

Mechanisms of Resistance to Novel Immunotherapies in B-Cell Lymphomas: Focus on CAR T and Bispecific Antibodies

Simple Summary
Abstract
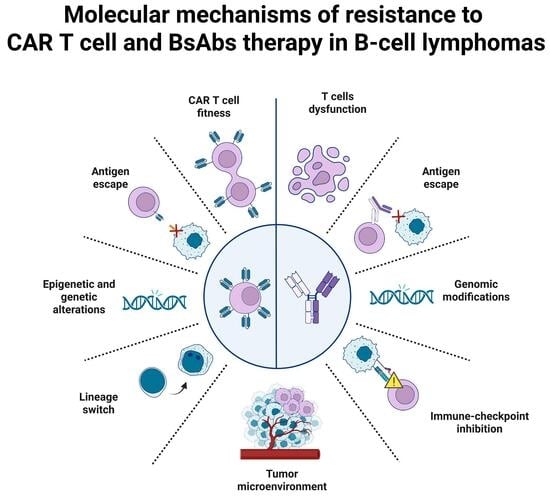
1. Introduction
2. Immune Evasion from CD19-CAR T Cell Therapy
2.1. CD19-CAR T Cell Fitness
2.2. Tumor-Intrinsic Determinants
2.2.1. CD19 Antigen Loss, Modification, and Reduction
2.2.2. Epigenetic Downregulation
2.2.3. Complex Genomic Alterations
2.2.4. Lineage Switch
2.3. Microenvironmental and Signaling Adaptations
3. BsAbs and Resistance Onset
3.1. BsAbs’ Properties
3.2. T Cell Exhaustion and the Role of the TME
3.3. Tumor-Intrinsic Factors
3.3.1. CD20 Antigen Loss and Alterations
3.3.2. Tumor Genomic Alterations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CD | Cluster of Differentiation |
| CAR | Chimeric Antigen Receptor |
| BsAbs | Bispecific antibodies |
| APOBEC | Apolipoprotein B mRNA-Editing Enzyme Catalytic Polypeptide-like |
| RHOA | Ras Homolog family member A |
| TP53 | Tumor Protein 53 |
| NHL | Non-Hodgkin Lymphoma |
| HL | Hodgkin Lymphoma |
| DLBCL | Diffuse Large B-Cell Lymphoma |
| FL | Follicular Lymphoma |
| MCL | Mantle Cell Lymphoma |
| B-NHL | B-Cell Non-Hodgkin Lymphoma |
| ORR | Overall response rate |
| LBCL | Large B-Cell Lymphoma |
| OS | Overall survival |
| TME | Tumor microenvironment |
| CRR | Complete response rate |
| PFS | Progression-free survival |
| FDA | Food and Drug Administration |
| EMA | European Medical Agency |
| scFv | Single-chain variable fragment |
| TCR | T Cell Receptor |
| IL | Interleukin |
| Cy/Flu | Cyclophosphamide/Fludarabine |
| MCP-1 | Monocyte Chemo-Attractant Protein-1 |
| CLL | Chronic Lymphocytic Leukemia |
| STAT | Signal Transducer and Activator of Transcription |
| PD-1 | Programmed cell Death protein 1 |
| IFN | Interferon |
| TET2 | Tet methylcytosine dioxygenase 2 |
| DNMT3A | DNA Methyltransferase-3 Alfa |
| HGBCL | High-Grade B-Cell Lymphoma |
| PMBCL | Primary Mediastinal B-Cell Lymphoma |
| IKZF1 | IKAROS Family Zinc Finger 1 |
| BCL2 | B-Cell Lymphoma 2 |
| BCL6 | B-Cell Lymphoma 6 |
| PAX5 | Paired box 5 |
| IRF8 | Interferon Regulatory Factor 8 |
| PD-L1 | Programmed Death-Ligand 1 |
| TMEM30A | Transmembrane protein 30A |
| PPM1D | Protein Phosphatase Magnesium-dependent 1 Delta |
| TAM | Tumor Associated Macrophage |
| MDCS | Myeloid-Derived Suppressor Cell |
| Treg | Regulatory T cell |
| TGFβ | Transforming Growth Factor Beta |
| R-CHOP | Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, Prednisone |
| ISRT | Involved Site Radiation Therapy |
| BTKi | Bruton’s Tyrosine Kinase inhibitor |
| MHC | Major Histocompatibility Complex |
| B-ALL | B-cell Acute Lymphoblastic Leukemia |
| SRSF3 | Serine and arginine Rich Splicing Factor 3 |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| Cas9 | CRISPR-Associated protein 9 |
| IHC | Immunohistochemistry |
| DEL | Double-Expressor Lymphoma |
| DHL | Double-Hit Lymphoma |
| WGS | Whole Genome Sequencing |
| PP2C | Protein Phosphatase 2C |
| AML | Acute Myeloid Leukemia |
| CCL | C-C motif ligand |
| LAG-3 | Lymphocyte Activation Gene 3 |
| CTLA-4 | Cytotoxic T Lymphocyte-Associated protein 4 |
| TIM-3 | T cell Immunoglobulin and Mucin domain-containing protein 3 |
| BiTE | Bispecific T cell Engager |
