
Chronic Lymphocytic Leukemia: Novel Therapeutic Targets Under Investigation



Simple Summary
Abstract
1. Introduction: Current Treatment Landscape in CLL
2. The Unmet Need in Double-Refractory CLL: A Clinical Crossroad
3. Novel Cellular Therapies in CLL
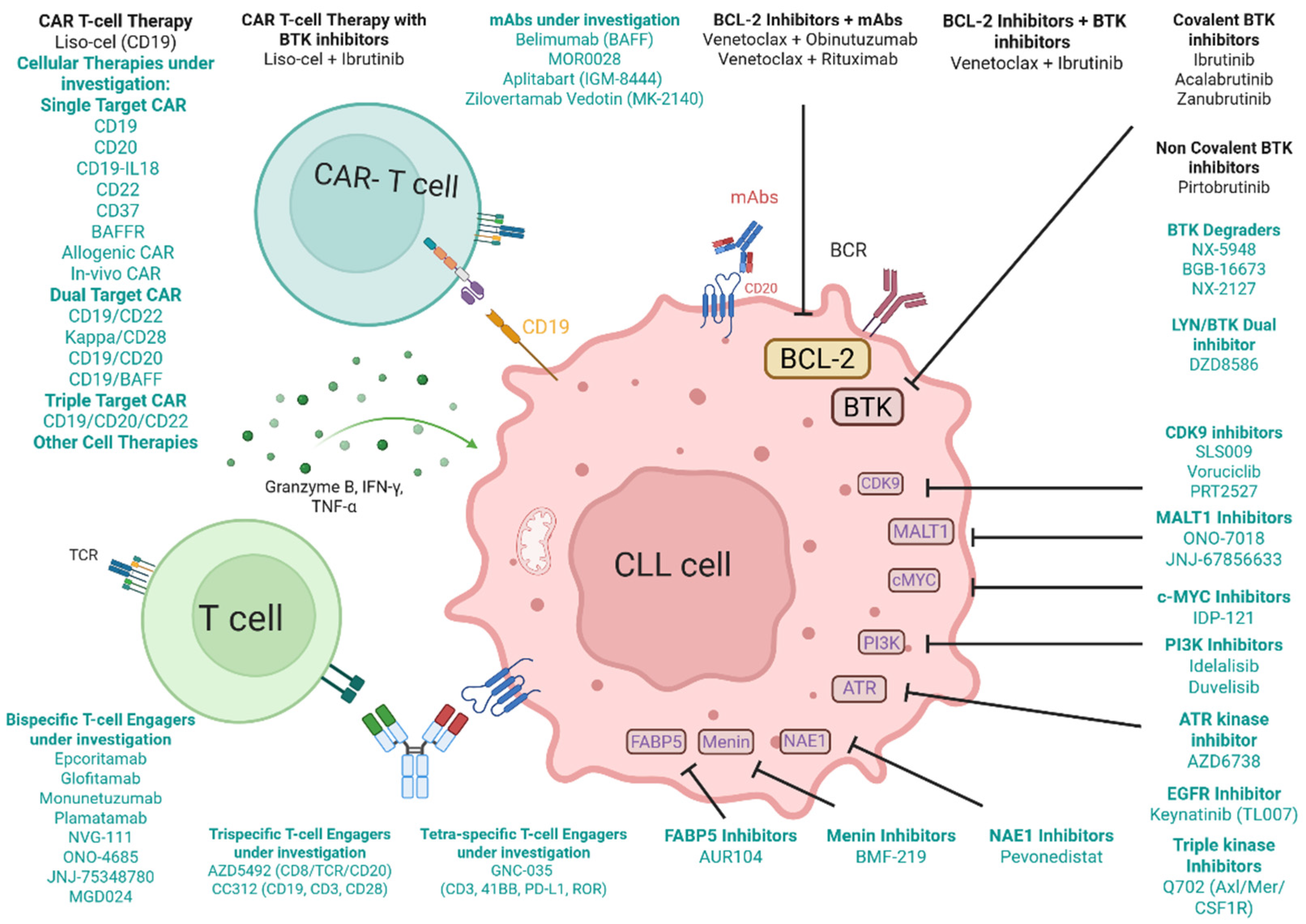
3.1. Single-Target CAR T-Cell Therapy
3.2. Dual-Target CAR T-Cell Therapy
3.3. Triple-Target CAR T-Cell Therapy
3.4. Other Cellular Therapies in Clinical Trials
4. T-Cell Engagers in CLL
4.1. Bispecific T-Cell Engagers
4.2. Trispecific Antibodies
4.3. Tetraspecific Antibodies
5. Monoclonal Antibodies and Antibody–Drug Conjugates in CLL
6. Small Molecule Inhibitors in CLL
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Leukemia—Chronic Lymphocytic Leukemia (CLL). Available online: https://seer.cancer.gov/statfacts/html/clyl.html (accessed on 1 April 2025).
- Tam, C.; Thompson, P.A. BTK inhibitors in CLL: Second-generation drugs and beyond. Blood Adv. 2024, 8, 2300–2309. [Google Scholar] [CrossRef] [PubMed]
- Aalipour, A.; Advani, R.H. Bruton’s tyrosine kinase inhibitors and their clinical potential in the treatment of B-cell malignancies: Focus on ibrutinib. Ther. Adv. Hematol. 2014, 5, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Lampson, B.L.; Davids, M.S. The Development and Current Use of BCL-2 Inhibitors for the Treatment of Chronic Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2017, 12, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Barr, P.M.; Owen, C.; Robak, T.; Tedeschi, A.; Bairey, O.; Burger, J.A.; Hillmen, P.; Coutre, S.E.; Dearden, C.; Grosicki, S.; et al. Up to 8-year follow-up from RESONATE-2: First-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022, 6, 3440–3450. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Hanson, C.A.; Paietta, E.M.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Long-term outcomes for ibrutinib-rituximab and chemoimmunotherapy in CLL: Updated results of the E1912 trial. Blood 2022, 140, 112–120. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood 2019, 133, 2031–2042. [Google Scholar] [CrossRef]
- Tam, C.S.; Brown, J.R.; Kahl, B.S.; Ghia, P.; Giannopoulos, K.; Jurczak, W.; Šimkovič, M.; Shadman, M.; Österborg, A.; Laurenti, L.; et al. Zanubrutinib versus bendamustine and rituximab in untreated chronic lymphocytic leukaemia and small lymphocytic lymphoma (SEQUOIA): A randomised, controlled, phase 3 trial. Lancet Oncol. 2022, 23, 1031–1043. [Google Scholar] [CrossRef]
- Brown, J.R.; Eichhorst, B.; Lamanna, N.; O’Brien, S.M.; Tam, C.S.; Qiu, L.; Jurczak, W.; Zhou, K.; Šimkovič, M.; Mayer, J.; et al. Sustained benefit of zanubrutinib vs ibrutinib in patients with R/R CLL/SLL: Final comparative analysis of ALPINE. Blood 2024, 144, 2706–2717. [Google Scholar] [CrossRef]
- Seymour, J.F.; Byrd, J.C.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.; Furman, R.R.; O’Brien, S.; Brown, J.R.; Munir, T.; Mato, A.; et al. Detailed safety profile of acalabrutinib vs ibrutinib in previously treated chronic lymphocytic leukemia in the ELEVATE-RR trial. Blood 2023, 142, 687–699. [Google Scholar] [CrossRef]
- Liu, T.-M.; Woyach, J.A.; Zhong, Y.; Lozanski, A.; Lozanski, G.; Dong, S.; Strattan, E.; Lehman, A.; Zhang, X.; Jones, J.A.; et al. Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood 2015, 126, 61–68. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Rodriguez-Blanco, G.; Sumpton, D.; Cloix, C.; Mullin, M.; Mackay, G.M.; O’Neill, K.; Lemgruber, L.; Luo, X.; Tait, S.W.G. Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Korycka-Wolowiec, A.; Wolowiec, D.; Kubiak-Mlonka, A.; Robak, T. Venetoclax in the treatment of chronic lymphocytic leukemia. Expert Opin. Drug Metab. Toxicol. 2019, 15, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; Serna, J.D.l.; et al. Venetoclax–Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Al-Sawaf, O.; Robrecht, S.; Zhang, C.; Olivieri, S.; Chang, Y.M.; Fink, A.M.; Tausch, E.; Schneider, C.; Ritgen, M.; Kreuzer, K.-A.; et al. Venetoclax-obinutuzumab for previously untreated chronic lymphocytic leukemia: 6-year results of the randomized phase 3 CLL14 study. Blood 2024, 144, 1924–1935. [Google Scholar] [CrossRef]
- Condoluci, A.; Rossi, D. Mechanisms of resistance to venetoclax. Blood 2022, 140, 2094–2096. [Google Scholar] [CrossRef]
- Kotmayer, L.; László, T.; Mikala, G.; Kiss, R.; Lévay, L.; Hegyi, L.L.; Gróf, S.; Nagy, T.; Barna, G.; Farkas, P.; et al. Landscape of BCL2 Resistance Mutations in a Real-World Cohort of Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia Treated with Venetoclax. Int. J. Mol. Sci. 2023, 24, 5802. [Google Scholar] [CrossRef]
- Aronson, J.H.; Skånland, S.S.; Roeker, L.E.; Thompson, M.C.; Mato, A.R. Approach to a patient with “double refractory” chronic lymphocytic leukemia: “Double, double toil and trouble” (Shakespeare). Am. J. Hematol. 2022, 97 (Suppl. 2), S19–S25. [Google Scholar] [CrossRef]
- Yoon, J.T.; Zhou, Y.; Mikhaleva, M.; Choi, D.S.; Fernandes, S.M.; Armand, P.; Bessnow, A.C.; Crombie, J.L.; Fisher, D.C.; Jacobsen, E.D.; et al. Characteristics and Outcomes of Patients with Double Refractory and Double Exposed Chronic Lymphocytic Leukemia. Blood Adv. 2025, 9, 2808–2817. [Google Scholar] [CrossRef]
- Thompson, M.C.; Roeker, L.E.; Coombs, C.C.; Jensen, J.L.; Kamdar, M.; Skarbnik, A.; Pagel, J.M.; Bailey, N.; Pu, J.J.; Jacobs, R.; et al. Addressing a New Challenge in Chronic Lymphocytic Leukemia: Outcomes of Therapies after Exposure to Both a Covalent Bruton’s Tyrosine Kinase Inhibitor and Venetoclax. Blood 2021, 138, 2628. [Google Scholar] [CrossRef]
