
The Role of Organoids in Advancing Colorectal Cancer Research: Insights and Future Directions


Simple Summary
Abstract
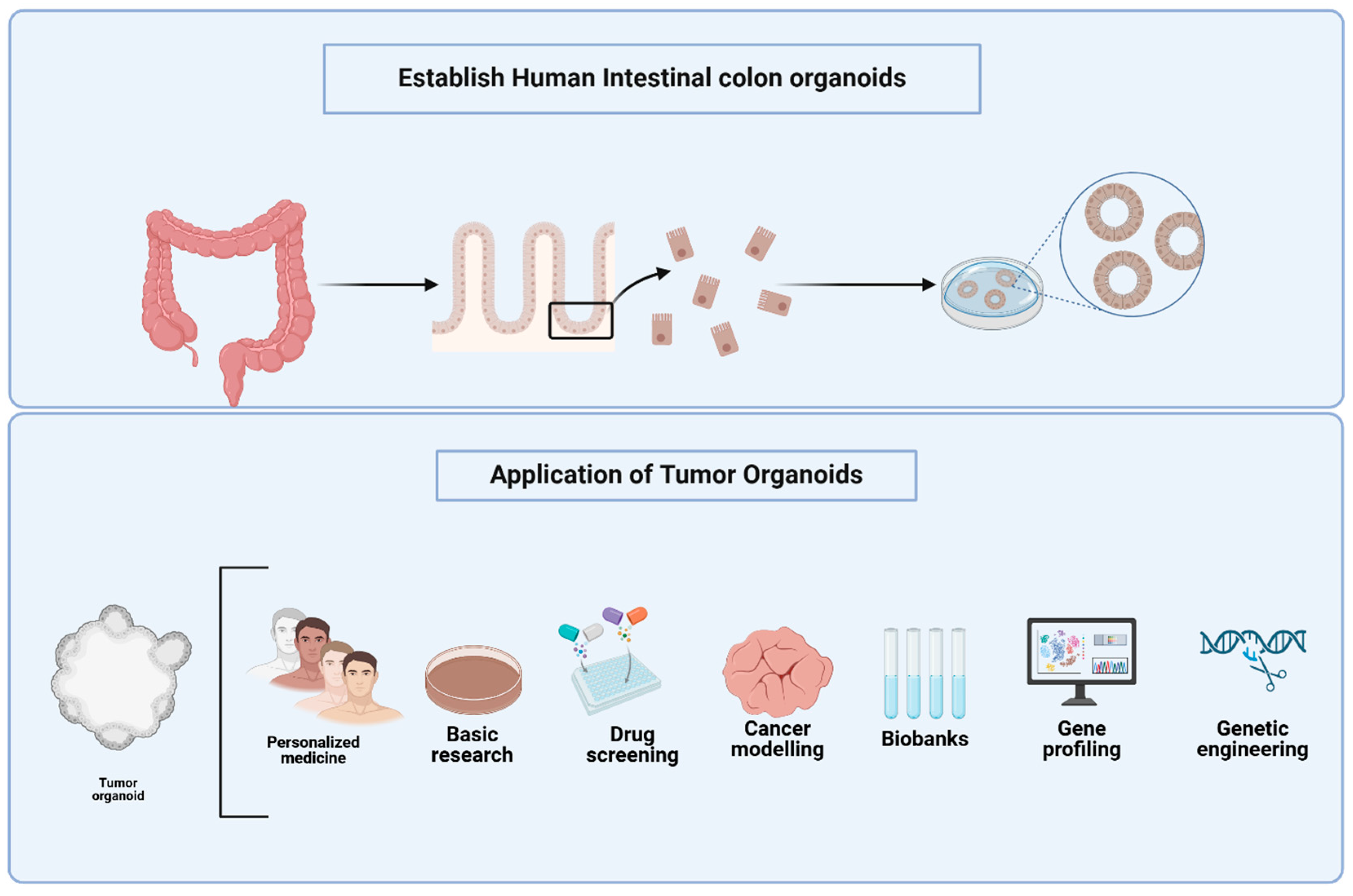
1. Introduction
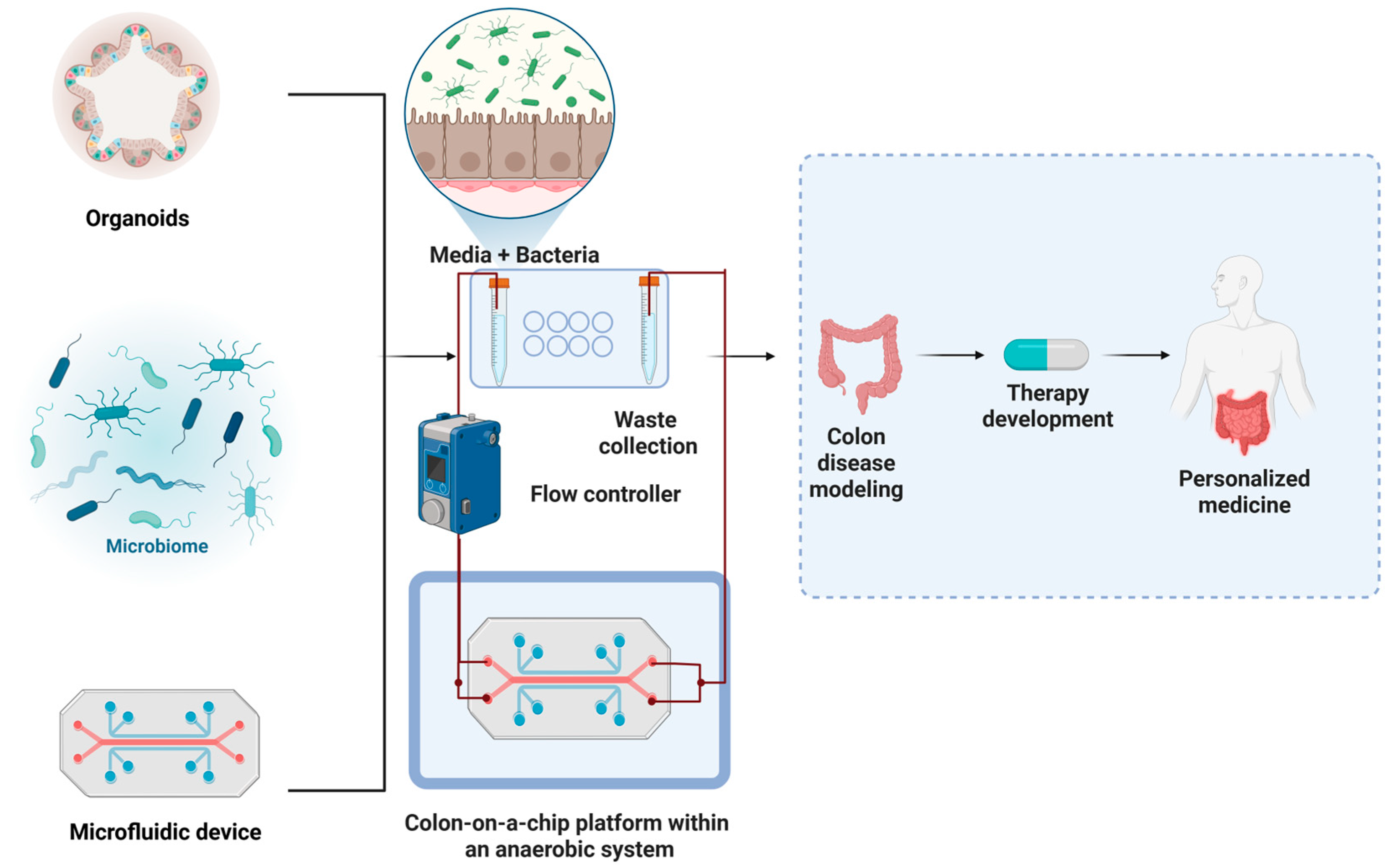
2. Organoid Platforms to Study Host–Microbiota Crosstalk in CRC
3. Advancements in CRC Organoid Models for Tumor Heterogeneity, Drug Screening, and Co-Culture Systems
4. Microfluidic Platforms for Modeling the Tumor Microenvironment and Drug Responses
5. Future Directions and Implications
- -
- Development of specialized CRC-specific organoid models that would include normal colon and polyps from cancer-free individuals and CRC patients. Such organoid models will enable researchers to study the progression from benign lesions to malignant tumors, facilitating the identification of early biomarkers for CRC detection and intervention [25].
- -
- Establishment of reproducible long-term cultured CRC organoids. This will shed light on tumor evolution and resistance mechanisms and will also help identify genetic markers associated with therapy response or resistance that facilitate the development of more effective personalized therapies [109]. The utilization of CRC organoids to study drug resistance patterns can reveal critical insights into why certain patients do not respond to standard therapies. This knowledge could guide the development of combination therapies or novel agents targeting resistant phenotypes.
- -
- Creation of composite organoids that incorporate various CRC-relevant cell types, including immune cells and stromal components. This will help mimic the TME more accurately, enhancing our understanding of tumor-immune interactions and informing immunotherapy strategies. However, implementing these advanced models presents several challenges. Technically, co-culturing multiple cell types requires careful optimization of media conditions, timing of cell addition, and maintenance protocols to ensure viability and physiological relevance. Moreover, inter-laboratory variability and lack of standardized protocols can limit reproducibility and scalability. To address these barriers, future efforts should focus on the development of standardized co-culture methodologies and shared organoid biobanks. Training programs and collaborative platforms can also help disseminate technical expertise. Integration of automation (e.g., microfluidics, bioprinting) and advanced imaging technologies may reduce manual labor and improve consistency [110,111]. These advancements will be crucial for translating composite organoid models into robust platforms for studying CRC biology and guiding immunotherapy development.