| ADDC | Antibody-Dependent Cell-mediated Cytotoxicity |
| MS4A1 | Membrane Spanning 4-domains A1 |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| FOXP3 | Forkhead box Protein 3 |
| WES | Whole Exome Sequencing |
| KMT2D | Lysine methyltransferase 2D |
| IGLL5 | Immunoglobulin Lambda Like polypeptide |
| CARD11 | Caspase Recruitment Domain family member 11 |
| HIST1H1E | H1 Histone family member 4 |
| CREBBP | CREB-Binding Protein |
References
- Silkenstedt, E.; Salles, G.; Campo, E.; Dreyling, M. B-Cell Non-Hodgkin Lymphomas. Lancet 2024, 403, 1791–1807. [Google Scholar] [CrossRef]
- Shanbhag, S.; Ambinder, R.F. Hodgkin Lymphoma: A Review and Update on Recent Progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.; Dabaja, B.; Wu, S.; Kelly, K.; Berkahn, L.; Pavlovsky, A.; Sureda, A.; LaCasce, A. Classic Hodgkin Lymphoma. Hematol. Oncol. 2024, 42, e3239. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. Mechanisms of B-Cell Lymphoma Pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Eyre, T.A.; Cwynarski, K.; d’Amore, F.; De Leval, L.; Dreyling, M.; Eichenauer, D.A.; Ferreri, A.J.M.; Giné, E.; Kersten, M.J.; Ladetto, M.; et al. Lymphomas: ESMO Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2025, S0923753425009111. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, N.; Wang, G.; Cao, Y. Predictive Short/Long-Term Efficacy Biomarkers and Resistance Mechanisms of CD19-Directed CAR-T Immunotherapy in Relapsed/Refractory B-Cell Lymphomas. Front. Immunol. 2023, 14, 1110028. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maus, M.V.; Hinrichs, C.S. CAR T Cells and T-Cell Therapies for Cancer: A Translational Science Review. J. Am. Med. Assoc. 2024, 332, 1924. [Google Scholar] [CrossRef]
- Di Blasi, R.; Le Gouill, S.; Bachy, E.; Cartron, G.; Beauvais, D.; Le Bras, F.; Gros, F.-X.; Choquet, S.; Bories, P.; Feugier, P.; et al. Outcomes of Patients with Aggressive B-Cell Lymphoma after Failure of Anti-CD19 CAR T-Cell Therapy: A DESCAR-T Analysis. Blood 2022, 140, 2584–2593. [Google Scholar] [CrossRef]
- Ruella, M.; Korell, F.; Porazzi, P.; Maus, M.V. Mechanisms of Resistance to Chimeric Antigen Receptor-T Cells in Haematological Malignancies. Nat. Rev. Drug Discov. 2023, 22, 976–995. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.R.; Zabor, E.C.; Bedelu, Y.; Yang, X.; Karimi, Y.H.; Nedved, A.N.; Wang, Y.; Dave, N.K.; Landsburg, D.J.; Baron, K.; et al. Real-World Outcomes of Patients with Aggressive B-Cell Lymphoma Treated with Epcoritamab or Glofitamab. Blood J. 2025, blood.2025029117. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yang, Y.; Wang, G.; Liu, M. Current Landscape and Future Directions of Bispecific Antibodies in Cancer Immunotherapy. Front. Immunol. 2022, 13, 1035276. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.-E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 Study of the Bispecific T-Cell Engager (BiTE) Antibody Blinatumomab in Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Kyvsgaard, E.R.; Grauslund, M.; Sjø, L.; Melchior, L.C.; Grantzau, T.L.; Gjerdrum, L.M.R.; Trab, T.; Andersen, L.S.; Gang, A.O.; Breinholt, M.; et al. NOTCH1 Mutations Are Associated With Therapy-Resistance in Patients With B-Cell Lymphoma Treated With CD20xCD3 Bispecific Antibodies. Am. J. Hematol. 2025, 100, 712–716. [Google Scholar] [CrossRef]
