
Translational Insights into Interferon Alpha’s Effects on Immunomolecular Dynamics in Philadelphia-Negative Myeloproliferative Neoplasms


 , and
, and 
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Patients and Study Design
2.2. Plasma Cytokine Profiling
2.3. RT-qPCR Primer Design and Validation
2.4. Gene Expression Analysis in Peripheral Blood Mononuclear Cells (PBMCs)
2.5. Correlation Analysis Between Gene Expression and Cytokines
2.6. Statistical Analysis
2.7. Ethical Statement
3. Results
3.1. Baseline Characteristics of the Cohort
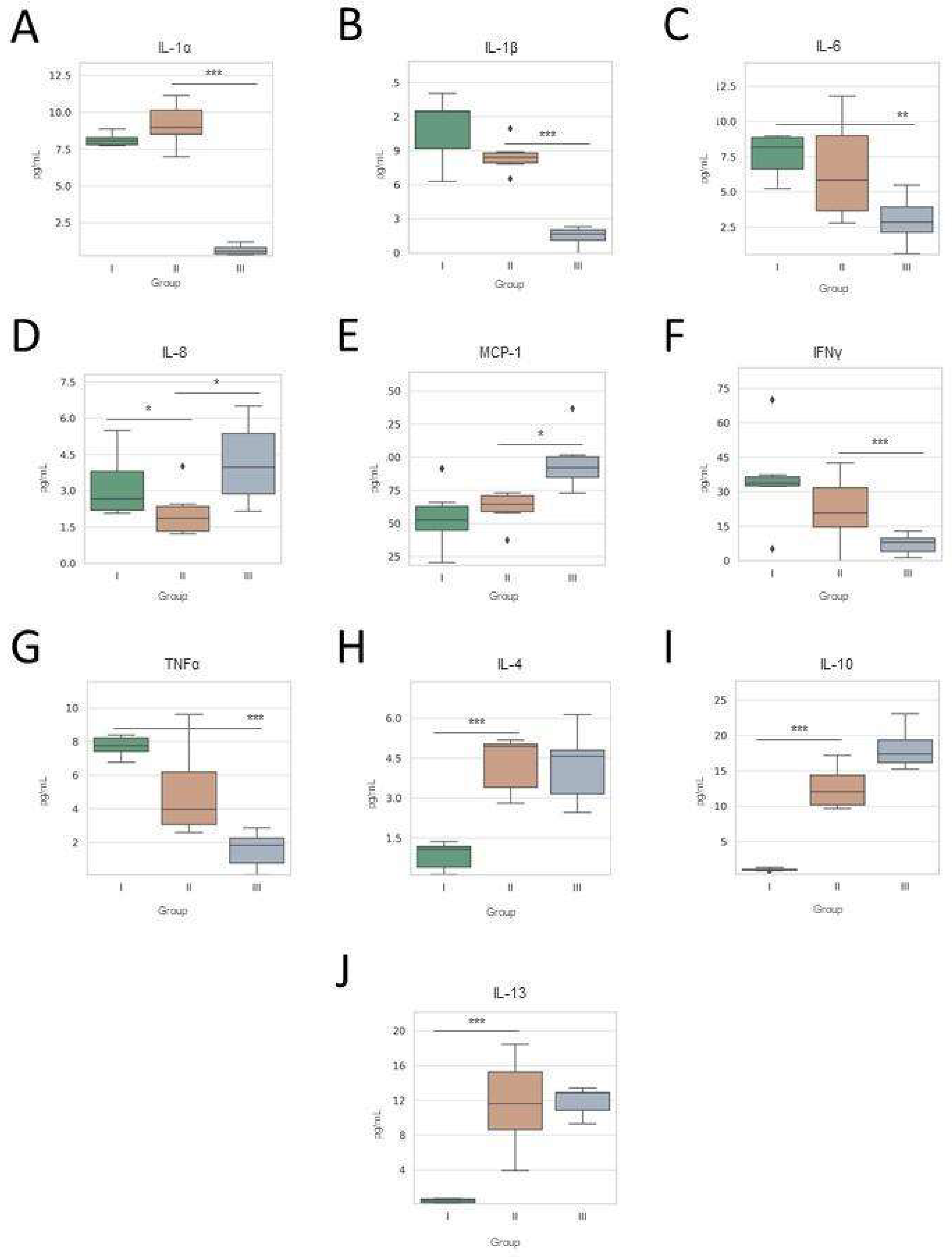
3.2. Plasma Cytokine Profiling Across Treatment Time Points
3.3. Gene Expression Profiles Across IFNα Exposure Groups
3.4. Correlations Between Gene Expression and Cytokine Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukaemia |
| CALR | Calreticulin |
| cDNA | Complementary DNA |
| CHIP | Clonal haematopoiesis of Indeterminate Potential |
| CML | Chronic myeloid leukaemia |
| CTLA-4 | Cytotoxic T Lymphocyte Associated Protein 4 |
| EBV | Epstein-Barr virus |
| ET | Essential thrombocythemia |
| HCMV | Cytomegalovirus |
| HSC | Haematopoietic stem cell |
| IFNγ | Interferon-gamma |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| JAK2 | Janus Tyrosine Kinase 2 |
| MCP-1 | Monocyte Chemoattractant protein-1 |
| MDS | Myelodysplastic syndromes |
| MPNs | Myeloproliferative neoplasms |
| NK | Natural killers |
| PBMC | Peripheral blood mononuclear cell |
| PD-1 | Programmed Death 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| Ph-neg MPNs | Philadelphia-negative myeloproliferative neoplasms |
| PMF | Primary myelofibrosis |
| PV | Polycythaemia vera |
| qPCR | Quantitative real-time PCR |
| TNF-α | Tumour necrosis factor-alpha |
| STAT | Signal Transducer and Activator of Transcription |
| VZV | Varicella Zoster Virus |
References
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 1599–1613. [Google Scholar] [CrossRef]
- Strickland, M.; Quek, L.; Psaila, B. The immune landscape in BCR-ABL negative myeloproliferative neoplasms: Inflammation, infections and opportunities for immunotherapy. Br. J. Haematol. 2021, 196, 1149–1158. [Google Scholar] [CrossRef]
- Sukari, A.; Nagasaka, M.; Al-Hadidi, A.; Lum, L.G. Cancer Immunology and Immunotherapy. Anticancer Res. 2016, 36, 5593–5606. [Google Scholar] [CrossRef]
- Camacho, V.; Kuznetsova, V.; Welner, R.S. Inflammatory Cytokines Shape an Altered Immune Response During Myeloid Malignancies. Front. Immunol. 2021, 12, 772408. [Google Scholar] [CrossRef]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Understanding Society Scientific Group; Crawley, C.; Craig, J.; Scott, M.A.; et al. Leukemia-Associated Somatic Mutations Drive Distinct Patterns of Age-Related Clonal Hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef]