- -
- Development of organoid-based models to assess the impact of environmental factors, known as exposomes, on CRC, which can provide insights into how diet, microbiota, and other external factors influence cancer development. This approach could lead to preventive strategies tailored to individual risk profiles.
- -
- Advancements, like automated bioprinting, machine learning integration and rapid data acquisition, in high-throughput screening modalities utilizing organoids will allow for rapid testing of drug efficacy across diverse patient-derived models. This will streamline the process of identifying effective treatments tailored to individual genetic backgrounds.
- -
- Technical innovation will need to be made to rapidly integrate multi-omics data—genomics, transcriptomics, proteomics—from organoid cultures. This will yield an inclusive understanding of CRC heterogeneity and treatment responses.
6. Potential Limitations
- -
- Present-day CRC organoid systems still cannot accurately replicate the intricate interactions and microenvironments of human organs.
- -
- The absence of vascularization within organoids limits their ability to support sustained growth and mimic certain physiological processes. Traditional organoids lack intrinsic vascularization, which limits their ability to support sustained growth, model angiogenesis, or replicate physiological nutrient and oxygen gradients. However, emerging microfluidic technologies and vascularized organoid platforms, such as tumor-on-a-chip models, have begun to address this limitation by incorporating perfused microvessels and endothelial networks. While promising, these systems are still under development and not yet standard in most organoid-based studies.
- -
- Issues related to variability and stability during long-term culture pose challenges to reproducibility and reliability in experimental studies.
- -
- Difficulty of scaling organoid production, which restricts their application in large-scale research or clinical settings.