- Thieblemont, C.; Karimi, Y.H.; Ghesquieres, H.; Cheah, C.Y.; Clausen, M.R.; Cunningham, D.; Jurczak, W.; Do, Y.R.; Gasiorowski, R.; Lewis, D.J.; et al. Epcoritamab in Relapsed/Refractory Large B-Cell Lymphoma: 2-Year Follow-up from the Pivotal EPCORE NHL-1 Trial. Leukemia 2024, 38, 2653–2662. [Google Scholar] [CrossRef]
- Phillips, T.J.; Trněný, M.; Carlo-Stella, C.; Zaucha, J.M.; Wrobel, T.; Offner, F.; Dickinson, M.J.; Tani, M.; Crump, M.; Bartlett, N.L.; et al. Glofitamab Induces High Response Rates and Durable Remissions in Patients (Pts) with Heavily Pretreated Relapsed/Refractory (R/R) Mantle Cell Lymphoma (MCL), Including Those with a Poor Prognosis: Subgroup Results from a Phase I/II Study. Blood 2024, 144, 1631. [Google Scholar] [CrossRef]
- Lejeune, M.; Köse, M.C.; Duray, E.; Einsele, H.; Beguin, Y.; Caers, J. Bispecific, T-Cell-Recruiting Antibodies in B-Cell Malignancies. Front. Immunol. 2020, 11, 762. [Google Scholar] [CrossRef]
- Heine, R.; Thielen, F.W.; Koopmanschap, M.; Kersten, M.J.; Einsele, H.; Jaeger, U.; Sonneveld, P.; Sierra, J.; Smand, C.; Uyl-de Groot, C.A. Health Economic Aspects of Chimeric Antigen Receptor T-Cell Therapies for Hematological Cancers: Present and Future. HemaSphere 2021, 5, e524. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T Design: Elements and Their Synergistic Function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Gambella, M.; Carlomagno, S.; Raiola, A.M.; Giannoni, L.; Ghiggi, C.; Setti, C.; Giordano, C.; Luchetti, S.; Serio, A.; Bo, A.; et al. CD19-Targeted Immunotherapies for Diffuse Large B-Cell Lymphoma. Front. Immunol. 2022, 13, 837457. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Tomasik, J.; Jasiński, M.; Basak, G.W. Next Generations of CAR-T Cells—New Therapeutic Opportunities in Hematology? Front. Immunol. 2022, 13, 1034707. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of Resistance to CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Bashor, C.J.; Hilton, I.B.; Bandukwala, H.; Smith, D.M.; Veiseh, O. Engineering the next Generation of Cell-Based Therapeutics. Nat. Rev. Drug Discov. 2022, 21, 655–675. [Google Scholar] [CrossRef]
- Neelapu, S.S. CAR-T Efficacy: Is Conditioning the Key? Blood 2019, 133, 1799–1800. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of Non-Hodgkin’s Lymphoma with a Defined Ratio of CD8+ and CD4+ CD19-Specific Chimeric Antigen Receptor–Modified T Cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The Response to Lymphodepletion Impacts PFS in Patients with Aggressive Non-Hodgkin Lymphoma Treated with CD19 CAR T Cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Van Der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Labanieh, L.; Mackall, C.L. CAR Immune Cells: Design Principles, Resistance and the next Generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef]
- Rossi, J.; Paczkowski, P.; Shen, Y.-W.; Morse, K.; Flynn, B.; Kaiser, A.; Ng, C.; Gallatin, K.; Cain, T.; Fan, R.; et al. Preinfusion Polyfunctional Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated with Clinical Outcomes in NHL. Blood 2018, 132, 804–814. [Google Scholar] [CrossRef]
- TET2 Tet Methylcytosine Dioxygenase 2 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/54790 (accessed on 23 October 2025).
