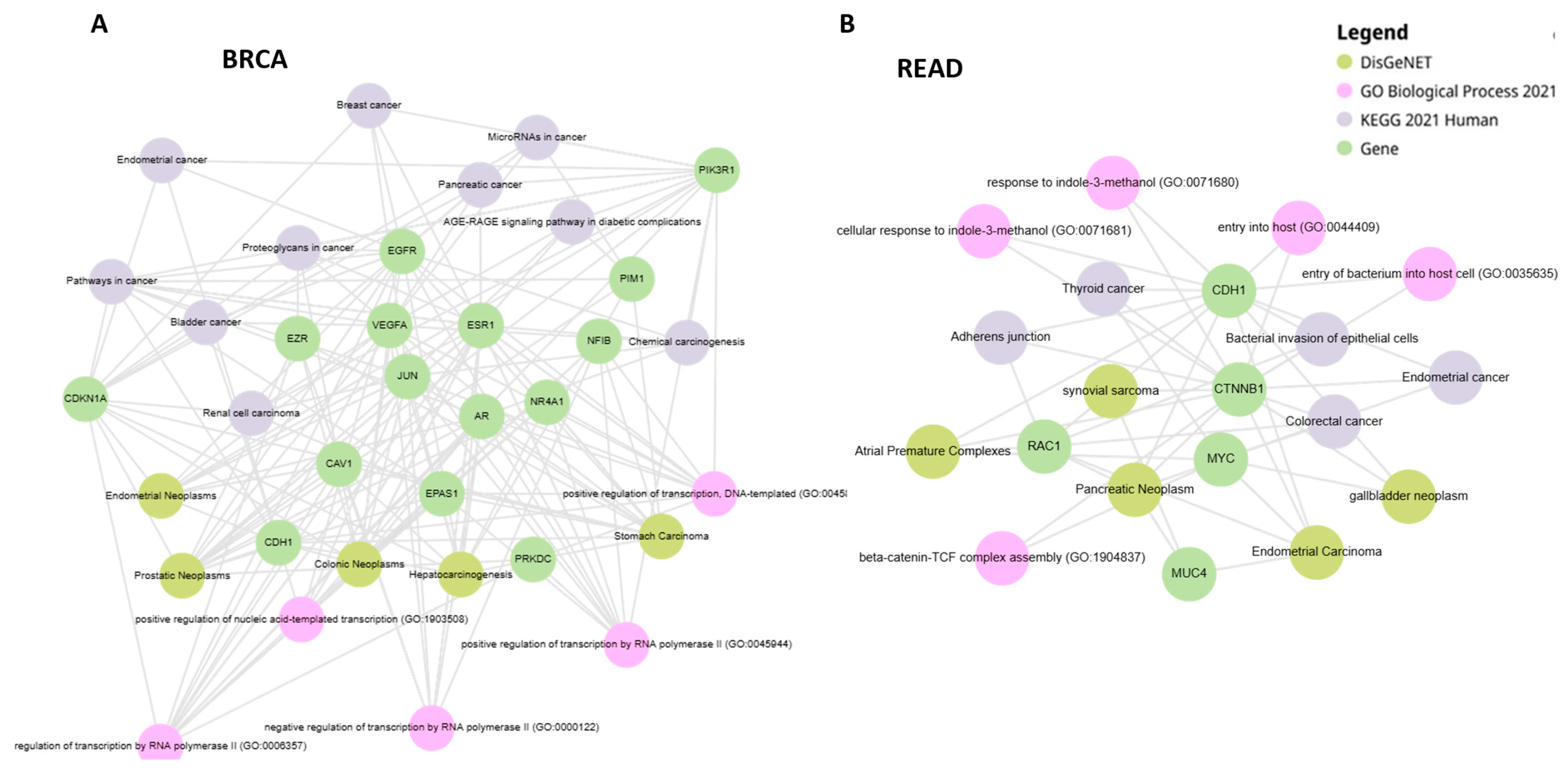
2.3. PALADIN—Functional Enrichment Analysis
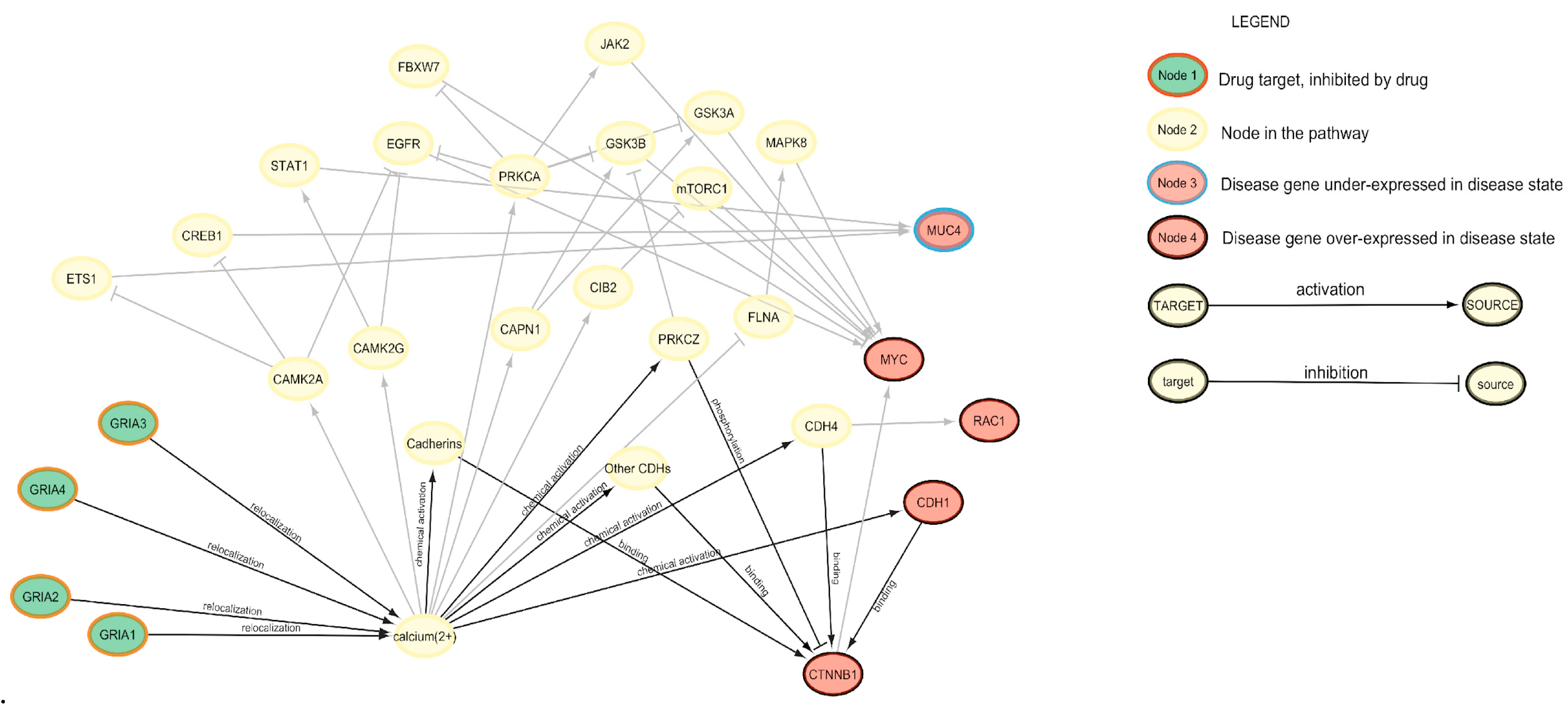
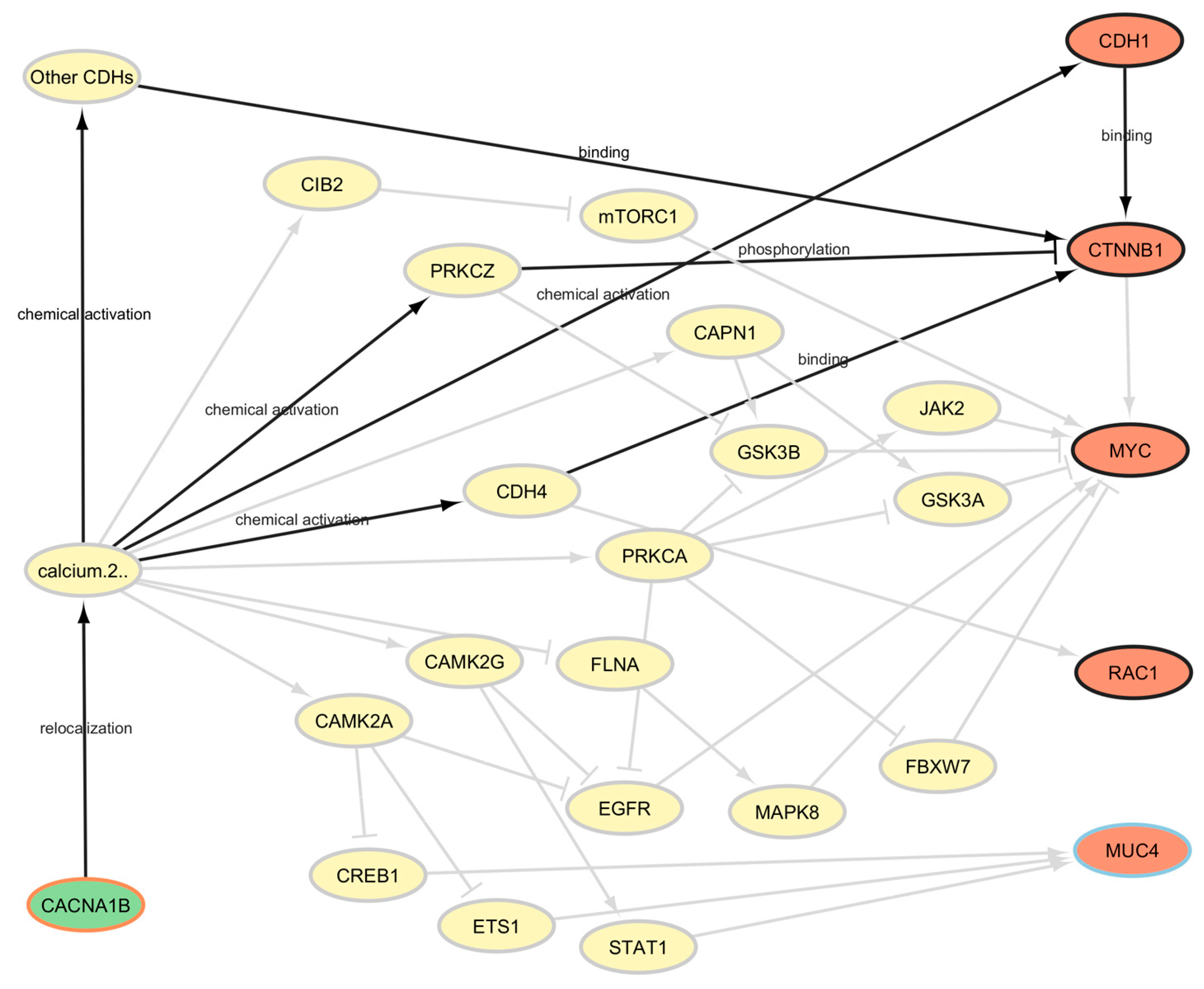
2.4. PALADIN—Pathways Analyzer for Off-LAbel inDIcatioNs
3. Results and Discussion
3.1. PALADIN Analysis
3.2. SAveRUNNER Analysis and Comparison
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PALADIN | Pathways Analyzer for off-LAbel inDIcatioNs |
| DR | Drug Repurposing |
| TCGA | The Cancer Genome Atlas |
| HGCN | HUGO Gene Nomenclature |
| UCEC | Uterine Corpus Endometrial Carcinoma |
| THYM | Thymoma |
| THCA | Thyroid carcinoma |
| STAD | Stomach Adenocarcinoma |
| SKCM | Skin Cutaneous Melanoma |
| READ | Rectum Adenocarcinoma |
| PRAD | Prostate Adenocarcinoma |
| PAAD | Pancreatic Adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| LUAD | Lung Adenocarcinoma |
| LIHC | Liver Hepatocellular Carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| KIRC | Kidney renal clear cell carcinoma |
| KICH | Kidney chromophobe |
| ESCA | Esophageal carcinoma |
| COAD | Colon adenocarcinoma |
| BRCA | Breast cancer |
| BLCA | Bladder cancer |
| SAveRUNNER | Searching off-lAbel dRUg aNd NEtwoRk |
| SIGNOR | SIGnaling Network Open Resource |
| DEGs | Differentially Expressed Genes |
| SANTA | Spatial Analysis of Network Associations |
| iml | Interpretable Machine Learning |
References
- Available online: https://www.efpia.eu/media/rm4kzdlx/the-pharmaceutical-industry-in-figures-2023.pdf (accessed on 19 March 2025).