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Yang, T.; Li, X.; Montazeri, Z.; Little, J.; Farrington, S.M.; Ioannidis, J.P.; Dunlop, M.G.; Campbell, H.; Timofeeva, M.; Theodoratou, E. Gene–environment interactions and colorectal cancer risk: An umbrella review of systematic reviews and meta-analyses of observational studies. Int. J. Cancer 2019, 145, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.; Iqbal, S.; Pereira, K.; Thirumaran, R. P-185 Global and regional trends in the incidence and mortality of Colorectal Cancer (CRC): Analysis of data from the GLOBOCAN database and SEER database. Ann. Oncol. 2023, 34, S81–S82. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Brusco, L.; Shaw, K.; Horombe, C.; Kopetz, S.; Davies, M.A.; Routbort, M.; Piha-Paul, S.A.; Janku, F.; Ueno, N. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J. Clin. Oncol. 2015, 33, 2753. [Google Scholar] [CrossRef]
- Luo, L.; Ma, Y.; Zheng, Y.; Su, J.; Huang, G. Application progress of organoids in colorectal cancer. Front. Cell Dev. Biol. 2022, 10, 815067. [Google Scholar] [CrossRef]
- Karim, B.O.; Huso, D.L. Mouse models for colorectal cancer. Am. J. Cancer Res. 2013, 3, 240. [Google Scholar]
- Courau, T.; Bonnereau, J.; Chicoteau, J.; Bottois, H.; Remark, R.; Assante Miranda, L.; Toubert, A.; Blery, M.; Aparicio, T.; Allez, M. Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J. Immunother. Cancer 2019, 7, 1–14. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Xu, H.; Jiao, Y.; Qin, S.; Zhao, W.; Chu, Q.; Wu, K. Organoid technology in disease modelling, drug development, personalized treatment and regeneration medicine. Exp. Hematol. Oncol. 2018, 7, 1–12. [Google Scholar] [CrossRef]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid technology and applications in cancer research. J. Hematol. Oncol. 2018, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Morra, L.; Schlenker, R.; Sulcova, J.; Fahrni, L.; Waldhauer, I.; Lehmann, S.; Reisländer, T.; Agarkova, I.; Kelm, J.M. A novel three-dimensional heterotypic spheroid model for the assessment of the activity of cancer immunotherapy agents. Cancer Immunol. Immunother. 2017, 66, 129–140. [Google Scholar] [CrossRef]
- Dolznig, H.; Rupp, C.; Puri, C.; Haslinger, C.; Schweifer, N.; Wieser, E.; Kerjaschki, D.; Garin-Chesa, P. Modeling colon adenocarcinomas in vitro: A 3D co-culture system induces cancer-relevant pathways upon tumor cell and stromal fibroblast interaction. Am. J. Pathol. 2011, 179, 487–501. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.-L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, G.; Treacy, O.; Lynch, K.; Naicker, S.D.; Leonard, N.A.; Lohan, P.; Dunne, P.D.; Ritter, T.; Egan, L.J.; Ryan, A.E. Stromal cell PD-L1 inhibits CD8+ T-cell antitumor immune responses and promotes colon cancer. Cancer Immunol. Res. 2018, 6, 1426–1441. [Google Scholar] [CrossRef]
- Tauriello, D.V.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Son, G.M.; Kwon, M.-S.; Shin, D.-H.; Shin, N.; Ryu, D.; Kang, C.-D. Comparisons of cancer-associated fibroblasts in the intratumoral stroma and invasive front in colorectal cancer. Medicine 2019, 98, e15164. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: The road less traveled, changing anticancer therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 1–12. [Google Scholar] [CrossRef]
- Kawano, S.; Kojima, M.; Higuchi, Y.; Sugimoto, M.; Ikeda, K.; Sakuyama, N.; Takahashi, S.; Hayashi, R.; Ochiai, A.; Saito, N. Assessment of elasticity of colorectal cancer tissue, clinical utility, pathological and phenotypical relevance. Cancer Sci. 2015, 106, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wan, Z.; Kamm, R.D. Vascularized organoids on a chip: Strategies for engineering organoids with functional vasculature. Lab. A Chip 2021, 21, 473–488. [Google Scholar] [CrossRef]
- Sánchez-Salazar, M.G.; Crespo-López Oliver, R.; Ramos-Meizoso, S.; Jerezano-Flores, V.S.; Gallegos-Martínez, S.; Bolívar-Monsalve, E.J.; Ceballos-González, C.F.; Trujillo-de Santiago, G.; Álvarez, M.M. 3D-Printed Tumor-on-Chip for the Culture of Colorectal Cancer Microspheres: Mass Transport Characterization and Anti-Cancer Drug Assays. Bioengineering 2023, 10, 554. [Google Scholar] [CrossRef] [PubMed]
- Sakshaug, B.C.; Folkesson, E.; Haukaas, T.H.; Visnes, T.; Flobak, Å. Systematic review: Predictive value of organoids in colorectal cancer. Sci. Rep. 2023, 13, 18124. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, S.; Lin, X.; Chen, G.; Kang, J.; Ma, Z.; Wang, Y.; Li, Z.; Xiao, X.; He, A. Application of Organoids in Carcinogenesis Modeling and Tumor Vaccination. Front. Oncol. 2022, 12, 855996. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, R.; Sha, D.; Gao, H.; Wang, S.; Zhou, J.; Wang, X.; Li, F.; Li, X.; Song, W. Advances in the application of colorectal cancer organoids in precision medicine. Front. Oncol. 2024, 14, 1506606. [Google Scholar] [CrossRef]
- Wang, Q.; Yuan, F.; Zuo, X.; Li, M. Breakthroughs and challenges of organoid models for assessing cancer immunotherapy: A cutting-edge tool for advancing personalised treatments. Cell Death Discov. 2025, 11, 1–13. [Google Scholar] [CrossRef]