- Norman, L.; Tarrant, P.; Chevassut, T. TET2 Inhibits Differentiation of Embryonic Stem Cells but Does Not Overcome Methylation-Induced Gene Silencing. Bone Marrow Res. 2014, 2014, 986571. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 Promotes the Therapeutic Efficacy of CD19-Targeted T Cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Ali, A.; Zhang, Y.; DiPersio, J. The Invisible Hand: How Epigenetics Shapes CAR T Cell Destiny. Mol. Ther. 2024, 32, 1614–1616. [Google Scholar] [CrossRef]
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T Cells Prevents Exhaustion and Enhances Antitumor Activity. Sci. Transl. Med. 2021, 13, eabh0272. [Google Scholar] [CrossRef] [PubMed]
- PubChem DNMT3A—DNA Methyltransferase 3 Alpha (Human). Available online: https://pubchem.ncbi.nlm.nih.gov/gene/DNMT3A/human (accessed on 23 September 2025).
- Mazinani, M.; Rahbarizadeh, F. CAR-T Cell Potency: From Structural Elements to Vector Backbone Components. Biomark. Res. 2022, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Kymriah. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah (accessed on 23 September 2025).
- Kymriah|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kymriah (accessed on 23 September 2025).
- Yescarta. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta (accessed on 23 September 2025).
- Yescarta|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta (accessed on 23 September 2025).
- Tecartus. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/tecartus (accessed on 23 September 2025).
- Tecartus|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/tecartus (accessed on 23 September 2025).
- Breyanzi. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/breyanzi (accessed on 23 September 2025).
- Breyanzi|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/breyanzi (accessed on 23 September 2025).
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P.; et al. Chronic Lymphocytic Leukaemia: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2021, 32, 23–33. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, X.; Tian, Y.; Li, F.; Zhao, X.; Liu, J.; Yao, C.; Zhang, Y. Point Mutation in CD19 Facilitates Immune Escape of B Cell Lymphoma from CAR-T Cell Therapy. J. Immunother. Cancer 2020, 8, e001150. [Google Scholar] [CrossRef]
- Jain, M.D.; Ziccheddu, B.; Coughlin, C.A.; Faramand, R.; Griswold, A.J.; Reid, K.M.; Menges, M.; Zhang, Y.; Cen, L.; Wang, X.; et al. Whole-Genome Sequencing Reveals Complex Genomic Features Underlying Anti-CD19 CAR T-Cell Treatment Failures in Lymphoma. Blood 2022, 140, 491–503. [Google Scholar] [CrossRef]
- IKZF1 IKAROS Family Zinc Finger 1 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/10320 (accessed on 23 September 2025).
- Domizi, P.; Sarno, J.; Jager, A.; Merchant, M.; Pacheco, K.Z.B.; Yamada-Hunter, S.A.; Rotiroti, M.C.; Liu, Y.; Baskar, R.; Reynolds, W.D.; et al. IKAROS Levels Are Associated with Antigen Escape in CD19- and CD22-Targeted Therapies for B-Cell Malignancies. Nat. Commun. 2025, 16, 3800. [Google Scholar] [CrossRef]
- MYC MYC Proto-Oncogene, bHLH Transcription Factor [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/4609 (accessed on 23 September 2025).
- BCL2 BCL2 Apoptosis Regulator [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/596 (accessed on 23 October 2025).
- BCL6 BCL6 Transcription Repressor [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/604 (accessed on 23 September 2025).
- BCL6 Gene—GeneCards|BCL6 Protein|BCL6 Antibody. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=BCL6 (accessed on 23 September 2025).