- Gomez, E.B.; Ebata, K.; Randeria, H.S.; Rosendahl, M.S.; Cedervall, E.P.; Morales, T.H.; Hanson, L.M.; Brown, N.E.; Gong, X.; Stephens, J.; et al. Preclinical characterization of pirtobrutinib, a highly selective, noncovalent (reversible) BTK inhibitor. Blood 2023, 142, 62–72. [Google Scholar] [CrossRef]
- Mato, A.R.; Woyach, J.A.; Brown, J.R.; Ghia, P.; Patel, K.; Eyre, T.A.; Munir, T.; Lech-Maranda, E.; Lamanna, N.; Tam, C.S.; et al. Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023, 389, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Desikan, S.P.; Nguyen, B.; Won, H.; Tantawy, S.I.; McNeely, S.; Marella, N.; Ebata, K.; Woyach, J.A.; Patel, K.; et al. Genomic Evolution and Resistance during Pirtobrutinib Therapy in Covalent BTK-Inhibitor (cBTKi) Pre-Treated Chronic Lymphocytic Leukemia Patients: Updated Analysis from the BRUIN Study. Blood 2023, 142, 326. [Google Scholar] [CrossRef]
- Naeem, A.; Utro, F.; Wang, Q.; Cha, J.; Vihinen, M.; Martindale, S.; Zhou, Y.; Ren, Y.; Tyekucheva, S.; Kim, A.S.; et al. Pirtobrutinib targets BTK C481S in ibrutinib-resistant CLL but second-site BTK mutations lead to resistance. Blood Adv. 2023, 7, 1929–1943. [Google Scholar] [CrossRef] [PubMed]
- Roeker, L.E. A fresh look at covalent BTK inhibitor resistance. Blood 2024, 144, 1029–1031. [Google Scholar] [CrossRef]
- Montoya, S.; Bourcier, J.; Noviski, M.; Lu, H.; Thompson, M.C.; Chirino, A.; Jahn, J.; Sondhi, A.K.; Gajewski, S.; Tan, Y.S.M.; et al. Kinase-impaired BTK mutations are susceptible to clinical-stage BTK and IKZF1/3 degrader NX-2127. Science 2024, 383, eadi5798. [Google Scholar] [CrossRef]
- Hampel, P.J.; Rabe, K.G.; Call, T.G.; Ding, W.; Leis, J.F.; Kenderian, S.S.; Muchtar, E.; Wang, Y.; Koehler, A.B.; Parrondo, R.; et al. Combined ibrutinib and venetoclax for treatment of patients with ibrutinib-resistant or double-refractory chronic lymphocytic leukaemia. Br. J. Haematol. 2022, 199, 239–244. [Google Scholar] [CrossRef]
- Rogers, K.A.; McLaughlin, E.; Wei, L.; Bhat, S.A.; Crouse, A.; Grever, M.R.; Jones, D.; Kittai, A.S.; Lozanski, G.; Moran, M.; et al. Initial Results of a Phase 2 Study of Venetoclax Added to Ibrutinib to Eliminate Ibrutinib Resistance Mutations in CLL. Blood 2023, 142, 1899. [Google Scholar] [CrossRef]
- Roeker, L.E.; Woyach, J.A.; Cheah, C.Y.; Coombs, C.C.; Shah, N.N.; Wierda, W.G.; Patel, M.R.; Lamanna, N.; Tsai, D.E.; Nair, B.; et al. Fixed-duration pirtobrutinib plus venetoclax with or without rituximab in relapsed/refractory CLL: The phase 1b BRUIN trial. Blood 2024, 144, 1374–1386. [Google Scholar] [CrossRef]
- Siddiqi, T.; Maloney, D.G.; Kenderian, S.S.; Brander, D.M.; Dorritie, K.; Soumerai, J.; Riedell, P.A.; Shah, N.N.; Nath, R.; Fakhri, B.; et al. Lisocabtagene maraleucel in chronic lymphocytic leukaemia and small lymphocytic lymphoma (TRANSCEND CLL 004): A multicentre, open-label, single-arm, phase 1–2 study. Lancet 2023, 402, 641–654. [Google Scholar] [CrossRef]
- Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef]
- Camerini, E.; Amsen, D.; Kater, A.P.; Peters, F.S. The complexities of T-cell dysfunction in chronic lymphocytic leukemia. Semin. Hematol. 2024, 61, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Shaim, H.; Thompson, P.A.; Burger, J.A.; Keating, M.; Estrov, Z.; Harris, D.; Kim, E.; Ferrajoli, A.; Daher, M.; et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia 2018, 32, 960–970. [Google Scholar] [CrossRef]
- Hanna, B.S.; Yazdanparast, H.; Demerdash, Y.; Roessner, P.M.; Schulz, R.; Lichter, P.; Stilgenbauer, S.; Seiffert, M. Combining ibrutinib and checkpoint blockade improves CD8+ T-cell function and control of chronic lymphocytic leukemia in E-TCL1 mice. Haematologica 2021, 106, 968–977. [Google Scholar] [CrossRef]
- Wierda, W.G.; Dorritie, K.; Gauthier, J.; Nath, R.; Kipps, T.J.; Riedell, P.A.; Eradat, H.A.; Kenderian, S.S.; Kharfan-Dabaja, M.A.; Shah, N.N.; et al. Lisocabtagene Maraleucel (liso-cel) Combined with Ibrutinib (ibr) for Patients (pts) with Relapsed or Refractory (R/R) Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL): Primary Results from the Open-Label, Phase 1/2 Transcend CLL 004 St. Blood 2024, 144, 887. [Google Scholar] [CrossRef]
- Gribben, J.G. How and when I do allogeneic transplant in CLL. Blood 2018, 132, 31–39. [Google Scholar] [CrossRef]
- Roeker, L.E.; Dreger, P.; Brown, J.R.; Lahoud, O.B.; Eyre, T.A.; Brander, D.M.; Skarbnik, A.; Coombs, C.C.; Kim, H.T.; Davids, M.; et al. Allogeneic stem cell transplantation for chronic lymphocytic leukemia in the era of novel agents. Blood Adv. 2020, 4, 3977–3989. [Google Scholar] [CrossRef]
- Ruella, M.; Korell, F.; Porazzi, P.; Maus, M.V. Mechanisms of resistance to chimeric antigen receptor-T cells in haematological malignancies. Nat. Rev. Drug Discov. 2023, 22, 976–995. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, A.; Karadimitris, A.; Ruella, M. Building upon the success of CART19: Chimeric antigen receptor T cells for hematologic malignancies. Leuk. Lymphoma 2018, 59, 2040–2055. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Schilhabel, A.; Walter, P.J.; Cramer, P.; Von Tresckow, J.; Kohlscheen, S.; Szczepanowski, M.; Laqua, A.; Fischer, K.; Eichhorst, B.; Böttcher, S.; et al. CD20 Expression as a Possible Novel Prognostic Marker in CLL: Application of EuroFlow Standardization Technique and Normalization Procedures in Flow Cytometric Expression Analysis. Cancers 2022, 14, 4917. [Google Scholar] [CrossRef]
- Tan Su Yin, E.; Hu, Y.X.; Huang, H. The breakthrough and the future: CD20 chimeric antigen receptor T-cell therapy for hematologic malignancies. ImmunoMedicine 2022, 2, e1039. [Google Scholar] [CrossRef]