- Esposito, A.; Agostini, A.; Quero, G.; Piro, G.; Priori, L.; Caggiano, A.; Scaglione, G.; Battaglia, A.; Calegari, M.; Salvatore, L. Colorectal cancer patients-derived immunity-organoid platform unveils cancer-specific tissue markers associated with immunotherapy resistance. Cell Death Dis. 2024, 15, 1–12. [Google Scholar] [CrossRef]
- Wu, Y.; Sha, Y.; Guo, X.; Gao, L.; Huang, J.; Liu, S.-B. Organoid models: Applications and research advances in colorectal cancer. Front. Oncol. 2025, 15, 1432506. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Turkington, C.J.; Varadan, A.C.; Grenier, S.F.; Grasis, J.A. The viral Janus: Viruses as aetiological agents and treatment options in colorectal cancer. Front. Cell. Infect. Microbiol. 2021, 10, 601573. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Clevers, H. Gut microbiota in colorectal cancer: Associations, mechanisms, and clinical approaches. Annu. Rev. Cancer Biol. 2022, 6, 65–84. [Google Scholar] [CrossRef]
- Maisonneuve, C.; Irrazabal, T.; Martin, A.; Girardin, S.E.; Philpott, D.J. The impact of the gut microbiome on colorectal cancer. Annu. Rev. Cancer Biol. 2018, 2, 229–249. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Y.; Yang, L.; Wang, Z. Effect of gut microbiota in the colorectal cancer and potential target therapy. Discov. Oncol. 2022, 13, 51. [Google Scholar] [CrossRef]
- Bartfeld, S. Modeling infectious diseases and host-microbe interactions in gastrointestinal organoids. Dev. Biol. 2016, 420, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Miyamoto, K.; Maslowski, K.M.; Ohno, H.; Kanai, T.; Sato, T. Development of a scalable coculture system for gut anaerobes and human colon epithelium. Gastroenterology 2020, 159, 388–390.e385. [Google Scholar] [CrossRef]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef]
- Pignatelli, P.; Nuccio, F.; Piattelli, A.; Curia, M.C. The role of Fusobacterium nucleatum in oral and colorectal carcinogenesis. Microorganisms 2023, 11, 2358. [Google Scholar] [CrossRef]
- Barbáchano, A.; Fernández-Barral, A.; Bustamante-Madrid, P.; Prieto, I.; Rodríguez-Salas, N.; Larriba, M.J.; Muñoz, A. Organoids and colorectal cancer. Cancers 2021, 13, 2657. [Google Scholar] [CrossRef]
- Ahn, J.-S.; Kang, M.-J.; Seo, Y.; Kim, H.-S. Intestinal organoids as advanced modeling platforms to study the role of host-microbiome interaction in homeostasis and disease. BMB Rep. 2023, 56, 15. [Google Scholar] [CrossRef]
- Nalluri, H.; Subramanian, S.; Staley, C. Intestinal organoids: A model to study the role of microbiota in the colonic tumor microenvironment. Future Microbiol. 2020, 15, 1583–1594. [Google Scholar] [CrossRef]
- Kromann, E.H.; Cearra, A.P.; Neves, J.F. Organoids as a tool to study homeostatic and pathological immune–epithelial interactions in the gut. Clin. Exp. Immunol. 2024, 218, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Moskal, K.; Khurana, N.; Siegert, L.; Lee, Y.S.; Clevers, H.; Elinav, E.; Puschhof, J. Modeling cancer–microbiome interactions in vitro: A guide to co-culture platforms. Int. J. Cancer 2024, 156, 2053–2067. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.S.; Tocchi, A.; Holly, M.K.; Parks, W.C.; Smith, J.G. A small intestinal organoid model of non-invasive enteric pathogen–epithelial cell interactions. Mucosal Immunol. 2015, 8, 352–361. [Google Scholar] [CrossRef]
- Puschhof, J.; Pleguezuelos-Manzano, C.; Martinez-Silgado, A.; Akkerman, N.; Saftien, A.; Boot, C.; de Waal, A.; Beumer, J.; Dutta, D.; Heo, I. Intestinal organoid cocultures with microbes. Nat. Protoc. 2021, 16, 4633–4649. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, T.; Bakhshi, B.; Rasekhi, A.; Fazeli, M.S.; Fallah, F. Significant Presence of Clostridioides difficile in Colorectal Cancer Patients by TaqMan Real-Time PCR. Iran. J. Med. Microbiol. 2024, 18, 106–112. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Salesse, L.; Garcia-Weber, D.; Marinelli, L.; Beguet-Crespel, F.; Brochard, V.; Le Gléau, C.; Jamet, A.; Doré, J.; Blottière, H.M. Fusobacterium nucleatum promotes inflammatory and anti-apoptotic responses in colorectal cancer cells via ADP-heptose release and ALPK1/TIFA axis activation. Gut Microbes 2024, 16, 2295384. [Google Scholar] [CrossRef]
- Alzahabi, M.; Haddad, J.; Bishai, S.K. Streptococcus lutetiensis prosthetic shoulder infection assisting in the diagnosis of invasive adenocarcinoma of the colon. JSES Rev. Rep. Tech. 2024, 4, 559–562. [Google Scholar] [CrossRef]
- Sadeghi, M.; Mestivier, D.; Sobhani, I. Contribution of pks+ Escherichia coli (E. coli) to Colon Carcinogenesis. Microorganisms 2024, 12, 1111. [Google Scholar] [CrossRef]
- Liang, R.; Li, P.; Yang, N.; Xiao, X.; Gong, J.; Zhang, X.; Bai, Y.; Chen, Y.; Xie, Z.; Liao, Q. Parabacteroides distasonis-Derived Outer Membrane Vesicles Enhance Antitumor Immunity Against Colon Tumors by Modulating CXCL10 and CD8+ T Cells. Drug Des. Dev. Ther. 2024, 18, 1833–1853. [Google Scholar] [CrossRef]