- Karmali, R.; Shouse, G.; Torka, P.; Moyo, T.K.; Romancik, J.; Barta, S.K.; Bhansali, R.; Cohen, J.B.; Shah, N.N.; Zurko, J.; et al. Double Hit & Double Expressor Lymphomas: A Multicenter Analysis of Survival Outcomes with CD19-Directed CAR T-Cell Therapy. Blood Cancer J. 2025, 15, 43. [Google Scholar] [CrossRef]
- Jain, M.D.; Ziccheddu, B.; Coughlin, C.A.; Faramand, R.; Griswold, A.J.; Reid, K.M.; Landgren, O.; Locke, F.L.; Maura, F.; Davila, M.L.; et al. Genomic Drivers of Large B-Cell Lymphoma Resistance to CD19 CAR-T Therapy. Blood 2021, 138, 42. [Google Scholar] [CrossRef]
- Petljak, M.; Dananberg, A.; Chu, K.; Bergstrom, E.N.; Striepen, J.; Von Morgen, P.; Chen, Y.; Shah, H.; Sale, J.E.; Alexandrov, L.B.; et al. Mechanisms of APOBEC3 Mutagenesis in Human Cancer Cells. Nature 2022, 607, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Chiarle, R. RHO Family GTPases in the Biology of Lymphoma. Cells 2019, 8, 646. [Google Scholar] [CrossRef]
- Sworder, B.J.; Kurtz, D.M.; Alig, S.K.; Frank, M.J.; Shukla, N.; Garofalo, A.; Macaulay, C.W.; Shahrokh Esfahani, M.; Olsen, M.N.; Hamilton, J.; et al. Determinants of Resistance to Engineered T Cell Therapies Targeting CD19 in Large B Cell Lymphomas. Cancer Cell 2023, 41, 210–225.e5. [Google Scholar] [CrossRef] [PubMed]
- Simovic-Lorenz, M.; Ernst, A. Chromothripsis in Cancer. Nat. Rev. Cancer 2025, 25, 79–92. [Google Scholar] [CrossRef]
- PPM1D Protein Phosphatase, Mg2+/Mn2+ Dependent 1D [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/8493 (accessed on 23 September 2025).
- Seipel, K.; Frey, M.; Nilius, H.; Akhoundova, D.; Banz, Y.; Bacher, U.; Pabst, T. Low-Frequency PPM1D Gene Mutations Affect Treatment Response to CD19-Targeted CAR T-Cell Therapy in Large B-Cell Lymphoma. Curr. Oncol. 2023, 30, 10463–10476. [Google Scholar] [CrossRef]
- Ong, S.Y.; Chen, Y.; Tan, M.S.Y.; Ho, A.Y.L.; Hwang, W.Y.K.; Lim, F.L.W.I. Current Perspectives on Resistance to Chimeric Antigen Receptor T-cell Therapy and Strategies to Improve Efficacy in B-cell Lymphoma. Eur. J. Haematol. 2024, 112, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xie, S.; Li, Y.; Wang, J.; Xiao, J.; Huang, K.; Wang, X.; Wu, Y.; Ma, L.; Nie, D. Lineage Switch from Lymphoma to Myeloid Neoplasms: First Case Series from a Single Institution. Open Med. 2022, 17, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Lucero, O.M.; Parker, K.; Funk, T.; Dunlap, J.; Press, R.; Gardner, R.A.; Chang, B.H. Phenotype Switch in Acute Lymphoblastic Leukaemia Associated with 3 Years of Persistent CAR T Cell Directed—CD 19 Selective Pressure. Br. J. Haematol. 2019, 186, 333–336. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef]
- Baker, D.J.; Arany, Z.; Baur, J.A.; Epstein, J.A.; June, C.H. CAR T Therapy beyond Cancer: The Evolution of a Living Drug. Nature 2023, 619, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Rejeski, K.; Jain, M.D.; Smith, E.L. Mechanisms of Resistance and Treatment of Relapse after CAR T-Cell Therapy for Large B-Cell Lymphoma and Multiple Myeloma. Transplant. Cell. Ther. 2023, 29, 418–428. [Google Scholar] [CrossRef]
- Jain, M.D.; Zhao, H.; Wang, X.; Atkins, R.; Menges, M.; Reid, K.; Spitler, K.; Faramand, R.; Bachmeier, C.; Dean, E.A.; et al. Tumor Interferon Signaling and Suppressive Myeloid Cells Are Associated with CAR T-Cell Failure in Large B-Cell Lymphoma. Blood 2021, 137, 2621–2633. [Google Scholar] [CrossRef]