- Xu, J.; Luo, W.; Li, C.; Mei, H. Targeting CD22 for B-cell hematologic malignancies. Exp. Hematol. Oncol. 2023, 12, 90. [Google Scholar] [CrossRef]
- Shadman, M.; Caimi, P.F.; O’Brien, S.M.; Reagan, P.M.; Dezube, B.; Navaratnarajah, P.; Gaur, T.; Petrossian, S.; Germani, A.; Till, B.G.; et al. Efficacy and Safety of a Third Generation CD20 CAR-T (MB-106) for Treatment of Relapsed/Refractory Indolent B-Cell Non-Hodgkin Lymphoma: Phase-1 Results from a Multicenter Trial. Blood 2023, 142, 2102. [Google Scholar] [CrossRef]
- Svoboda, J.; Landsburg, D.J.; Nasta, S.D.; Barta, S.K.; Chong, E.A.; Lariviere, M.J.; Shea, J.; Cervini, A.; Hexner, E.O.; Marshall, A.; et al. Safety and efficacy of armored huCART19-IL18 in patients with relapsed/refractory lymphomas that progressed after anti-CD19 CAR T cells. J. Clin. Oncol. 2024, 42, 7004. [Google Scholar] [CrossRef]
- Svoboda, J.; Landsburg, D.J.; Gerson, J.; Nasta, S.D.; Barta, S.K.; Chong, E.A.; Cook, M.; Frey, N.V.; Shea, J.; Cervini, A.; et al. Enhanced CAR T-Cell Therapy for Lymphoma after Previous Failure. N. Engl. J. Med. 2025, 392, 1824–1835. [Google Scholar] [CrossRef]
- Scarfò, I.; Ormhøj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood 2018, 132, 1495–1506. [Google Scholar] [CrossRef]
- Kipps, T.J. ROR1: An orphan becomes apparent. Blood 2022, 140, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Daneshmanesh, A.H.; Hojjat-Farsangi, M.; Khan, A.; Jeddi-Tehrani, M.; Akhondi, M.; Bayat, A.; Ghods, R.; Mahmoudi, A.-R.; Hadavi, R.; Österborg, A.; et al. Monoclonal antibodies against ROR1 induce apoptosis of chronic lymphocytic leukemia (CLL) cells. Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund UK 2012, 26, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Graham, C.E.; Berger, T.R.; Ritchey, J.; Horick, N.K.; El-Jawahri, A.; Scarfò, I.; Schmidts, A.; Haradhvala, N.J.; Wehrli, M.; et al. Phase 1 study of CAR-37 T cells in patients with relapsed or refractory CD37+ lymphoid malignancies. Blood 2024, 144, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; MacKay, F.; Steiner, V.; Hofmann, K.; Bodmer, J.L.; Holler, N.; Ambrose, C.; Lawton, P.; Bixler, S.; Acha-Orbea, H.; et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J. Exp. Med. 1999, 189, 1747–1756. [Google Scholar] [CrossRef]
- Luo, Y.; Qie, Y.; Gadd, M.E.; Manna, A.; Rivera-Valentin, R.; To, T.; Li, S.; Yassine, F.; Murthy, H.S.; Dronca, R.; et al. Translational development of a novel BAFF-R CAR-T therapy targeting B-cell lymphoid malignancies. Cancer Immunol. Immunother. 2023, 72, 4031–4047. [Google Scholar] [CrossRef]
- Shahid, S.; Prockop, S.E.; Flynn, G.C.; Mauguen, A.; White, C.O.; Bieler, J.; McAvoy, D.; Hosszu, K.; Cancio, M.I.; Jakubowski, A.A.; et al. Allogeneic off-the-shelf CAR T-cell therapy for relapsed or refractory B-cell malignancies. Blood Adv. 2025, 9, 1644–1657. [Google Scholar] [CrossRef]
- Magnani, C.F.; Gaipa, G.; Lussana, F.; Belotti, D.; Gritti, G.; Napolitano, S.; Matera, G.; Cabiati, B.; Buracchi, C.; Borleri, G.; et al. Sleeping Beauty–engineered CAR T cells achieve antileukemic activity without severe toxicities. J. Clin. Investig. 2020, 130, 6021–6033. [Google Scholar] [CrossRef]
- Michels, K.R.; Sheih, A.; Hernandez, S.A.; Brandes, A.H.; Parrilla, D.; Irwin, B.; Perez, A.M.; Ting, H.-A.; Nicolai, C.J.; Gervascio, T.; et al. Preclinical proof of concept for VivoVec, a lentiviral-based platform for in vivo CAR T-cell engineering. J. Immunother. Cancer 2023, 11, e006292. [Google Scholar] [CrossRef]
- Parker, M.; Ulrich-Lewis, J.; Tang, W.; Nicolai, C.; Michels, K.; Hernandez, S.; Parrilla, D.; Cooper, S.; Perez, A.; McDonnell, M.; et al. Vivovec™ Surface-Engineered Lentiviral Particles Mediate In Vivo CAR T Generation with Potent and Highly Durable Activity in Non-Human Primates and Tumor-Bearing Humanized Mice. Blood 2023, 142, 765. [Google Scholar] [CrossRef]
- Nicolai, C.J.; Parker, M.H.; Qin, J.; Tang, W.; Ulrich-Lewis, J.T.; Gottschalk, R.J.; Cooper, S.E.; Hernandez Lopez, S.A.; Parrilla, D.; Mangio, R.S.; et al. In vivo CAR T-cell generation in nonhuman primates using lentiviral vectors displaying a multidomain fusion ligand. Blood 2024, 144, 977–987. [Google Scholar] [CrossRef]
- Roddie, C.; Lekakis, L.J.; Marzolini, M.A.V.; Ramakrishnan, A.; Zhang, Y.; Hu, Y.; Peddareddigari, V.G.R.; Khokhar, N.Z.; Chen, R.W.; Basilico, S.; et al. Dual targeting of CD19 and CD22 with Bicistronic CAR-T cells in Patients with Relapsed/Refractory Large B Cell Lymphoma. Blood 2023, 141, 2470–2482. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Li, W.; Cai, J.; Yang, X.; Yang, L.; Yang, J.; Wang, T.; Li, Y.; Zhou, Z.; Lu, X.; et al. Safety and Efficacy of Bicistronic CD19/CD22 CAR T Cell Therapy in Childhood B Cell Acute Lymphoblastic Leukemia. Blood 2024, 144, 681. [Google Scholar] [CrossRef]
- Tong, C.; Zhang, Y.; Liu, Y.; Ji, X.; Zhang, W.-Y.; Guo, Y.; Han, X.; Ti, D.; Dai, H.; Wang, C.; et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B cell lymphoma. Blood 2020, 136, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, Z.; Awuah, D.; Chang, W.-C.; Cheng, W.A.; Vyas, V.; Cha, S.-C.; Anderson, A.J.; Zhang, T.; Wang, Z.; et al. CD19/BAFF-R dual-targeted CAR T cells for the treatment of mixed antigen-negative variants of acute lymphoblastic leukemia. Leukemia 2022, 36, 1015–1024. [Google Scholar] [CrossRef]
- Lemal, R.; Tournilhac, O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J. Immunother. Cancer 2019, 7, 202. [Google Scholar] [CrossRef]