- Pasquier, F.; Pegliasco, J.; Martin, J.-E.; Marti, S.; Plo, I. New approaches to standard of care in early-phase myeloproliferative neoplasms: Can interferon-α alter the natural history of the disease? Haematologica 2024, 110, 850–862. [Google Scholar] [CrossRef]
- Ianotto, J.-C.; Chauveau, A.; Boyer-Perrard, F.; Gyan, E.; Laribi, K.; Cony-Makhoul, P.; Demory, J.-L.; de Renzis, B.; Dosquet, C.; Rey, J.; et al. Benefits and pitfalls of pegylated interferon-α2a therapy in patients with myeloproliferative neoplasm-associated myelofibrosis: A French Intergroup of Myeloproliferative neoplasms (FIM) study. Haematologica 2017, 103, 438–446. [Google Scholar] [CrossRef]
- Alves, R.; McArdle, S.E.B.; Vadakekolathu, J.; Gonçalves, A.C.; Freitas-Tavares, P.; Pereira, A.; Almeida, A.M.; Sarmento-Ribeiro, A.B.; Rutella, S. Flow cytometry and targeted immune transcriptomics identify distinct profiles in patients with chronic myeloid leukemia receiving tyrosine kinase inhibitors with or without interferon-α. J. Transl. Med. 2020, 18, 2. [Google Scholar] [CrossRef]
- Vachhani, P.; Mascarenhas, J.; Bose, P.; Hobbs, G.; Yacoub, A.; Palmer, J.M.; Gerds, A.T.; Masarova, L.; Kuykendall, A.T.; Rampal, R.K.; et al. Interferons in the treatment of myeloproliferative neoplasms. Ther. Adv. Hematol. 2024, 15. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Cassinat, B.; Chevret, S.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Grandchamp, B.; Chomienne, C.; Fenaux, P. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008, 112, 3065–3072. [Google Scholar] [CrossRef]
- Huang, C.-E.; Wu, Y.-Y.; Hsu, C.-C.; Chen, Y.-J.; Tsou, H.-Y.; Li, C.-P.; Lai, Y.-H.; Lu, C.-H.; Chen, P.-T.; Chen, C.-C. Real-world experience with Ropeginterferon-alpha 2b (Besremi) in Philadelphia-negative myeloproliferative neoplasms. J. Formos. Med. Assoc. 2021, 120, 863–873. [Google Scholar] [CrossRef]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef]
- Melikyan, A.L.; Subortseva, I.N.; Gilyazitdinova, E.A.; Koloshejnova, T.I.; Pustovaya, E.I.; Egorova, E.K.; Kovrigina, A.M.; Sudarikov, A.B.; Abdullaev, A.O.; Lomaia, E.G.; et al. Cepeginterferon alfa-2b in the treatment of chronic myeloproliferative diseases. Ter. Arkh. 2018, 90, 23–29. [Google Scholar] [CrossRef]
- Verger, E.; Cassinat, B.; Chauveau, A.; Dosquet, C.; Giraudier, S.; Schlageter, M.-H.; Ianotto, J.-C.; Yassin, M.A.; Al-Dewik, N.; Carillo, S.; et al. Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood 2015, 126, 2585–2591. [Google Scholar] [CrossRef]