- Scholler, N.; Perbost, R.; Locke, F.L.; Jain, M.D.; Turcan, S.; Danan, C.; Chang, E.C.; Neelapu, S.S.; Miklos, D.B.; Jacobson, C.A.; et al. Tumor Immune Contexture Is a Determinant of Anti-CD19 CAR T Cell Efficacy in Large B Cell Lymphoma. Nat. Med. 2022, 28, 1872–1882. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Newsam, A.D.; Coughlin, C.A.; Trabolsi, A.; Schatz, J.H. Functional Drivers of Resistance to Anti-CD19 CAR-T Cell Therapy in Diffuse Large B Cell Lymphoma. Leuk. Lymphoma 2023, 64, 2217–2224. [Google Scholar] [CrossRef]
- Chen, P.-H.; Lipschitz, M.; Weirather, J.L.; Jacobson, C.; Armand, P.; Wright, K.; Hodi, F.S.; Roberts, Z.J.; Sievers, S.A.; Rossi, J.; et al. Activation of CAR and Non-CAR T Cells within the Tumor Microenvironment Following CAR T Cell Therapy. J. Clin. Investig. Insight 2020, 5, e134612. [Google Scholar] [CrossRef]
- Klein, C.; Brinkmann, U.; Reichert, J.M.; Kontermann, R.E. The Present and Future of Bispecific Antibodies for Cancer Therapy. Nat. Rev. Drug Discov. 2024, 23, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Pretelli, G.; Desai, J.; Garralda, E.; Siu, L.L.; Steiner, T.M.; Au, L. Bispecific Antibodies: Advancing Precision Oncology. Trends Cancer 2024, 10, 893–919. [Google Scholar] [CrossRef] [PubMed]
- Falchi, L.; Vardhana, S.A.; Salles, G.A. Bispecific Antibodies for the Treatment of B-Cell Lymphoma: Promises, Unknowns, and Opportunities. Blood 2023, 141, 467–480. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Stuhler, G.; Bargou, R. Bispecific and Multispecific Antibodies in Oncology: Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2024, 21, 539–560. [Google Scholar] [CrossRef]
- Radhakrishnan, V.S.; Davies, A.J. Bispecific Antibodies in Indolent B-Cell Lymphomas. Front. Immunol. 2024, 14, 1295599. [Google Scholar] [CrossRef] [PubMed]
- FDA, D.I.S.C.O. Burst Edition: FDA Approval of Lunsumio (Mosunetuzumab-Axgb) for Adult Patients with Relapsed or Refractory Follicular Lymphoma After Two or More Lines of Systemic Therapy. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-disco-burst-edition-fda-approval-lunsumio-mosunetuzumab-axgb-adult-patients-relapsed-or (accessed on 23 September 2025).
- Lunsumio|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lunsumio (accessed on 23 September 2025).
- FDA, D.I.S.C.O. Burst Edition: FDA Approval of Epkinly (Epcoritamab-Bysp) for Relapsed or Refractory Diffuse Large B-Cell Lymphoma and High-Grade B-Cell Lymphoma|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-disco-burst-edition-fda-approval-epkinly-epcoritamab-bysp-relapsed-or-refractory-diffuse-large-b (accessed on 23 September 2025).
- Tepkinly|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/tepkinly (accessed on 23 September 2025).
- FDA Grants Accelerated Approval to Glofitamab-Gxbm for Selected Relapsed or Refractory Large B-Cell Lymphomas. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-grants-accelerated-approval-glofitamab-gxbm-selected-relapsed-or-refractory-large-b-cell (accessed on 23 September 2025).
- Columvi|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/columvi (accessed on 23 September 2025).
- Ordspono|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/ordspono (accessed on 23 September 2025).
- Goebeler, M.-E.; Bargou, R.C. T Cell-Engaging Therapies—BiTEs and Beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Van De Donk, N.W.C.J.; Zweegman, S. T-Cell-Engaging Bispecific Antibodies in Cancer. Lancet 2023, 402, 142–158. [Google Scholar] [CrossRef]
- Singh, A.; Dees, S.; Grewal, I.S. Overcoming the Challenges Associated with CD3+ T-Cell Redirection in Cancer. Br. J. Cancer 2021, 124, 1037–1048. [Google Scholar] [CrossRef]
- MS4A1 Gene—GeneCards|CD20 Protein|CD20 Antibody. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=MS4A1 (accessed on 22 October 2025).