- Schneider, D.; Xiong, Y.; Wu, D.; Hu, P.; Alabanza, L.; Steimle, B.; Mahmud, H.; Anthony-Gonda, K.; Krueger, W.; Zhu, Z.; et al. Trispecific CD19-CD20-CD22–targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci. Transl. Med. 2021, 13, eabc6401. [Google Scholar] [CrossRef]
- Mehta, R.S.; Chen, X.; Antony, J.; Boyiadzis, M.; Szabolcs, P. Generating Peripheral Blood Derived Lymphocytes Reacting Against Autologous Primary AML Blasts. J. Immunother. 2016, 39, 71–80. [Google Scholar] [CrossRef]
- imbruvica. RESONATE-2 Primary Data. Available online: https://www.imbruvicahcp.com/cll/efficacy/resonate-2/long-term-data?gclid=cb11759b5baa1c144c71126529fdff0d&gclsrc=3p.ds&msclkid=cb11759b5baa1c144c71126529fdff0d&utm_source=bing&utm_medium=cpc&utm_campaign=BI-USA-ENG-PS-Imbruvica-GP-PH-RN-HCP_CLL%20Condition&utm_term=cll&utm_content=CLL%20Condition (accessed on 10 April 2025).
- Vera, J.; Savoldo, B.; Vigouroux, S.; Biagi, E.; Pule, M.; Rossig, C.; Wu, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. T lymphocytes redirected against the κ light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood 2006, 108, 3890–3897. [Google Scholar] [CrossRef]
- Roskrow, M.A.; Suzuki, N.; Gan, Y.-J.; Sixbey, J.W.; Ng, C.Y.C.; Kimbrough, S.; Hudson, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Epstein-Barr Virus (EBV)-Specific Cytotoxic T Lymphocytes for the Treatment of Patients With EBV-Positive Relapsed Hodgkin’s Disease. Blood 1998, 91, 2925–2934. [Google Scholar] [CrossRef]
- Marin, D.; Li, Y.; Basar, R.; Rafei, H.; Daher, M.; Dou, J.; Mohanty, V.; Dede, M.; Nieto, Y.; Uprety, N.; et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19+ B cell tumors: A phase 1/2 trial. Nat. Med. 2024, 30, 772–784. [Google Scholar] [CrossRef]
- Page, A.; Chuvin, N.; Valladeau-Guilemond, J.; Depil, S. Development of NK cell-based cancer immunotherapies through receptor engineering. Cell. Mol. Immunol. 2024, 21, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Epcoritamab: First Approval. Drugs 2023, 83, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Kater, A.P.; Christensen, J.H.; Bentzen, H.H.; Niemann, C.U.; Hutchings, M.; Chen, J.; Rios, M.; Palenski, T.; Li, T.; Mato, A.R. Subcutaneous Epcoritamab in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia: Preliminary Results from the Epcore CLL-1 Trial. Blood 2021, 138, 2627. [Google Scholar] [CrossRef]
- Kang, C. Mosunetuzumab: First Approval. Drugs 2022, 82, 1229–1234. [Google Scholar] [CrossRef]
- Shirley, M. Glofitamab: First Approval. Drugs 2023, 83, 935–941. [Google Scholar] [CrossRef]
- Patel, K.; Riedell, P.A.; Tilly, H.; Ahmed, S.; Michot, J.-M.; Ghesquieres, H.; Schiano de Collela, J.M.; Chanan-Khan, A.; Bouabdallah, K.; Tessoulin, B.; et al. A Phase 1 Study of Plamotamab, an Anti-CD20 x Anti-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Non-Hodgkin’s Lymphoma: Recommended Dose Safety/Efficacy Update and Escalation Exposure-Response Analysis. Blood 2022, 140, 9470–9472. [Google Scholar] [CrossRef]
- Song, Y.; Li, Z.; Li, L.; Qian, Z.; Zhou, K.; Fan, L.; Tan, P.; Giri, P.; Li, Z.; Kenealy, M.; et al. GB261, an Fc-Function Enabled and CD3 Affinity De-Tuned CD20/CD3 Bispecific Antibody, Demonstrated a Highly Advantageous Safety/Efficacy Balance in an Ongoing First-in-Human Dose-Escalation Study in Patients with Relapsed/Refractory Non-Hodgkin Lymphoma. Blood 2023, 142, 1719. [Google Scholar] [CrossRef]
- Granger, D.; Gohil, S.; Barbarulo, A.; Baccaro, A.; Muczynski, V.; Chester, K.; Germaschewski, F.; Batten, T.; Brown, K.; Cook, S.; et al. NVG-111, a novel ROR1xCD3 bispecific antibody for non-Hodgkin lymphoma. J. Clin. Oncol. 2021, 39, 7549. [Google Scholar] [CrossRef]
- Shin, H.G.; Yang, H.R.; Yoon, A.; Lee, S. Bispecific Antibody-Based Immune-Cell Engagers and Their Emerging Therapeutic Targets in Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 5686. [Google Scholar] [CrossRef]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Zhao, L.; Li, S.; Wei, X.; Qi, X.; Liu, D.; Liu, L.; Wen, F.; Zhang, J.-s.; Wang, F.; Liu, Z.-l.; et al. A novel CD19/CD22/CD3 trispecific antibody enhances therapeutic efficacy and overcomes immune escape against B-ALL. Blood 2022, 140, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Rolin, C.; Zimmer, J.; Seguin-Devaux, C. Bridging the gap with multispecific immune cell engagers in cancer and infectious diseases. Cell. Mol. Immunol. 2024, 21, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shah, K.; Barat, B.; Lam, C.-Y.K.; Gorlatov, S.; Ciccarone, V.; Tamura, J.; Moore, P.A.; Diedrich, G. Multispecific, Multivalent Antibody-Based Molecules Engineered on the DART® and TRIDENTTM Platforms. Curr. Protoc. Immunol. 2020, 129, e95. [Google Scholar] [CrossRef] [PubMed]
- Khalili, J.S.; Xiao, S.; Zhu, Y. Abstract 5679: Tetra-specific antibody GNC-035: Guidance and navigation control (GNC) molecule development for treatment of ROR1+ malignancies. Cancer Res. 2023, 83, 5679. [Google Scholar] [CrossRef]
- Tandler, C.; Schmidt, M.; Heitmann, J.S.; Hierold, J.; Schmidt, J.; Schneider, P.; Dörfel, D.; Walz, J.; Salih, H.R. Neutralization of B-Cell Activating Factor (BAFF) by Belimumab Reinforces Small Molecule Inhibitor Treatment in Chronic Lymphocytic Leukemia. Cancers 2020, 12, 2725. [Google Scholar] [CrossRef]
- Jurczak, W.; Zinzani, P.L.; Gaidano, G.; Goy, A.; Provencio, M.; Nagy, Z.; Robak, T.; Maddocks, K.; Buske, C.; Ambarkhane, S.; et al. Phase IIa study of the CD19 antibody MOR208 in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma. Ann. Oncol. 2018, 29, 1266–1272. [Google Scholar] [CrossRef]