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, J.-P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rousseau, J.F.; Ding, Y.; Song, M.; Lu, W. Understanding Drug Repurposing from the Perspective of Biomedical Entities and Their Evolution: Bibliographic Research Using Aspirin. JMIR Public Health Surveill. 2020, 8, e16739. [Google Scholar] [CrossRef]
- Rehman, W.; Arfons, L.M.; Lazarus, H.M. The rise, fall and subsequent triumph of thalidomide: Lessons learned in drug development. Ther. Adv. Hematol. 2011, 2, 291–308. [Google Scholar] [CrossRef]
- Available online: https://www.elsevier.com/industry/drug-repurposing (accessed on 19 March 2025).
- Ng, Y.L.; Salim, C.K.; Chu, J.J.H. Drug repurposing for COVID-19: Approaches, challenges and promising candidates. Pharmacol. Ther. 2021, 228, 107930. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef]
- March-Vila, E.; Pinzi, L.; Sturm, N.; Tinivella, A.; Engkvist, O.; Chen, H.; Rastelli, G. On the Integration of In Silico Drug Design Methods for Drug Repurposing. Front. Pharmacol. 2017, 8, 298. [Google Scholar] [CrossRef]
- Fiscon, G.; Paci, P. SAveRUNNER: An R-based tool for drug repurposing. BMC Bioinform. 2021, 22, 150. [Google Scholar] [CrossRef]
- Fiscon, G.; Conte, F.; Farina, L.; Paci, P. SAveRUNNER: A network-based algorithm for drug repurposing and its application to COVID-19. PLoS Comput. Biol. 2021, 17, e1008686. [Google Scholar] [CrossRef]
- Fiscon, G.; Conte, F.; Amadio, S.; Volonté, C.; Paci, P. Drug Repurposing: A Network-based Approach to Amyotrophic Lateral Sclerosis. Neurotherapeutics 2021, 18, 1678–1691. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, M.; Paci, P.; Fiscon, G. A network-based bioinformatic analysis for identifying potential repurposable active molecules in different types of human cancers. In Proceedings of the 2023 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Istanbul, Turkiye, 5–8 December 2023; pp. 3626–3631. [Google Scholar]
- Amadio, S.; Conte, F.; Esposito, G.; Fiscon, G.; Paci, P.; Volonté, C. Repurposing Histaminergic Drugs in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 6347. [Google Scholar] [CrossRef] [PubMed]
- Fiscon, G.; Sibilio, P.; Funari, A.; Conte, F.; Paci, P. Identification of Potential Repurposable Drugs in Alzheimer’s Disease Exploiting a Bioinformatics Analysis. J. Pers. Med. 2022, 12, 1731. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://xenabrowser.net/datapages/ (accessed on 19 March 2025).