- How, J.; Garcia, J.S.; Mullally, A. Biology and therapeutic targeting of molecular mechanisms in MPNs. Blood 2023, 141, 1922–1933. [Google Scholar] [CrossRef]
- Melo-Cardenas, J.; Bezavada, L.; Crawford, J.C.; Gurbuxani, S.; Cotton, A.; Kang, G.; Gossett, J.M.; Marinaccio, C.; Weinberg, R.S.; Hoffman, R.; et al. IL-13/IL-4 signaling contributes to fibrotic progression of the myeloproliferative neoplasms. Blood 2022, 140, 2805–2817. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, H.; Bai, J.; Li, Y.; Li, L.; Zhang, L. Associations of the circulating levels of cytokines with the risk of myeloproliferative neoplasms: A bidirectional mendelian-randomization study. BMC Cancer 2024, 24, 531. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation ofJAK2in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef]
- Li, Y.; Sun, T.; Chen, J.; Zhang, L. Identification of Novel Risk Variants of Inflammatory Factors Related to Myeloproliferative Neoplasm: A Bidirectional Mendelian Randomization Study. Glob. Med Genet. 2024, 11, 48–58. [Google Scholar] [CrossRef]
- Jewell, A.P.; Worman, C.P.; Lydyard, P.M.; Yong, K.L.; Giles, F.J.; Goldstone, A.H. Interferon-alpha up-regulates bcl-2 expression and protects B-CLL cells from apoptosis in vitro and in vivo. Br. J. Haematol. 1994, 88, 268–274. [Google Scholar] [CrossRef]
- Shi, W.; Yao, X.; Fu, Y.; Wang, Y. Interferon-α and its effects on cancer cell apoptosis (Review). Oncol. Lett. 2022, 24, 235. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master Switch from Tumor-Promoting Inflammation to Antitumor Immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Holmström, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: Minimal residual disease and cure? Semin. Immunopathol. 2018, 41, 5–19. [Google Scholar] [CrossRef]
- Tassiulas, I.; Hu, X.; Ho, H.; Kashyap, Y.; Paik, P.; Hu, Y.; Lowell, C.A.; Ivashkiv, L.B. Amplification of IFN-α-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nat. Immunol. 2004, 5, 1181–1189. [Google Scholar] [CrossRef]
- Kalmer, M.M.; Feldberg, K.; Gezer, D.; Isfort, S.; Brümmendorf, T.H.; Chatain, N.; Koschmieder, S. STAT1 Transcriptional Response Predicts Molecular Responses of PB-Derived Clonogenic Cells from MPN Patients to Interferon Alpha. Blood 2019, 134, 1679. [Google Scholar] [CrossRef]
- Convertino, I.; Tuccori, M.; Ferraro, S.; Valdiserra, G.; Cappello, E.; Focosi, D.; Blandizzi, C. Exploring pharmacological approaches for managing cytokine storm associated with pneumonia and acute respiratory distress syndrome in COVID-19 patients. Crit. Care 2020, 24, 331. [Google Scholar] [CrossRef]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Reddy, E.P. JAK/STAT Pathways in Cytokine Signaling and Myeloproliferative Disorders: Approaches for Targeted Therapies. Genes Cancer 2010, 1, 979–993. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Samad, A.; Ahmad, I.; Hasan, A.; Alhashmi, M.H.; Ayub, A.; Al-Abbasi, F.A.; Kumer, A.; Tabrez, S. STAT3 Signaling Pathway in Health and Disease. Medcomm 2025, 6, e70152. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, Z.J.; Shenoy, A.; Chen, A.; Heller, N.M.; Spangler, J.B. Engineering the IL-4/IL-13 axis for targeted immune modulation. Immunol. Rev. 2023, 320, 29–57. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Cervantes, F.; Mesa, R.; Passamonti, F.; Verstovsek, S.; Vannucchi, A.M.; Gotlib, J.; Dupriez, B.; Pardanani, A.; Harrison, C.; et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood 2013, 122, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Teng, G.; Zhang, Y.; Wang, Y.; Du, C.; Li, Y.; Hu, N.; Xiang, G.; Shao, Z.; Bai, J. Efficacy and safety of pegylated interferon alpha-2b for patients with myeloproliferative neoplasm. Chin. Med. J. 2023, 103, 3645–3651. [Google Scholar] [CrossRef]
- Dunbar, A.; Kosiorek, H.E.; Krishnan, A.; McGovern, E.; Park, Y.; Patel, M.A.; Weinberg, R.S.; Yacoub, A.; Dueck, A.C.; Mascarenhas, J.; et al. Identifying Cytokine Biomarkers of Response to Pegylated-Interferon Therapy in Polycythemia Vera and Essential Thrombocythemia: Correlative Analysis from the MPN-RC 111/112 Trials. Blood 2019, 134 (Suppl. S1), 1661. [Google Scholar] [CrossRef]
- Saleiro, D.; Wen, J.Q.; Kosciuczuk, E.M.; Eckerdt, F.; Beauchamp, E.M.; Oku, C.V.; Blyth, G.T.; Fischietti, M.; Ilut, L.; Colamonici, M.; et al. Discovery of a signaling feedback circuit that defines interferon responses in myeloproliferative neoplasms. Nat. Commun. 2022, 13, 1750. [Google Scholar] [CrossRef]
- Skov, V.; Riley, C.; Thomassen, M.; Kjær, L.; Larsen, T.S.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. The Impact of Interferon on Interferon-Related Genes in Polycythemia Vera and Allied Neoplasms. Blood 2018, 132, 4328. [Google Scholar] [CrossRef]
- Zhan, H.; Yu, W.; Xu, H.; Rogers, O.; Moliterno, A.; Dang, C.; Spivak, J.L. Differential Regulation of MicroRNA Expression in Polycythemia Vera CD34+ Cells. Blood 2010, 116, 4785. [Google Scholar] [CrossRef]
- Sundar, K.; Song, J.; Kim, S.J.; Shah, B.; Workman, K.; Smith, B.; Brooks, E.; Matinkhah, G.; Tantravahi, S.K.; Tashi, T.; et al. Exploratory Study on Impact of Comorbid Sleep Apnea Treatment in Patients with Polycythemia Vera and Essential Thrombocythemia. Blood 2023, 142, 6414. [Google Scholar] [CrossRef]
- He, F.; Laranjeira, A.B.; Kong, T.; Lin, S.; Ashworth, K.J.; Liu, A.; Lasky, N.M.; Fisher, D.A.; Cox, M.J.; Fulbright, M.C.; et al. Multiomic profiling reveals metabolic alterations mediating aberrant platelet activity and inflammation in myeloproliferative neoplasms. J. Clin. Investig. 2024, 134, e172256. [Google Scholar] [CrossRef]