- Shastry, R.P.; Ghate, S.D.; Hameed, A.; Rao, R.S.P.; Bhandary, Y.P.; Shetty, R. Emergence of rare and low abundant anaerobic gut Firmicutes is associated with a significant downfall of Klebsiella in human colon cancer. Microb. Pathog. 2024, 193, 106726. [Google Scholar] [CrossRef] [PubMed]
- Gobert, A.P.; Finley, J.; Asim, M.; Barry, D.P.; Allaman, M.M.; Hawkins, C.V.; Williams, K.J.; Delagado, A.G.; Mirmira, R.G.; Zhao, S. Analysis of the effect of hypusination in myeloid cells on colitis and colitis-associated cancer. Heliyon 2024, 10, e33838. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Tang, Z.; Yan, L.; Zhang, L.; Zheng, Q.; Zeng, X.; Hu, Q.; Tian, Q.; Liang, L.; Zhao, X. Norepinephrine may promote the progression of Fusobacterium nucleatum related colorectal cancer via quorum sensing signalling. Virulence 2024, 15, 2350904. [Google Scholar] [CrossRef]
- Abdulamir, A.S.; Hafidh, R.R.; Bakar, F.A. The association of Streptococcus bovis/gallolyticus with colorectal tumors: The nature and the underlying mechanisms of its etiological role. J. Exp. Clin. Cancer Res. 2011, 30, 1–13. [Google Scholar] [CrossRef]
- Xuefeng, X.; Hou, M.-X.; Yang, Z.-W.; Agudamu, A.; Wang, F.; Su, X.-L.; Li, X.; Shi, L.; Terigele, T.; Bao, L.-L. Epithelial–mesenchymal transition and metastasis of colon cancer cells induced by the FAK pathway in cancer-associated fibroblasts. J. Int. Med. Res. 2020, 48, 0300060520931242. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xie, S.; Liu, Y.; Zou, S.; Zhu, X. The application of patient-derived xenograft models in gynecologic cancers. J. Cancer 2020, 11, 5478. [Google Scholar] [CrossRef]
- Weeber, F.; Van De Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-van Hooijdonk, C.G.; van der Velden, D.L.; Peeper, D.S.; Cuppen, E.P. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA 2015, 112, 13308–13311. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Janakiraman, H.; Zhu, Y.; Becker, S.A.; Wang, C.; Cross, A.; Curl, E.; Lewin, D.; Hoffman, B.J.; Warren, G.W.; Hill, E.G. Modeling rectal cancer to advance neoadjuvant precision therapy. Int. J. Cancer 2020, 147, 1405–1418. [Google Scholar] [CrossRef]
- Padmanaban, A.M.; Ganesan, K.; Ramkumar, K.M. A Co-Culture System for Studying Cellular Interactions in Vascular Disease. Bioengineering 2024, 11, 1090. [Google Scholar] [CrossRef]
- Luo, X.; Fong, E.L.S.; Zhu, C.; Lin, Q.X.X.; Xiong, M.; Li, A.; Li, T.; Benoukraf, T.; Yu, H.; Liu, S. Hydrogel-based colorectal cancer organoid co-culture models. Acta Biomater. 2021, 132, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Mosa, M.H.; Michels, B.E.; Menche, C.; Nicolas, A.M.; Darvishi, T.; Greten, F.R.; Farin, H.F. A Wnt-induced phenotypic switch in cancer-associated fibroblasts inhibits EMT in colorectal cancer. Cancer Res. 2020, 80, 5569–5582. [Google Scholar] [CrossRef] [PubMed]
- Drápela, S.; Bouchal, J.; Jolly, M.K.; Culig, Z.; Souček, K. ZEB1: A critical regulator of cell plasticity, DNA damage response, and therapy resistance. Front. Mol. Biosci. 2020, 7, 36. [Google Scholar] [CrossRef]
- Lazarova, D.; Bordonaro, M. ZEB1 mediates drug resistance and EMT in p300-deficient CRC. J. Cancer 2017, 8, 1453. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ji, S.; Ma, Y.; Xing, X.; Zhou, Y.; Xu, X.; Song, J.; Wang, S.; Jiang, W.; Wang, X. Vimentin plays an important role in the promotion of breast cancer cell migration and invasion by leucine aminopeptidase 3. Cytotechnology 2020, 72, 639–647. [Google Scholar] [CrossRef]
- Strating, E.; Verhagen, M.P.; Wensink, E.; Dünnebach, E.; Wijler, L.; Aranguren, I.; De la Cruz, A.S.; Peters, N.A.; Hageman, J.H.; van der Net, M.M. Co-cultures of colon cancer cells and cancer-associated fibroblasts recapitulate the aggressive features of mesenchymal-like colon cancer. Front. Immunol. 2023, 14, 1053920. [Google Scholar] [CrossRef]
- Ryu, K.-B.; Seo, J.-a.; Lee, K.; Choi, J.; Yoo, G.; Ha, J.-h.; Ahn, M.R. Drug-Resistance Biomarkers in Patient-Derived Colorectal Cancer Organoid and Fibroblast Co-Culture System. Curr. Issues Mol. Biol. 2024, 46, 5794–5811. [Google Scholar] [CrossRef]
- Parseh, B.; Khosravi, A.; Fazel, A.; Ai, J.; Ebrahimi-Barough, S.; Verdi, J.; Shahbazi, M. 3-Dimensional model to study apoptosis induction of activated natural killer cells conditioned medium using patient-derived colorectal cancer organoids. Front. Cell Dev. Biol. 2022, 10, 895284. [Google Scholar] [CrossRef]
- Chen, Y.; Vandereyken, M.; Newton, I.P.; Moraga, I.; Näthke, I.S.; Swamy, M. Loss of adenomatous polyposis coli function renders intestinal epithelial cells resistant to the cytokine IL-22. PLoS Biol. 2019, 17, e3000540. [Google Scholar] [CrossRef]