- Zhang, L.; Fan, S.; Vera, J.; Lai, X. A network medicine approach for identifying diagnostic and prognostic biomarkers and exploring drug repurposing in human cancer. Comput. Struct. Biotechnol. J. 2022, 21, 34–45. [Google Scholar] [CrossRef]
- Available online: https://www.cancer.gov/ccg/research/genome-sequencing/tcga (accessed on 19 March 2025).
- GTEx Expression Data. Available online: https://www.gtexportal.org/home/ (accessed on 20 August 2020).
- Zhou, Y.; Zhang, Y.; Zhao, D.; Yu, X.; Shen, X.; Zhou, Y.; Wang, S.; Qiu, Y.; Chen, Y.; Zhu, F. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2023, 52, D1465–D1477. [Google Scholar] [CrossRef]
- Available online: https://signor.uniroma2.it/score.php (accessed on 19 March 2025).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Harrell, F., Jr. Hmisc: Harrell Miscellaneous. R Package Version 5.2-4. 2025. Available online: https://cran.r-project.org/web/packages/Hmisc/index.html (accessed on 19 March 2025).
- Cornish, A.J.; Markowetz, F. SANTA: Quantifying the Functional Content of Molecular Networks. PLoS Comput. Biol. 2014, 10, e1003808. [Google Scholar] [CrossRef]
- Available online: https://cran.r-project.org/web/packages/iml/vignettes/intro.html (accessed on 19 March 2025).
- Kuhn, M. Caret: Classification and Regression Training. R Package Version 60-30. 2014. Available online: http://CRAN.R-project.org/package=caret (accessed on 17 April 2019).
- Available online: https://giuseppec.github.io/iml/reference/FeatureImp.html (accessed on 25 March 2025).
- McHugh, M.L. Interrater reliability: The kappa statistic. Biochem. Medica 2012, 22, 276–282. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, J.E.; Xie, Z.; Marino, G.B.; Nguyen, N.; Clarke, D.J.B.; Ma’ayan, A. Enrichr-KG: Bridging enrichment analysis across multiple libraries. Nucleic Acids Res. 2023, 51, W168–W179. [Google Scholar] [CrossRef] [PubMed]
- Surdo, P.L.; Iannuccelli, M.; Contino, S.; Castagnoli, L.; Licata, L.; Cesareni, G.; Perfetto, L. SIGNOR 3.0, the SIGnaling network open resource 3.0: 2022 update. Nucleic Acids Res. 2022, 51, D631–D637. [Google Scholar] [CrossRef]
- Ilakiyalakshmi, M.; Napoleon, A.A. Review on recent development of quinoline for anticancer activities. Arab. J. Chem. 2022, 15, 104168. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, C.; Zhang, N.; Fan, R.; Ye, Y.; Xu, J. Recent Advances in the Development of Pyrazole Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 12724. [Google Scholar] [CrossRef]
- Xu, G.; Fang, Z.; Clark, L.H.; Sung, W.; Yin, Y.; Zhang, R.; Sullivan, S.A.; Tran, A.-Q.; Kong, W.; Wang, J.; et al. Topiramate exhibits anti-tumorigenic and metastatic effects in ovarian cancer cells. Am. J. Transl. Res. 2018, 10, 1663–1676. [Google Scholar]
- Tuan, N.M.; Lee, C.H. Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent. Molecules 2019, 24, 3659. [Google Scholar] [CrossRef]
- Available online: https://adisinsight.springer.com/drugs/800000236 (accessed on 19 March 2025).