- Antonia, R.J.; Karelehto, E.; Toriguchi, K.; Matli, M.; Warren, R.S.; Pfeffer, L.M.; Donner, D.B. STAT3 regulates inflammatory cytokine production downstream of TNFR1 by inducing expression of TNFAIP3/A20. J. Cell. Mol. Med. 2022, 26, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. IL-13 Effector Functions. Annu. Rev. Immunol. 2003, 21, 425–456. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Ph-Neg MPN Patients (n = 44) |
|---|---|
| Number of patients | 44 |
| Age (years), median (range) | 67 (43–81) |
| Sex (Male/Female) | 27/17 |
| Diagnosis | |
| 22 |
| 16 |
| 6 |
| Driver mutation | |
| 34 (77.3%) |
| 5 (11.4%) |
| 2 (4.5%) |
| 3 (6.8%) |
| Haemoglobin (g/dL), mean (range) | 12.3 (9.6–15.1) |
| Platelet count (×109/L), mean (range) | 426 (130–870) |
| Leukocyte count (×109/L), mean (range) | 9.4 (3.2–17.8) |
| EBV, HCMV or VZV reactivation | 5 (11.4%) |
| Prior cytoreductive therapy | 6 (13.6%) |
| IFNα treatment duration (months), median (range) | 8.2 (1–36) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Delgado, R.; Luque-Lupiáñez, E.; Mora-Infante, D.; Ortíz-Flores, R.M.; Cidoncha-Morcillo, B.; Torres-González, J.; Fontalba-Navas, A.; Escamilla-Sánchez, A. Translational Insights into Interferon Alpha’s Effects on Immunomolecular Dynamics in Philadelphia-Negative Myeloproliferative Neoplasms. Cancers 2025, 17, 2273. https://doi.org/10.3390/cancers17142273
García-Delgado R, Luque-Lupiáñez E, Mora-Infante D, Ortíz-Flores RM, Cidoncha-Morcillo B, Torres-González J, Fontalba-Navas A, Escamilla-Sánchez A. Translational Insights into Interferon Alpha’s Effects on Immunomolecular Dynamics in Philadelphia-Negative Myeloproliferative Neoplasms. Cancers. 2025; 17(14):2273. https://doi.org/10.3390/cancers17142273
Chicago/Turabian StyleGarcía-Delgado, Regina, Elena Luque-Lupiáñez, David Mora-Infante, Rodolfo Matías Ortíz-Flores, Borja Cidoncha-Morcillo, Julio Torres-González, Andrés Fontalba-Navas, and Alejandro Escamilla-Sánchez. 2025. "Translational Insights into Interferon Alpha’s Effects on Immunomolecular Dynamics in Philadelphia-Negative Myeloproliferative Neoplasms" Cancers 17, no. 14: 2273. https://doi.org/10.3390/cancers17142273
APA StyleGarcía-Delgado, R., Luque-Lupiáñez, E., Mora-Infante, D., Ortíz-Flores, R. M., Cidoncha-Morcillo, B., Torres-González, J., Fontalba-Navas, A., & Escamilla-Sánchez, A. (2025). Translational Insights into Interferon Alpha’s Effects on Immunomolecular Dynamics in Philadelphia-Negative Myeloproliferative Neoplasms. Cancers, 17(14), 2273. https://doi.org/10.3390/cancers17142273