- Subtil, B.; Iyer, K.K.; Poel, D.; Bakkerus, L.; Gorris, M.A.; Escalona, J.C.; Dries, K.v.d.; Cambi, A.; Verheul, H.M.; de Vries, I.J.M. Dendritic cell phenotype and function in a 3D co-culture model of patient-derived metastatic colorectal cancer organoids. Front. Immunol. 2023, 14, 1105244. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Huang, Y.; Luo, Y.; Tang, J.; Yu, M.; Zhang, Y.; Zhong, M. SIRT1 induces the accumulation of TAMs at colorectal cancer tumor sites via the CXCR4/CXCL12 axis. Cell. Immunol. 2022, 371, 104458. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 2018, 174, 1586–1598.e1512. [Google Scholar] [CrossRef]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor organoid–T-cell coculture systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Feng, H.; Zhao, J.-k.; Schiergens, T.S.; Wang, P.-x.; Ou, B.-c.; Al-Sayegh, R.; Li, M.-l.; Lu, A.-g.; Yin, S.; Thasler, W.E. Bone marrow-derived mesenchymal stromal cells promote colorectal cancer cell death under low-dose irradiation. Br. J. Cancer 2018, 118, 353–365. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.; Van Boxtel, R.; De Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, C.; Hu, Y.; Mu, L.; Liu, S.; Huang, K.; Li, Q.; Li, X.; Tao, D.; Qin, J. Differentiated cancer cell-originated lactate promotes the self-renewal of cancer stem cells in patient-derived colorectal cancer organoids. Cancer Lett. 2020, 493, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Gevers, S.; Kok, R.N.; Burgering, L.M.; Neikes, H.; Akkerman, N.; Betjes, M.A.; Ludikhuize, M.C.; Gulersonmez, C.; Stigter, E.C. Lactate controls cancer stemness and plasticity through epigenetic regulation. Cell Metab. 2025. [Google Scholar] [CrossRef]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef] [PubMed]
- Wensink, G.E.; Elias, S.G.; Mullenders, J.; Koopman, M.; Boj, S.F.; Kranenburg, O.W.; Roodhart, J.M. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. NPJ Precis. Oncol. 2021, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M. Patient-derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell Stem Cell 2020, 26, 17–26.e16. [Google Scholar] [CrossRef]
- Hsu, K.-S.; Adileh, M.; Martin, M.L.; Makarov, V.; Chen, J.; Wu, C.; Bodo, S.; Klingler, S.; Sauvé, C.-E.G.; Szeglin, B.C. Colorectal cancer develops inherent radiosensitivity that can be predicted using patient-derived organoids. Cancer Res. 2022, 82, 2298–2312. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.W.; Min, D.W.; Kim, H.P.; An, Y.; Kim, S.; Youk, J.; Chun, J.; Im, J.P.; Song, S.H.; Ju, Y.S. Patient-derived organoids as a preclinical platform for precision medicine in colorectal cancer. Mol. Oncol. 2022, 16, 2396–2412. [Google Scholar] [CrossRef]
- Geevimaan, K.; Guo, J.-Y.; Shen, C.-N.; Jiang, J.-K.; Fann, C.S.; Hwang, M.-J.; Shui, J.-W.; Lin, H.-T.; Wang, M.-J.; Shih, H.-C. Patient-derived organoid serves as a platform for personalized chemotherapy in advanced colorectal cancer patients. Front. Oncol. 2022, 12, 883437. [Google Scholar] [CrossRef]
- Bradley, C.A. Gastrointestinal cancer: Organoids predict clinical responses. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 189–190. [Google Scholar] [CrossRef]
- De Oliveira, T.; Goldhardt, T.; Edelmann, M.; Rogge, T.; Rauch, K.; Kyuchukov, N.D.; Menck, K.; Bleckmann, A.; Kalucka, J.; Khan, S. Effects of the novel PFKFB3 inhibitor KAN0438757 on colorectal cancer cells and its systemic toxicity evaluation in vivo. Cancers 2021, 13, 1011. [Google Scholar] [CrossRef]
- Ganesh, K.; Wu, C.; O’Rourke, K.P.; Szeglin, B.C.; Zheng, Y.; Sauvé, C.-E.G.; Adileh, M.; Wasserman, I.; Marco, M.R.; Kim, A.S. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 2019, 25, 1607–1614. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Zafari, N.; Velayati, M.; Damavandi, S.; Pourali, G.; Mobarhan, M.G.; Nassiri, M.; Hassanian, S.M.; Khazaei, M.; Ferns, G.A.; Avan, A. Metabolic pathways regulating colorectal cancer: A potential therapeutic approach. Curr. Pharm. Des. 2022, 28, 2995–3009. [Google Scholar] [CrossRef]
- Ban, H.S.; Kim, B.-K.; Lee, H.; Kim, H.M.; Harmalkar, D.; Nam, M.; Park, S.-K.; Lee, K.; Park, J.-T.; Kim, I. The novel hypoxia-inducible factor-1α inhibitor IDF-11774 regulates cancer metabolism, thereby suppressing tumor growth. Cell Death Dis. 2017, 8, e2843. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, L.; Zhu, L.T.; Wang, Y.; Pan, D.; Yao, J.; You, Q.D.; Guo, Q.L. Wogonin reverses hypoxia resistance of human colon cancer HCT116 cells via downregulation of HIF-1α and glycolysis, by inhibiting PI3K/Akt signaling pathway. Mol. Carcinog. 2014, 53, E107–E118. [Google Scholar] [CrossRef]
- Carr, R.M.; Qiao, G.; Qin, J.; Jayaraman, S.; Prabhakar, B.S.; Maker, A.V. Targeting the metabolic pathway of human colon cancer overcomes resistance to TRAIL-induced apoptosis. Cell Death Discov. 2016, 2, 1–13. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, Z.; Zou, X.; Lan, Y.; Sun, X.; Wang, X.; Zhao, S.; Jiang, C.; Liu, H. Mechanisms underlying 3-bromopyruvate-induced cell death in colon cancer. J. Bioenerg. Biomembr. 2015, 47, 319–329. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, N.; Zhang, D.; Zhang, K.; Zheng, W.; Bao, Y.; Yang, W. PFKFB3 inhibition attenuates oxaliplatin-induced autophagy and enhances its cytotoxicity in colon cancer cells. Int. J. Mol. Sci. 2019, 20, 5415. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.J.; Yin, P.H.; Xu, K.; Wang, Y.Z.; Shi, F.; Gao, J.; Fu, X.L. New strategies for targeting glucose metabolism–mediated acidosis for colorectal cancer therapy. J. Cell. Physiol. 2019, 234, 348–368. [Google Scholar] [CrossRef]