- Tatsuoka, J.; Sano, E.; Hanashima, Y.; Yagi, C.; Yamamuro, S.; Sumi, K.; Hara, H.; Takada, K.; Kanemaru, K.; Komine-Aizawa, S.; et al. Anti-tumor effects of perampanel in malignant glioma cells. Oncol. Lett. 2022, 24, 421. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bowie, D. The many faces of the AMPA-type ionotropic glutamate receptor. Neuropharmacology 2022, 208, 108975. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2023, 52, D1265–D1275. [Google Scholar] [CrossRef]
- Gysi, D.M.; Do Valle, Í.; Zitnik, M.; Ameli, A.; Gan, X.; Varol, O.; Ghiassian, S.D.; Patten, J.J.; Davey, R.A.; Loscalzo, J.; et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2025581118. [Google Scholar] [CrossRef]
- Sun, K.-X.; Tan, W.-S.; Wang, H.-Y.; Gao, J.-M.; Wang, S.-Y.; Xie, M.-L.; Deng, W.-L. Hesperidin Suppressed Colorectal Cancer through Inhibition of Glycolysis. Chin. J. Integr. Med. 2024, 1–12. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, W.; Zhang, S.; Wang, X.; Tang, Z.; Gu, J.; Li, J.; Huang, J. CACNA1B (Cav2.2) Overexpression and Its Association with Clinicopathologic Characteristics and Unfavorable Prognosis in Non-Small Cell Lung Cancer. Dis. Markers 2017, 2017, 6136401. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Samples | Tumor Samples | Cancer Acronym | Cancer Type |
|---|---|---|---|
| 101 | 180 | UCEC | Uterine corpus |
| Endometrial carcinoma | |||
| 339 | 119 | THYM | Thymoma |
| 338 | 512 | THCA | Thyroid carcinoma |
| 211 | 413 | STAD | Stomach adenocarcinoma |
| 557 | 468 | SKCM | Skin cutaneous melanoma |
| 317 | 92 | READ | Rectum adenocarcinoma |
| 151 | 495 | PRAD | Prostate adenocarcinoma |
| 171 | 179 | PAAD | Pancreatic adenocarcinoma |
| 338 | 498 | LUSC | Lung squamous |
| Cell carcinoma | |||
| 347 | 513 | LUAD | Lung adenocarcinoma |
| 160 | 369 | LIHC | Liver hepatocellular |
| Carcinoma | |||
| 60 | 288 | KIRP | Kidney renal papillary cell carcinoma |
| 100 | 530 | KIRC | Kidney renal clear cell carcinoma |
| 53 | 66 | KICH | Kidney chromophobe |
| 286 | 182 | ESCA | Esophageal carcinoma |
| 348 | 288 | COAD | Colon adenocarcinoma |
| 291 | 1099 | BRCA | Breast cancer |
| 28 | 407 | BLCA | Bladder cancer |
| 4196 | 6698 | TOTAL |
| Disease | Kappa log2fc | Kappa Sign Genes | Accuracy log2fc | Accuracy Sign Genes | AUC log2fc | AUC Sign Genes |
|---|---|---|---|---|---|---|
| KICH | 0.8799 | 0.966 | 0.9403 | 0.9832 | 0.9841 | 0.9994 |
| BLCA | 0.1423 | 0.3485 | 0.9352 | 0.9386 | 0.9471 | 0.8906 |
| BRCA | 0.9428 | 0.9539 | 0.9809 | 0.9847 | 0.9974 | 0.9987 |
| COAD | 0.9169 | 0.974 | 0.9588 | 0.9871 | 0.992 | 0.999 |
| ESCA | 0.9277 | 0.9005 | 0.9654 | 0.9521 | 0.983 | 0.9913 |
| KIRC | 0.7563 | 0.9253 | 0.9251 | 0.9803 | 0.9783 | 0.9923 |
| KIRP | 0.7814 | 0.9208 | 0.9282 | 0.9776 | 0.9854 | 0.997 |
| LIHC | 0.8763 | 0.8351 | 0.9474 | 0.931 | 0.989 | 0.9756 |
| LUAD | 0.931 | 0.9623 | 0.967 | 0.9819 | 0.9969 | 0.9979 |
| LUSC | 0.9501 | 0.9675 | 0.9761 | 0.9843 | 0.997 | 0.996 |
| PAAD | 0.8634 | 0.9514 | 0.9317 | 0.9757 | 0.9861 | 0.989 |
| PRAD | 0.6914 | 0.8429 | 0.8872 | 0.9446 | 0.9046 | 0.9781 |
| READ | 0.7492 | 0.8917 | 0.8985 | 0.9601 | 0.9888 | 0.9971 |