- Mateu-Albero, T.; Marcos-Jimenez, A.; Delgado-Wicke, P.; Terrón, F.; Loscertales, J.; López-Matencio, J.M.S.; Muñoz-Calleja, C.; Cuesta-Mateos, C. Evaluation of the novel therapeutic anti-CCR7 antibody CAP-100 as an add-on therapy in chronic lymphocytic leukemia patients receiving venetoclax. Hematol. Oncol. 2023, 41, 869–876. [Google Scholar] [CrossRef]
- Wang, B.T.; Kothambawala, T.; Wang, L.; Matthew, T.J.; Calhoun, S.E.; Saini, A.K.; Kotturi, M.F.; Hernandez, G.; Humke, E.W.; Peterson, M.S.; et al. Multimeric Anti-DR5 IgM Agonist Antibody IGM-8444 Is a Potent Inducer of Cancer Cell Apoptosis and Synergizes with Chemotherapy and BCL-2 Inhibitor ABT-199. Mol. Cancer Ther. 2021, 20, 2483–2494. [Google Scholar] [CrossRef]
- Spurgeon, S.; Mei, M.; Barr, P.M.; Barrientos, J.; de Vos, S.; Furman, R.; Patel, K.; Thompson, P.; Choi, M.Y.; Kallam, A.; et al. P1200: ZILOVERTAMAB VEDOTIN (MK-2140) IN RELAPSED OR REFRACTORY (R/R) NON-HODGKIN LYMPHOMA (NHL): UPDATED RESULTS FROM THE PHASE 1 WAVELINE-001 STUDY. HemaSphere 2023, 7, e54506b2. [Google Scholar] [CrossRef]
- Kocsis, A.; Gajwani, R.; Gross, J.; Gumley, A.I.; Lawrie, S.M.; Schwannauer, M.; Schultze-Lutter, F.; Grent-‘T-Jong, T.; Uhlhaas, P.J. Altered Autonomic Function in Individuals at Clinical High Risk for Psychosis. Front. Psychiatry 2020, 11, 580503. [Google Scholar] [CrossRef]
- DiEugenio, J. Sonrotoclax/Zanubrutinib Leads to Durable Responses in R/R CLL/SLL. Available online: https://www.onclive.com/view/sonrotoclax-zanubrutinib-leads-to-durable-responses-in-r-r-cll-sll (accessed on 1 April 2025).
- Soumerai, J.D.; Cheah, C.Y.; Anderson, M.A.; Lasica, M.; Verner, E.; Opat, S.S.; Ma, S.; Weinkove, R.; Cordoba, R.; Ghia, P.; et al. Sonrotoclax and Zanubrutinib as Frontline Treatment for CLL Demonstrates High MRD Clearance Rates with Good Tolerability: Data from an Ongoing Phase 1/1b Study BGB-11417-101. Blood 2024, 144, 1012. [Google Scholar] [CrossRef]
- Davids, M.S.; Ailawadhi, S.; Chanan-Khan, A.A.; Mudenda, B.; Nogaieva, L.; Kryachok, I.; Usenko, G.; Ivanov, V.; Kyselova, O.; Perekhrestenko, T.; et al. Lisaftoclax (APG-2575) Demonstrates Activity and Safety When Given with Accelerated Ramp-up and then Combined with Acalabrutinib or Rituximab in Patients (pts) with Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL), Including Those with Prior Exposure to Venetoclax. Blood 2024, 144, 4614. [Google Scholar] [CrossRef]
- Woyach, J.A.; Flinn, I.W.; Awan, F.T.; Eradat, H.; Brander, D.; Tees, M.; Parikh, S.A.; Phillips, T.J.; Ghori, R.; Reddy, N.M.; et al. Efficacy and Safety of Nemtabrutinib, a Wild-Type and C481S-Mutated Bruton Tyrosine Kinase Inhibitor for B-Cell Malignancies: Updated Analysis of the Open-Label Phase 1/2 Dose-Expansion Bellwave-001 Study. Blood 2022, 140, 7004–7006. [Google Scholar] [CrossRef]
- Xu, W.; Song, Y.; Wang, T.; Yang, S.; Liu, L.; Hu, Y.; Zhang, W.; Zhou, J.; Gao, S.; Ding, K.; et al. Orelabrutinib Monotherapy in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma: Updated Long Term Results of Phase II Study. Blood 2021, 138, 2638. [Google Scholar] [CrossRef]
- Woyach, J.A.; Brander, D.M.; Hu, B.; Rogers, K.A.; Omer, Z.; Stephens, D.M.; Sitlinger, A.; Tan, F.; Chen, Y.; Anthony, S.P.; et al. LP-168 (Rocbrutinib), a Novel Covalent and Non-Covalent Next-Generation Inhibitor of Bruton’s Tyrosine Kinase: Updates on the Phase 1 Trial and Initial Results of the CLL Gatekeeper Mutation Cohort. Blood 2024, 144, 4622. [Google Scholar] [CrossRef]
- Woyach, J.A.; Stephens, D.M.; Brander, D.M.; Kittai, A.S.; Hu, B.; Sitlinger, A.; Curran, E.K.; Tan, F.; Chen, Y.; Anthony, S.P.; et al. Initial Results of a Phase 1 Dose Escalation Study of LP-168, a Novel Covalent and Non-Covalent Next-Generation Inhibitor of Bruton’s Tyrosine Kinase. Blood 2023, 142, 328. [Google Scholar] [CrossRef]
- Shah, N.N.; Omer, Z.; Collins, G.P.; Forconi, F.; Danilov, A.; Byrd, J.C.; El-Sharkawi, D.; Searle, E.; Alencar, A.J.; Ma, S.; et al. Efficacy and Safety of the Bruton’s Tyrosine Kinase (BTK) Degrader NX-5948 in Patients with Relapsed/Refractory (R/R) Chronic Lymphocytic Leukemia (CLL): Updated Results from an Ongoing Phase 1a/b Study. Blood 2024, 144, 884. [Google Scholar] [CrossRef]
- Thompson, M.C.; Parrondo, R.D.; Frustaci, A.M.; Allan, J.N.; Ghia, P.; Mocanu, I.; Tam, C.S.; Trotman, J.; Ahn, I.E.; Stilgenbauer, S.; et al. Preliminary Efficacy and Safety of the Bruton Tyrosine Kinase Degrader BGB-16673 in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma: Results from the Phase 1 CaDAnCe-101 Study. Blood 2024, 144, 885. [Google Scholar] [CrossRef]
- Danilov, A.; Tees, M.T.; Patel, K.; Wierda, W.G.; Patel, M.; Flinn, I.W.; Latif, T.; Ai, W.; Thompson, M.C.; Wang, M.L.; et al. A First-in-Human Phase 1 Trial of NX-2127, a First-in-Class Bruton’s Tyrosine Kinase (BTK) Dual-Targeted Protein Degrader with Immunomodulatory Activity, in Patients with Relapsed/Refractory B Cell Malignancies. Blood 2023, 142, 4463. [Google Scholar] [CrossRef]
- Montoya, S.; Thompson, M.C. Non-Covalent Bruton’s Tyrosine Kinase Inhibitors in the Treatment of Chronic Lymphocytic Leukemia. Cancers 2023, 15, 3648. [Google Scholar] [CrossRef]
- Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.; et al. Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N. Engl. J. Med. 2022, 386, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Eyre, T.A.; Riches, J.C. The Evolution of Therapies Targeting Bruton Tyrosine Kinase for the Treatment of Chronic Lymphocytic Leukaemia: Future Perspectives. Cancers 2023, 15, 2596. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Cheah, C.Y.; Parrondo, R.; Thompson, M.C.; Stevens, D.A.; Lasica, M.; Wang, M.L.; Kumar, A.; Trotman, J.; Alwan, M.; et al. First Results from a Phase 1, First-in-Human Study of the Bruton’s Tyrosine Kinase (BTK) Degrader Bgb-16673 in Patients (Pts) with Relapsed or Refractory (R/R) B-Cell Malignancies (BGB-16673-101). Blood 2023, 142, 4401. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Vanhaesebroeck, B. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 2003, 3, 317–330. [Google Scholar] [CrossRef]