- Cerny, T.; Borisch, B.; Introna, M.; Johnson, P.; Rose, A.L. Mechanism of Action of Rituximab. Anti-Cancer Drugs 2002, 13, S3–S10. [Google Scholar] [CrossRef]
- Elice, F.; Sommaggio, R.; Cappuzzello, E.; Riva, M.; Tisi, M.C.; Bernardi, M.; Bozza, A.; Catanzaro, D.; Chieregato, K.; Merlo, A.; et al. A Rescue Approach in Refractory Diffuse Large B-Cell Lymphoma with Obinutuzumab-Redirected Cytokine-Induced Killer Cells: A First-in-Human Case Report. Haematologica 2024, 109, 3037–3041. [Google Scholar] [CrossRef]
- Kase, A.M.; Kharfan-Dabaja, M.A.; Donaldson, A.; Elliott, J.; Sher, T. Immunotherapy beyond Cellular Therapy in Follicular Lymphoma: A Case of Complete Remission after Failure of Two CAR-T. Clin. Case Rep. 2022, 10, e05572. [Google Scholar] [CrossRef]
- Grigg, S.; Minson, A.; Prins, E.; Dickinson, M.J. Relapse after Glofitamab Has a Poor Prognosis and Rates of CD20 Loss Are High. Br. J. Haematol. 2024, 205, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Huw, L.-Y.; Bolen, C.R.; Maximov, V.; Polson, A.G.; Hatzi, K.; Lasater, E.A.; Assouline, S.E.; Bartlett, N.L.; Budde, L.E.; et al. Loss of CD20 Expression as a Mechanism of Resistance to Mosunetuzumab in Relapsed/Refractory B-Cell Lymphomas. Blood 2024, 143, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Rushton, C.K.; Arthur, S.E.; Alcaide, M.; Cheung, M.; Jiang, A.; Coyle, K.M.; Cleary, K.L.S.; Thomas, N.; Hilton, L.K.; Michaud, N.; et al. Genetic and Evolutionary Patterns of Treatment Resistance in Relapsed B-Cell Lymphoma. Blood Adv. 2020, 4, 2886–2898. [Google Scholar] [CrossRef]
- Ang, Z.; Paruzzo, L.; Hayer, K.E.; Schmidt, C.; Torres Diz, M.; Xu, F.; Zankharia, U.; Zhang, Y.; Soldan, S.; Zheng, S.; et al. Alternative Splicing of Its 5′-UTR Limits CD20 mRNA Translation and Enables Resistance to CD20-Directed Immunotherapies. Blood 2023, 142, 1724–1739. [Google Scholar] [CrossRef]
- Nakamaki, T.; Fukuchi, K.; Nakashima, H.; Ariizumi, H.; Maeda, T.; Saito, B.; Yanagisawa, K.; Tomoyasu, S.; Homma, M.; Shiozawa, E.; et al. CD 20 Gene Deletion Causes a CD 20-negative Relapse in Diffuse Large B-cell Lymphoma. Eur. J. Haematol. 2012, 89, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.-M.; Buet-Elfassy, A.; Annereau, M.; Lazarovici, J.; Danu, A.; Sarkozy, C.; Chahine, C.; Bigenwald, C.; Bosq, J.; Rossignol, J.; et al. Clinical Significance of the Loss of CD20 Antigen on Tumor Cells in Patients with Relapsed or Refractory Follicular Lymphoma. Cancer Drug Resist. 2021, 4, 710–718. [Google Scholar] [CrossRef]
- Duell, J.; Leipold, A.M.; Appenzeller, S.; Fuhr, V.; Rauert-Wunderlich, H.; Da Via, M.; Dietrich, O.; Toussaint, C.; Imdahl, F.; Eisele, F.; et al. Sequential Antigen Loss and Branching Evolution in Lymphoma after CD19- and CD20-Targeted T-Cell–Redirecting Therapy. Blood 2024, 143, 685–696. [Google Scholar] [CrossRef]
- Saleh, K.; Khoury, R.; Khalife, N.; Chahine, C.; Ibrahim, R.; Tikriti, Z.; Le Cesne, A. The Evolving Role of Bispecific Antibodies in Diffuse Large B-Cell Lymphoma. J. Pers. Med. 2024, 14, 666. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Huw, L.-Y.; Bolen, C.R.; Assouline, S.E.; Bartlett, N.L.; Budde, L.E.; Matasar, M.J.; Koeppen, H.; Piccione, E.C.; Wilson, D.; et al. Characterization of CD20 Expression Loss as a Mechanism of Resistance to Mosunetuzumab in Patients with Relapsed/Refractory B-Cell Non-Hodgkin Lymphomas. J. Clin. Oncol. 2022, 40, 7526. [Google Scholar] [CrossRef]