| SKCM | 0.9418 | 0.9736 | 0.971 | 0.9869 | 0.9947 | 0.997 |
| STAD | 0.9255 | 0.9255 | 0.9665 | 0.967 | 0.9943 | 0.9939 |
| THCA | 0.9315 | 0.8763 | 0.9673 | 0.9408 | 0.9937 | 0.9901 |
| THYM | 0.9887 | 0.9543 | 0.9956 | 0.9825 | 0.999 | 0.9976 |
| UCEC | 0.9572 | 0.9259 | 0.9804 | 0.9655 | 0.9981 | 0.9984 |
| Drug TTD ID | Distance to READ Module | FDA Indication | p-Value | Drug Names |
|---|---|---|---|---|
| D0U3ED | -5,660,390 | Diabetic neuropathy | 0.030376 | E-2007 |
| D01EVT | -1,436,887 | NA | 0.03374 | Ethinyl-pyrazole derivative 1 |
| D02RRE | -1,436,887 | NA | 0.03374 | Pyrazole derivative 78 |
| D03KYL | -1,436,887 | NA | 0.03374 | Quinoline derivative 4 |
| D03PRA | -1,436,887 | NA | 0.03374 | Heteroaryl-pyrazole derivative 2 |
| D04LRT | -1,436,887 | NA | 0.03374 | Quinoline derivative 9 |
| D04VAE | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-26 |
| D05CBS | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-37 |
| D06NZY | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-44 |
| D08JPW | -1,436,887 | NA | 0.03374 | N-substituted pyrazole derivative 1 |
| D08PHH | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-45 |
| D09JZA | -1,436,887 | NA | 0.03374 | Quinoline derivative 8 |
| D09NRH | -1,436,887 | NA | 0.03374 | Quinoline derivative 5 |
| D09OQZ | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-49 |
| D09WXH | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-25 |
| D0B6DN | -1,436,887 | NA | 0.03374 | Ethinyl-pyrazole derivative 3 |
| D0BP6R | -1,436,887 | NA | 0.03374 | Ethinyl-pyrazole derivative 2 |
| D0C2ZJ | -1,436,887 | NA | 0.03374 | Tetra-hydro-imidazo[1,5-d][1,4]oxazepin-3-yl derivative 3 |
| D0CZ6T | -1,436,887 | NA | 0.03374 | Tetra-hydro-imidazo[1,5-d][1,4]oxazepin-3-yl derivative 2 |
| D0E5QO | -1,436,887 | NA | 0.03374 | Quinoline derivative 7 |
| D0H2BU | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-53 |
| D0H2LD | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-43 |
| D0HD8F | -1,436,887 | NA | 0.03374 | Pyrazole derivative 76 |
| D0HG1W | -1,436,887 | NA | 0.03374 | 2-(substituted ethynyl)quinoline derivative 4 |
| D0I6LO | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-47 |
| D0IL4Z | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-38 |
| D0IY8T | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-46 |
| D0K0ZX | -1,436,887 | NA | 0.03374 | Heteroaryl-pyrazole derivative 3 |
| D0K1RU | -1,436,887 | NA | 0.03374 | Quinoline derivative 3 |
| D0KZ6Q | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-40 |
| D0LI2E | -1,436,887 | NA | 0.03374 | Tetra-hydro-imidazo[1,5-d][1,4]oxazepin-3-yl derivative 1 |
| D0MB0V | -1,436,887 | NA | 0.03374 | 2-(substituted ethynyl)quinoline derivative 2 |
| D0N0FA | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-51 |
| D0N1CC | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-42 |
| D0N9IA | -1,436,887 | NA | 0.03374 | Tetra-hydro-imidazo[1,5-d][1,4]oxazepin-3-yl derivative 4 |
| D0NE0W | -1,436,887 | NA | 0.03374 | Tetra-hydro-imidazo[1,5-d][1,4]oxazepin-3-yl derivative 6 |
| D0O6QC | -1,436,887 | NA | 0.03374 | Heteroaryl-pyrazole derivative 1 |