- Meadows, S.A.; Vega, F.; Kashishian, A.; Johnson, D.; Diehl, V.; Miller, L.L.; Younes, A.; Lannutti, B.J. PI3Kδ inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood 2012, 119, 1897–1900. [Google Scholar] [CrossRef]
- Hoellenriegel, J.; Meadows, S.A.; Sivina, M.; Wierda, W.G.; Kantarjian, H.; Keating, M.J.; Giese, N.; O’Brien, S.; Yu, A.; Miller, L.L.; et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood 2011, 118, 3603–3612. [Google Scholar] [CrossRef]
- Balakrishnan, K.; Peluso, M.; Fu, M.; Rosin, N.Y.; Burger, J.A.; Wierda, W.G.; Keating, M.J.; Faia, K.; O’Brien, S.; Kutok, J.L.; et al. The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia 2015, 29, 1811–1822. [Google Scholar] [CrossRef]
- Hanlon, A.; Brander, D.M. Managing toxicities of phosphatidylinositol-3-kinase (PI3K) inhibitors. Hematology 2020, 2020, 346–356. [Google Scholar] [CrossRef]
- Patel, K.; Pagel, J.M. Exploring a Future for PI3K Inhibitors in Chronic Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2019, 14, 292–301. [Google Scholar] [CrossRef]
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. Oncologist 2021, 26, 184-e366. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, L.; Andrs, M.; Rezacova, M.; Korabecny, J. Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): Clinical candidate for cancer therapy. Pharmacol. Ther. 2020, 210, 107518. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wu, T.; Hu, M.; Han, S.; Liu, Y.; Zheng, J.; Qin, J.; Zhang, L.; Yang, Z. Preclinical Study of DZD8586, a Non-Covalent LYN/BTK Dual Inhibitor with Excellent BBB Penetration, for the Treatment of B-Cell Non-Hodgkin Lymphoma (B-NHL). Blood 2023, 142, 2822. [Google Scholar] [CrossRef]
- Song, Y.; Zhou, K.; Jing, H.; Wu, J.; Yang, H.; Bai, Y.; Fang, K.; Liu, Z.; Zhu, J.; Cheah, C.Y. First Report of Phase 1 Studies of DZD8586, a BBB Penetrant LYN/BTK Dual Inhibitor, in Patients with B-Cell Non-Hodgkin Lymphoma (B-NHL). Blood 2023, 142, 4465. [Google Scholar] [CrossRef]
- Qiu, L.; Li, Y.; Zhou, K.; Jiang, M.; Liu, W.; Li, Z.; Zheng, M.; Li, Z.; Qian, W.; Lan, M.; et al. Phase 2 study of DZD8586, a non-covalent BBB penetrant LYN/BTK dual inhibitor, as monotherapy in relapsed/refractory diffuse large B-cell lymphoma (r/r DLBCL; TAI-SHAN9). J. Clin. Oncol. 2025, 43, e19050. [Google Scholar] [CrossRef]
- Liang, X.; Cao, Y.; Li, C.; Yu, H.; Yang, C.; Liu, H. MALT1 as a promising target to treat lymphoma and other diseases related to MALT1 anomalies. Med. Res. Rev. 2021, 41, 2388–2422. [Google Scholar] [CrossRef]
- Tojjari, A.; Saeed, A.; Sadeghipour, A.; Kurzrock, R.; Cavalcante, L. Overcoming Immune Checkpoint Therapy Resistance with SHP2 Inhibition in Cancer and Immune Cells: A Review of the Literature and Novel Combinatorial Approaches. Cancers 2023, 15, 5384. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Lim, S.O.; Yamaguchi, H. Oncogenic signaling pathways associated with immune evasion and resistance to immune checkpoint inhibitors in cancer. Semin. Cancer Biol. 2020, 65, 51–64. [Google Scholar] [CrossRef]
- Philippar, U.; Lu, T.; Vloemans, N.; Bekkers, M.; Van Nuffel, L.; Gaudiano, M.; Wnuk-Lipinska, K.; Van Der Leede, B.-j.; Amssoms, K.; Kimpe, K.; et al. Abstract 5690: Discovery of JNJ-67856633: A novel, first-in-class MALT1 protease inhibitor for the treatment of B cell lymphomas. Cancer Res. 2020, 80, 5690. [Google Scholar] [CrossRef]
- Senga, S.; Kobayashi, N.; Kawaguchi, K.; Ando, A.; Fujii, H. Fatty acid-binding protein 5 (FABP5) promotes lipolysis of lipid droplets, de novo fatty acid (FA) synthesis and activation of nuclear factor-kappa B (NF-κB) signaling in cancer cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Ma, J.; Hua, X. Epigenetic regulation by the menin pathway. Endocr.-Relat. Cancer 2017, 24, T147–T159. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Xu, M.; Kang, Z.; Zhang, M.; Luo, Y. Menin–MLL1 Interaction Small Molecule Inhibitors: A Potential Therapeutic Strategy for Leukemia and Cancers. Molecules 2023, 28, 3026. [Google Scholar] [CrossRef] [PubMed]
- Guiyedi, K.; Parquet, M.; Aoufouchi, S.; Chauzeix, J.; Rizzo, D.; Al Jamal, I.; Feuillard, J.; Gachard, N.; Peron, S. Increased c-MYC Expression Associated with Active IGH Locus Rearrangement: An Emerging Role for c-MYC in Chronic Lymphocytic Leukemia. Cancers 2024, 16, 3749. [Google Scholar] [CrossRef]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The Action Mechanism of the Myc Inhibitor Termed Omomyc May Give Clues on How to Target Myc for Cancer Therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef]
- Coudé, M.M.; Braun, T.; Berrou, J.; Dupont, M.; Bertrand, S.; Masse, A.; Raffoux, E.; Itzykson, R.; Delord, M.; Riveiro, M.E.; et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. [Google Scholar] [CrossRef]
- Fairlie, W.D.; Lee, E.F. Co-Operativity between MYC and BCL-2 Pro-Survival Proteins in Cancer. Int. J. Mol. Sci. 2021, 22, 2841. [Google Scholar] [CrossRef]