- Ikezoe, T.; Chen, S.S.; Tong, X.-J.; Heber, D.; Taguchi, H.; Koeffler, H.P. Oridonin induces growth inhibition and apoptosis of a variety of human cancer cells. Int. J. Oncol. 2003, 23, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Reidy, E.; Leonard, N.A.; Treacy, O.; Ryan, A.E. A 3D view of colorectal cancer models in predicting therapeutic responses and resistance. Cancers 2021, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Sugimura, R.; Zhang, Y.S.; Ruan, C.; Wen, C. Organoids in concert: Engineering in vitro models toward enhanced fidelity. Aggregate 2024, 5, e478. [Google Scholar] [CrossRef]
- Heydari, Z.; Najimi, M.; Mirzaei, H.; Shpichka, A.; Ruoss, M.; Farzaneh, Z.; Montazeri, L.; Piryaei, A.; Timashev, P.; Gramignoli, R. Tissue engineering in liver regenerative medicine: Insights into novel translational technologies. Cells 2020, 9, 304. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, Y.; Liu, Y.; Zhang, M.; Zhang, X. Microfluidic control of tumor and stromal cell spheroids pairing and merging for three-dimensional metastasis study. Anal. Chem. 2020, 92, 7638–7645. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-Y.; Lee, J.-H.; Shin, Y.; Chung, S.; Kuh, H.-J. Co-culture of tumor spheroids and fibroblasts in a collagen matrix-incorporated microfluidic chip mimics reciprocal activation in solid tumor microenvironment. PloS One 2016, 11, e0159013. [Google Scholar] [CrossRef]
- Sobrino, A.; Phan, D.T.; Datta, R.; Wang, X.; Hachey, S.J.; Romero-López, M.; Gratton, E.; Lee, A.P.; George, S.C.; Hughes, C.C. 3D microtumors in vitro supported by perfused vascular networks. Sci. Rep. 2016, 6, 31589. [Google Scholar] [CrossRef]
- Mao, Y.; Wang, W.; Yang, J.; Zhou, X.; Lu, Y.; Gao, J.; Wang, X.; Wen, L.; Fu, W.; Tang, F. Drug repurposing screening and mechanism analysis based on human colorectal cancer organoids. Protein Cell 2024, 15, 285–304. [Google Scholar] [CrossRef]
- Heydari, Z.; Moeinvaziri, F.; Agarwal, T.; Pooyan, P.; Shpichka, A.; Maiti, T.K.; Timashev, P.; Baharvand, H.; Vosough, M. Organoids: A novel modality in disease modeling. Bio-Des. Manuf. 2021, 4, 689–716. [Google Scholar] [CrossRef]
- Revokatova, D.; Bikmulina, P.; Heydari, Z.; Solovieva, A.; Vosough, M.; Shpichka, A.; Timashev, P. Getting Blood out of a Stone: Vascularization via Spheroids and Organoids in 3D Bioprinting. Cells 2025, 14, 665. [Google Scholar] [CrossRef] [PubMed]
- Hachey, S.J.; Sobrino, A.; Lee, J.G.; Jafari, M.D.; Klempner, S.J.; Puttock, E.J.; Edwards, R.A.; Lowengrub, J.S.; Waterman, M.L.; Zell, J.A. A human vascularized microtumor model of patient-derived colorectal cancer recapitulates clinical disease. Transl. Res. 2023, 255, 97–108. [Google Scholar] [CrossRef]
- Lee, J.; Jung, S.; Hong, H.K.; Jo, H.; Rhee, S.; Jeong, Y.-L.; Ko, J.; Cho, Y.B.; Jeon, N.L. Vascularized tissue on mesh-assisted platform (VT-MAP): A novel approach for diverse organoid size culture and tailored cancer drug response analysis. Lab. A Chip 2024, 24, 2208–2223. [Google Scholar] [CrossRef] [PubMed]
- Rajasekar, S.; Lin, D.S.; Abdul, L.; Liu, A.; Sotra, A.; Zhang, F.; Zhang, B. IFlowPlate—A customized 384-well plate for the culture of perfusable vascularized colon organoids. Adv. Mater. 2020, 32, 2002974. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.-y.; Du, Y.-x.; Cao, H.-m.; Su, L.-y.; Su, X.-l.; Li, X. The Biological Macromolecules constructed Matrigel for cultured organoids in biomedical and tissue engineering. Colloids Surf. B Biointerfaces 2024, 114435. [Google Scholar] [CrossRef]
- Mitrofanova, O.; Nikolaev, M.; Xu, Q.; Broguiere, N.; Cubela, I.; Camp, J.G.; Bscheider, M.; Lutolf, M.P. Bioengineered human colon organoids with in vivo-like cellular complexity and function. Cell Stem Cell 2024, 31, 1175–1186.e7. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Bacterium | Functions | Microbiota Details | Mechanism | Study Limitations | Ref. |
|---|---|---|---|---|---|
| Clostridioides difficile | Higher incidence of C. difficile in the CRC patients compared to the healthy individuals | Identified using TaqMan Real-time PCR, significant difference in colonic | Disrupts the gut microbiota and promotes cancer by inducing inflammation | The cross-sectional design restricts the ability to determine causal relationships and does not provide longitudinal data | [47] |
| Fusobacterium nucleatum | Fusobacterium nucleatum promotes inflammatory and anti-apoptotic responses in colorectal cancer cells via ADP-heptose release and ALPK1/TIFA axis activation | Gut bacterial metabolites, dysregulation associated with cancer | Drives tumor progression by triggering inflammation and inhibits cell death | Results from in vitro and animal studies may not accurately reflect the effects observed in humans | [48] |
| Streptococcus lutetiensis | Assisted in diagnosing invasive adenocarcinoma | Bacterial translocation due to disruption of colonic mucus | Promotes cancer by breaching the mucosal barrier | Clinical evidence is limited, and the underlying mechanism has not been fully elucidated in humans | [49] |