- Zucchinetti, C.; Signori, C.; Di Trani, M.; Pirosa, M.C.; Bruscaggin, A.; Korfi, K.; Bottos, A.; Calabretta, E.; Corrado, F.; Rossi, D.; et al. Early Assessment of Circulating Tumor (Ct)DNA and Analysis of TP53 Mutations in Patients with Relapsed/Refractory (R/R) Large B-Cell Lymphoma Treated with Glofitamab Monotherapy. Blood 2024, 144, 4356. [Google Scholar] [CrossRef]

{kind=link}
| Trade Name (Product Name) | Target | Construct | Approval | Indications * | Molecular Mechanisms of Resistance |
|---|---|---|---|---|---|
| Kymriah [40,41,42] (Tisagenlecleucel) | CD19 | CD8α/CD8α 4-1BB + CD3ζ | 2017 (FDA) 2018 (EMA) | Adult patients with r/r DLBCL, DLBCL arising from FL, FL, or High-Grade B-Cell Lymphoma (HGBCL) after two or more lines of therapy | T cell determinants
|
| Yescarta [40,43,44] (Axicabtagene ciloleucel) | CD19 | CD28/CD28 CD28 + CD3ζ | 2017 (FDA) 2018 (EMA) | Adult patients with r/r DLBCL, FL, HGBCL, or Primary Mediastinal B-Cell Lymphoma (PMBCL) after first-line therapy | |
| Tecartus [40,45,46] (Brexucabtagene autoleucel) | CD19 | CD28/CD28 CD28 + CD3ζ | 2020 (EMA, FDA) | Adult patients with r/r MCL after two or more lines of therapy | |
| Breyanzi [40,47,48] (Lisocabtagene maraleucel) | CD19 | IgG4/CD28 4-1BB + CD3ζ | 2021 (FDA) 2022 (EMA) | Adult patients with r/r HGBCL, FL grade 3B, MCL, PMBCL, CLL, or Small Lymphocytic Lymphoma (SLL) after first-line therapy |
| Trade Name (Product Name) | Target (CD20:CD3 Ratio) | Structure | Approval | Indications | Molecular Mechanisms of Resistance |
|---|---|---|---|---|---|
| Lunsumio [83,84,85] (Mosunetuzumab) | CD20 × CD3 (1:1) | IgG1 | 2022 (EMA, FDA) | Adult patients with r/r FL after two or more lines of therapy | BsAbs properties
|
| EMA: Tepkinly FDA: Epkinly [83,86,87] (Epcoritamab) | CD20 × CD3 (1:1) | IgG1 | 2023 (EMA, FDA) | Adult patients with r/r DLBCL, FL, or HGBCL after two or more lines of therapy | |
| Columvi [83,88,89] (Glofitamab) | CD20 × CD3 (2:1) | IgG1 | 2023 (EMA, FDA) | Adult patients with r/r DLBCL or r/r HGBCL from FL after two or more lines of therapy | |
| Ordspono [83,90] (Odronextamab) | CD20 × CD3 (1:1) | IgG4 | 2024 (EMA) | Adult patients with r/r DLBCL or FL after two or more lines of therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arena, G.; Chiarle, R. Mechanisms of Resistance to Novel Immunotherapies in B-Cell Lymphomas: Focus on CAR T and Bispecific Antibodies. Cancers 2025, 17, 3453. https://doi.org/10.3390/cancers17213453
Arena G, Chiarle R. Mechanisms of Resistance to Novel Immunotherapies in B-Cell Lymphomas: Focus on CAR T and Bispecific Antibodies. Cancers. 2025; 17(21):3453. https://doi.org/10.3390/cancers17213453
Chicago/Turabian StyleArena, Gloria, and Roberto Chiarle. 2025. "Mechanisms of Resistance to Novel Immunotherapies in B-Cell Lymphomas: Focus on CAR T and Bispecific Antibodies" Cancers 17, no. 21: 3453. https://doi.org/10.3390/cancers17213453
APA StyleArena, G., & Chiarle, R. (2025). Mechanisms of Resistance to Novel Immunotherapies in B-Cell Lymphomas: Focus on CAR T and Bispecific Antibodies. Cancers, 17(21), 3453. https://doi.org/10.3390/cancers17213453