- Anshabo, A.T.; Milne, R.; Wang, S.; Albrecht, H. CDK9: A Comprehensive Review of Its Biology, and Its Role as a Potential Target for Anti-Cancer Agents. Front. Oncol. 2021, 11, 678559. [Google Scholar] [CrossRef]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Torka, P.; Thiruvengadam, S.K.; Chen, L.; Wang, X.; Chen, C.; Vuong, D.; Qin, H.; Muir, A.; Orand, K.; Borja, I.; et al. Pevonedistat, a Nedd8-activating enzyme inhibitor, in combination with ibrutinib in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Blood Cancer J. 2023, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lin, Y.; Zhuang, Q.; Deng, H.; Liu, A.; Sun, J. BTK inhibitors resistance in B cell malignancies: Mechanisms and potential therapeutic strategies. Blood Rev. 2025, 71, 101273. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Shanafelt, T.D.; Lesnick, C.E.; Erlichman, C.; Leis, J.F.; Secreto, C.; Sassoon, T.R.; Call, T.G.; Bowen, D.A.; Conte, M.; et al. Akt inhibitor MK2206 selectively targets CLL B-cell receptor induced cytokines, mobilizes lymphocytes and synergizes with bendamustine to induce CLL apoptosis. Br. J. Haematol. 2014, 164, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Li, S.; Guo, J.; Yue, H.; Shi, L.; Liu, R.; Wang, J.; Shi, X. The role and mechanism of AZD5363 anti-leukemia activity in T-cell acute lymphoblastic leukemia. Eur. J. Pharmacol. 2024, 963, 176268. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updates 2008, 11, 32–50. [Google Scholar] [CrossRef]
- Mahadevan, D.; Qi, W.; Stejskal, A.; Cooke, L.; Garlich, J.R. SF1126, a Pan-PI3K Inhibitor Has Superior Preclinical Activity to CAL-101 a PI3K Delta-Specific Inhibitor in Aggressive B-Cell Non-Hodgkin’s Lymphoma. Blood 2011, 118, 2720. [Google Scholar] [CrossRef]
- Jeon, Y.; Kang, H.; Yang, Y.; Park, D.; Choi, B.; Kim, J.; Kim, J.; Nam, K. A Novel Selective Axl/Mer/CSF1R Kinase Inhibitor as a Cancer Immunotherapeutic Agent Targeting Both Immune and Tumor Cells in the Tumor Microenvironment. Cancers 2022, 14, 4821. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef]
- Martens, A.W.J.; Rietveld, J.M.; de Boer, R.; Peters, F.S.; Ngo, A.; van Mil, L.; de Heer, K.; Spaargaren, M.; Verkleij, C.P.M.; van de Donk, N.; et al. Redirecting T-cell Activity with Anti-BCMA/Anti-CD3 Bispecific Antibodies in Chronic Lymphocytic Leukemia and Other B-cell Lymphomas. Cancer Res. Commun. 2022, 2, 330–341. [Google Scholar] [CrossRef]
- de Weerdt, I.; Hofland, T.; Lameris, R.; Endstra, S.; Jongejan, A.; Moerland, P.D.; de Bruin, R.C.G.; Remmerswaal, E.B.M.; Ten Berge, I.J.M.; Liu, N.; et al. Improving CLL Vγ9Vδ2-T-cell fitness for cellular therapy by ex vivo activation and ibrutinib. Blood 2018, 132, 2260–2272. [Google Scholar] [CrossRef]
- Nunes, J.; Tafesse, R.; Mao, C.; Purcell, M.; Mo, X.; Zhang, L.; Long, M.; Cyr, M.G.; Rader, C.; Muthusamy, N. Siglec-6 as a therapeutic target for cell migration and adhesion in chronic lymphocytic leukemia. Nat. Commun. 2024, 15, 5180. [Google Scholar] [CrossRef]
- Maher, N.; Mouhssine, S.; Matti, B.F.; Alwan, A.F.; Gaidano, G. Molecular Mechanisms in the Transformation from Indolent to Aggressive B Cell Malignancies. Cancers 2025, 17, 907. [Google Scholar] [CrossRef]

{kind=link}
| Agent Name | Target | Study Design/Phase | Clinicaltrial.gov Identifier | Population, Sample Size (Estimated) |
|---|---|---|---|---|
| Single target CAR T-cell | ||||
| MB-106 | CD20 | I/II | NCT03277729 | 53, R/R B Cell-NHL, R/R CLL, Other |
| huCART19-IL18 | huCART19-IL18 | I | NCT04684563 | 72, CLL, NHL, ALL |
| CAR-37 T Cells | CD37 | I | NCT04136275 | 6, CD37+ hematologic malignancies |
| BAFFR-targeting CAR T Cells | BAFFR | I | NCT05370430 | 36, R/R B-NHL |
| BAFFR-targeting CAR T Cells | BAFFR | I | NCT06191887 | 26, R/R BAFFR-Expressing B-Cell Hematologic Malignancies |
| CARCIK-CD19 | CD19 (Allogenic) | I/II | NCT05869279 | 29, B-cell NHL, CLL |
| ALLO-501A, ALLO-647, ALPHA2 study | CD19 (Allogenic) | I/II | NCT04416984 | 160, Large B Cell Lymphoma, CLL, SLL |
| SC291-101, ARDENT study | CD19 (Allogenic) | I | NCT05878184 | 16, NHL, CLL |
| UB-VV111 | CD19 (in vivo) | I | NCT06528301 | 106, R/R LBCL, CLL |
| FT819 | CD19 | I | NCT04629729 | 54, R/R B-cell Lymphoma, CLL and Precursor B-cell ALL |
| Double target CAR T-cell | ||||
| ATLCAR.κ.28 | Kappa-CD28 | I | NCT04223765 | 20, R/R κ+ mantle cell and indolent NHL |
| CD19/CD22 Bicistronic CAR | CD19/CD22 | I/II | NCT05442515 | 116, ALL or related B-cell lymphoma |
| CD19/CD22 CAR | CD19/CD22 | I/II | NCT03614858 | 20, R/R B-Cell ALL. |
| CD19/CD22 CA | CD19/CD22 | I | NCT03448393 | 44, CD19+CD22+ B cell ALL, isolated CNS ALL, or lymphoma |
| CAR2219 CAR | CD19/CD22 | I/II | NCT06834529 | 20, R/R CD19/CD22 positive B cell Leukemia and Lymphoma |
| IL-7/IL-15 manufactured CD20/19 CAR | CD-20/19 | I/II | NCT04186520 | 100, R/R B Cell Malignancies |
| CD19-BAFF CAR | CD19-BAFF | I | NCT06346912 | 20, data B-cell ALL and B-cell NHL |
| Triple target CAR T-cell | ||||
| CD19/CD20/CD22 CAR | CD19/CD20/CD22 | I | NCT05418088 | 54, R/R NHL, ALL, CLL, B-PLL |
| Other Cellular Therapies | ||||
| IOV-2001 (Adoptive Cell Therapy) | autologous PBL | I/II | NCT04155710 | 7, R/R CLL/SLL |
| CHARKALL (Adoptive Transfer of Autologous T Lymphocytes) | Kappa-CD28 T-cells | I | NCT00881920 | 54, CLL, B cell NHL, MM who express Kappa-light chain |
| ATECRAB | autologous or syngeneic PBTLs and EBV-CTLs expressing CD19 | I | NCT00709033 | 3, B cell NHL, CLL |
| ANCHOR2 | KUR-502, Allogeneic NK T-Cells Expressing CD19 | I | NCT05487651 | 36, R/R B-cell NHL, ALL, CLL |
| NKX019 | NK (Intravenous allogenic) | I | NCT05020678 | 150, B-cell Malignancies |
| ANCHOR | Allogenic CD19- NK | I | NCT03774654 | 48, R/R B-cell Malignancies |
| Allogenic CD19-CAR-NK | Allogenic CD19- NK | I | NCT05739227 | 12, R/R B-cell Malignancies |