| Escherichia coli (pks+) | Production of colibactin, associated with CRC | Prevalence in healthy individuals, tumor development in the colon | Induces DNA damage that leads to cancer | The influence of other genotoxic bacteria cannot be fully ruled out as a confounding factor. | [50] |
| Parabacteroides distasonis | Enhance antitumor immunity by modulating CXCL10 and CD8+ T cells | Found in the gut, modulation of immune response to colon tumors | Stimulates immune cells to act against tumor growth | Results from animal studies may not directly translate to human colorectal cancer | [51] |
| Various gut Firmicutes | Effects of hypsunation in myeloid cells on colitis and cancer | Increased levels of DHPS and EIF5A Hyp in cells infiltrating the colon in Crohn’s disease patients | Alters the gut microbial ecosystem, disrupting balance and facilitating cancer development | Observational data cannot establish cause-and-effect relationships | [52] |
| Various gut microbiota | Effects of hypsunation in myeloid cells on colitis and cancer | Increased levels of DHPS and EIF5A Hyp in cells infiltrating the colon in Crohn’s disease patients | Alters immune cells activity to regulate inflammation and cancer | Functional significance in human patients not fully validated | [53] |
| Fusobacterium nucleatum | Promotes CRC progression via quorum sensing signaling | Interacts with host hormones, novel strategy for managing pathogenic | Enhances tumor growth by cell-to-cell signaling and hormone interaction | Mechanistic details of host-hormone interactions require further study | [54] |
| Streptococcus gallolyticus | Induces inflammatory responses, enhances carcinogenesis | Commonly found in CRC patients, associated with advanced disease | Activates inflammation that promotes tumor initiation and progression | The role of infection as a direct etiological factor in cancer is still undetermined | [55] |
| Drug | Target | Metabolic Effect in CRC | Therapeutic Outcome | Ref. |
|---|---|---|---|---|
| RO5126766 | RAF-MEK-ERK signaling pathway | Decreased GLUT1 expression, which results in lower glucose uptake | Reduces CRC xenograft growth | [95] |
| IDF-11774 (LW6) | hypoxia-inducible factor-1 (HIF-1) inhibitor | Upregulation of HIF-1α correlates with diminished glucose uptake, suppressed glycolysis, and ATP depletion in HCT116 cells | Suppression of HCT116 xenograft growth | [96] |
| Wogonin | Inhibiting PI3k/Akt signaling pathway | Hypoxia-induced inhibition of HIF-1α, glucose consumption, and lactate synthesis in HCT116 cells | Inhibition of HCT116 xenograft growth | [97] |
| tephrosin with 2-deoxy-D-glucose drug combination | HT-29 and SW-620 | TSN and 2-DG synergistically promoted intracellular ATP depletion and robust AMPK activation, ultimately suppressing the mTOR pathway | The addition of TSN to 2-DG exacerbated intracellular ATP depletion and prevented 2-DG-induced autophagy by inhibiting the activation of eEF-2K, leading to an increase in apoptosis | [98] |
| 3-bromopyruvate (3-BP) | Hexokinase II (HK-II) | Decreased ATP levels in SW480 and HT29 cell lines | 3BP promotes various forms of cell death through energy depletion in vitro, reducing resistance to drug-induced cell death. Its anti-tumor activity in vivo shows its potential as a therapeutic option for CRC | [99] |
| PFK-15 (1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one) | PFKFB3 (6-phosphofructo-2-kinase) | Inhibition of oxaliplatin-induced autophagy | Increased sensitivity of SW480 cells to oxaliplatin | [100] |
| WZB117 | Glucose Transporter 1 (GLUT1) | Induction of platelet-derived growth factor | Increased levels of glycolysis, resulting in higher intracellular lactate and acidic byproduct accumulation | [101] |
| Oridonin | Active diterpenoid | It deactivates phosphorylated AMPK and downregulates the GLUT1 and MCT-1 expression. | Suppresses glucose uptake, decreases lactate production, and triggers autophagy and cell mortality in CRC cells. | [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heydari, Z.; Devasahayam Arokia Balaya, R.; Sarkar, G.; Boardman, L. The Role of Organoids in Advancing Colorectal Cancer Research: Insights and Future Directions. Cancers 2025, 17, 2129. https://doi.org/10.3390/cancers17132129
Heydari Z, Devasahayam Arokia Balaya R, Sarkar G, Boardman L. The Role of Organoids in Advancing Colorectal Cancer Research: Insights and Future Directions. Cancers. 2025; 17(13):2129. https://doi.org/10.3390/cancers17132129
Chicago/Turabian StyleHeydari, Zahra, Rex Devasahayam Arokia Balaya, Gobinda Sarkar, and Lisa Boardman. 2025. "The Role of Organoids in Advancing Colorectal Cancer Research: Insights and Future Directions" Cancers 17, no. 13: 2129. https://doi.org/10.3390/cancers17132129
APA StyleHeydari, Z., Devasahayam Arokia Balaya, R., Sarkar, G., & Boardman, L. (2025). The Role of Organoids in Advancing Colorectal Cancer Research: Insights and Future Directions. Cancers, 17(13), 2129. https://doi.org/10.3390/cancers17132129