| D0O9TL | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-52 |
| D0OT4O | -1,436,887 | NA | 0.03374 | Quinoline derivative 6 |
| D0P7DA | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-10 |
| D0PJ8X | -1,436,887 | NA | 0.03374 | 2-(substituted ethynyl)quinoline derivative 1 |
| D0Q2FC | -1,436,887 | NA | 0.03374 | Tetra-hydro-imidazo[1,5-d][1,4]oxazepin-3-yl derivative 5 |
| D0QM1R | -1,436,887 | NA | 0.03374 | Pyrazole derivative 77 |
| D0RX0M | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-39 |
| D0S5SK | -1,436,887 | NA | 0.03374 | Pyrazole derivative 79 |
| D0T3DG | -1,436,887 | NA | 0.03374 | BCI-632 |
| D0V0BC | -1,436,887 | NA | 0.03374 | N-substituted pyrazole derivative 2 |
| D0WP5S | -1,436,887 | NA | 0.03374 | N-substituted pyrazole derivative 3 |
| D0XQ4T | -1,436,887 | NA | 0.03374 | 2-(substituted ethynyl)quinoline derivative 3 |
| D0Y5DN | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-41 |
| D0YZ7D | -1,436,887 | NA | 0.03374 | PMID25435285-Compound-50 |
| D00GEG | -1,415,097 | NA | 0.035841 | Ralfinamide |
| D01NLB | -1,415,097 | Pain | 0.035841 | Ziconotide |
| D07VDZ | -1,415,097 | Epilepsy; alcohol use disorders | 0.035841 | Topiramate |
| D08EOD | -1,415,097 | Epileptic seizures | 0.035841 | Methsuximide |
| D09JBP | -1,415,097 | Paramethadione syndrome; seizures | 0.035841 | Paramethadione |
| D0A3MJ | -1,415,097 | NA | 0.035841 | XEN007 |
| D0CQ0Z | -1,415,097 | Schizophrenia | 0.035841 | Penfluridol |
| D0I9HF | -1,415,097 | Capillary fragility | 0.035841 | Hesperidin |
| D0M8AB | -1,415,097 | Dietary shortage | 0.035841 | Glycine |
| D0N3SR | -1,415,097 | NA | 0.035841 | Cilnidipine |
| D0Q4XQ | -1,415,097 | Epilepsy | 0.035841 | Ethosuximide |
| D0R0FE | -1,415,097 | Hypertension; angina | 0.035841 | Verapamil |
| D0U4VT | -1,415,097 | Epileptic conditions; pancreatic cancer | 0.035841 | Trimethadione |
| D0U7GP | -1,415,097 | NA | 0.035841 | Rauwolfia Serpentina root |
| D0W8XT | -1,415,097 | Granted orphan drug status by FDA for ovarian cancer, pancreatic cancer, and glioblastoma multiforme | 0.035841 | Mibefradil |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galimi, A.; Fiscon, G. A Network-Based Approach Exploiting Transcriptomics and Interactomics Data for Predicting Drug Repurposing Solutions Across Human Cancers. Cancers 2025, 17, 1144. https://doi.org/10.3390/cancers17071144
Galimi A, Fiscon G. A Network-Based Approach Exploiting Transcriptomics and Interactomics Data for Predicting Drug Repurposing Solutions Across Human Cancers. Cancers. 2025; 17(7):1144. https://doi.org/10.3390/cancers17071144
Chicago/Turabian StyleGalimi, Alessio, and Giulia Fiscon. 2025. "A Network-Based Approach Exploiting Transcriptomics and Interactomics Data for Predicting Drug Repurposing Solutions Across Human Cancers" Cancers 17, no. 7: 1144. https://doi.org/10.3390/cancers17071144
APA StyleGalimi, A., & Fiscon, G. (2025). A Network-Based Approach Exploiting Transcriptomics and Interactomics Data for Predicting Drug Repurposing Solutions Across Human Cancers. Cancers, 17(7), 1144. https://doi.org/10.3390/cancers17071144