| NK cells combination with IL-2 and vactosertib | IL-2 and TGFβ receptor 1 NK | I | NCT05400122 | 12, Colorectal Cancer, Gastric/esophageal Cancer, and R/R Hematologic Malignancies |
| Agent Name | Engaging Targets | Study Design/Phase | Clinicaltrial.gov Identifier | Population, Sample Size |
|---|---|---|---|---|
| Bispecific T-cell Engagers | ||||
| NVG-111 | ROR1 ×CD3 | I | NCT04763083 | 90, R/R ROR1+ Malignancies- CLL, SLL, MCL, FL, DLBCL, NSCLC, malignant melanoma |
| ONO-4685 | PD-1 × CD3 | I | NCT06547528 | 108 T cell Lymphoma, CLL/SLL |
| JNJ-75348780 | CD3 × CD22 | I | NCT04540796 | 147, R/R B-cell NHL, CLL |
| MGD024 | CD123 × CD3 | I | NCT05362773 | 130, R/R hematological malignancies |
| Trispecific T-cell engagers | ||||
| TITANium, AZD5492 | CD8/TCR × CD20 | I/II | NCT06542250 | 174, R/R B-Cell Malignancies |
| CC312 | CD19 × CD3 × CD28 | I | NCT06037018 | 44, R/R CD19 Positive B-cell Hematologic Malignancies |
| Tetraspecific T-cell Engagers | ||||
| GNC-035 | CD3 × 41BB × PD-L1 × ROR | I/II | NCT05944978 | 40, R/R CLL, other hematological malignancies |
| Agent Name | Targets | Study Design/Phase | Clinicaltrial.gov Identifier | Population, Sample Size |
|---|---|---|---|---|
| Belimumab (BeliVeR) | BAFF | II | NCT05069051 | 120, R/R CLL |
| MOR00208 | CD19 | II | NCT02005289 | 41, R/R or Old untreated CLL, SLL or PLL |
| CAP-100 | Humanized C-C-chemokine Receptor 7 | I | NCT04704323 | 18, R/R CLL |
| Aplitabart (IGM-8444) | IgM DR5 | I | NCT04553692 | 272, R/R solid or hematologic cancers |
| Zilovertamab Vedotin (MK-2140), (waveLINE-006) | Antibody-drug conjugate: ROR1 and monomethyl auristatin E | II | NCT05458297 | 223, MCL, RTL, FL, CLL |
| Agent Name | Targets | Study Design/Phase | Clinicaltrial.gov Identifier | Population, Sample Size | Efficacy (ORR, PFS, OS, Median Follow-Up |
|---|---|---|---|---|---|
| Sonrotoclax | BCL-2 inhibitor | III | NCT04277637 | 46 RR CLL/SLL | Median follow up 19.3 months Sontroclax + zanabrutinib ORR 97% (CR 57%) for all doses, 100% ORR (CR 73%) for 320 mg dose [93] |
| I/II | NCT04277637 | 112 TN CLL/SLL | Median follow up 18.3 months ORR 100% Best uMRD 90% [94] | ||
| Lisaftoclax (APG-2575) | BCL-2 inhibitor | I | NCT04215809 | 176, 22 TN and 154 R/R CLL/SLL | ORR- lisaftoclax plus acalabrutinib—96.6% ORR—85.7%, 100%, and 66.7% in the ven-exposed, ven-exposed but BTKi-naïve, and ven- and BTKi-exposed pts, respectively [95]. |
| Nemtabrutinib | BTK (non-covalent) | I/II | NCT03162536 | 112 | ORR 56% (42–69) mPFS 26.3 months ORR in double exposed 58% (37–78) mPFS in double exposed 10.1 months [96] |
| Orelabrutinib | Irreversible BTK | II | NCT03493217 | 80, R/R CLL/SLL | ORR 93.8% (86.01–97.94) CR 23.8% [97] |
| LP-168 | Dual covalent + non-covalent BTK | I | NCT04775745 | R/R B-cell malignancies | ORR 54.5% Median follow up 12.6 months In Gatekeeper mutation CLL patients ORR 77.8%, median follow up 14 months [98,99] |
| NX-5948 | BTK Degrader | I | NCT05131022 | 87, B-cell malignancies, 34 CLL | ORR 76.7% [100] |
| BGB-16673 | BTK Degrader | I/II | NCT05006716 | 49, R/R CLL, WM, MCL, MZL, DLBCL, FL, or RT | ORR 77.6% [101] |
| NX-2127 | BTK + Ikaros/Aiolos degrader | I | NCT04830137 | 47, R/R B-cell malignancies | Median follow up 9.5 months [102]. ORR- not reported |
| Agent Name | Targets | Study Design/Phase | Clinicaltrial.gov Identifier | Population, Sample Size |
|---|---|---|---|---|
| ACY-1215 (Ricolinostat) | Histone deacetylase inhibitor, HDAC6 | I | NCT02787369 | 3, R/R CLL |
| AZD6738 (Ceralasertib) | ATR kinase | I | NCT03328273 | 11, R/R High-risk CLL |
| DZD8586 (TAI-SHAN8) | LYN/BTK | II | NCT06539182 | 155, R/R CLL |
| ONO-7018 | MALT1 | I | NCT05515406 | 108, R/R NHL or CLL |
| JNJ-67856633 | MALT1 | I | NCT04876092 | 45, B-cell NHL, CLL |
| JNJ-67856633 | MALT1 | I | NCT03900598 | 266, R/R B-cell NHL, CLL |
| AUR104 (VIJAY-1) | FABP5 | I | NCT06761586 | 42, R/R NHL or CLL |
| BMF-219 | Covalent Menin | I | NCT05153330 | 55, AML, ALL (With KMT2A/ MLL1r, NPM1 Mutations), DLBCL, MM, and CLL/SLL |
| IDP-121 (CASSANDRA) | c-MYC | I/II | NCT05908409 | 37, MM, DLBCL-NOS, HGBL-DH/TH, HGBL-NOS, CLL |
| MLN4924, TAK924 (Pevonedistat) | Nedd8-activating enzyme E1 regulatory subunit (NAE1) | I | NCT03479268 | 18, R/R CLL, NHL |
| SLS009 | CDK9 | I/II | NCT04588922 | 160, R/R AML, lymphoma/CLL/SLL |
| Voruciclib | CDK9 | I | NCT03547115 | 100, R/R B-Cell Malignancies or AML |
| PRT2527 | CDK9 | I | NCT05665530 | 86, R/R Hematologic Malignancies |
| Keynatinib (TL007) | EGFR | I | NCT04807881 | 75, R/R-PCNSL, CLL/SLL, MCL |
| Q702 | Axl/Mer/CSF1R Triple Kinase | I | NCT06712810 | Estimated enrollment 46, Hematological malignancies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayyar, M.; Menezes, R.C.B.d.; Ailawadhi, S.; Parrondo, R.D. Chronic Lymphocytic Leukemia: Novel Therapeutic Targets Under Investigation. Cancers 2025, 17, 2298. https://doi.org/10.3390/cancers17142298
Nayyar M, Menezes RCBd, Ailawadhi S, Parrondo RD. Chronic Lymphocytic Leukemia: Novel Therapeutic Targets Under Investigation. Cancers. 2025; 17(14):2298. https://doi.org/10.3390/cancers17142298
Chicago/Turabian StyleNayyar, Madhavi, Ricardo C. B. de Menezes, Sikander Ailawadhi, and Ricardo D. Parrondo. 2025. "Chronic Lymphocytic Leukemia: Novel Therapeutic Targets Under Investigation" Cancers 17, no. 14: 2298. https://doi.org/10.3390/cancers17142298
APA StyleNayyar, M., Menezes, R. C. B. d., Ailawadhi, S., & Parrondo, R. D. (2025). Chronic Lymphocytic Leukemia: Novel Therapeutic Targets Under Investigation. Cancers, 17(14), 2298. https://doi.org/10.3390/cancers17142298