
The Vimentin-Targeting Drug ALD-R491 Partially Reverts the Epithelial-to-Mesenchymal Transition and Vimentin Interactome of Lung Cancer Cells

 , ,
, ,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Antibodies and Cell Dyes
2.3. Western Blot
2.4. Immunoflourescence
2.5. Live-Cell Imaging
2.6. Image Analysis
2.7. Tracking Cell Migration
2.8. Monitoring Nuclear Division
2.9. Proteomic Analysis Using Label-Free Quantification Mass Spectrometry
2.10. Protein Identification, Relative Quantification, Bioinformatic Functional Profiling and Interaction Analysis
2.11. Statistical Analysis
3. Results
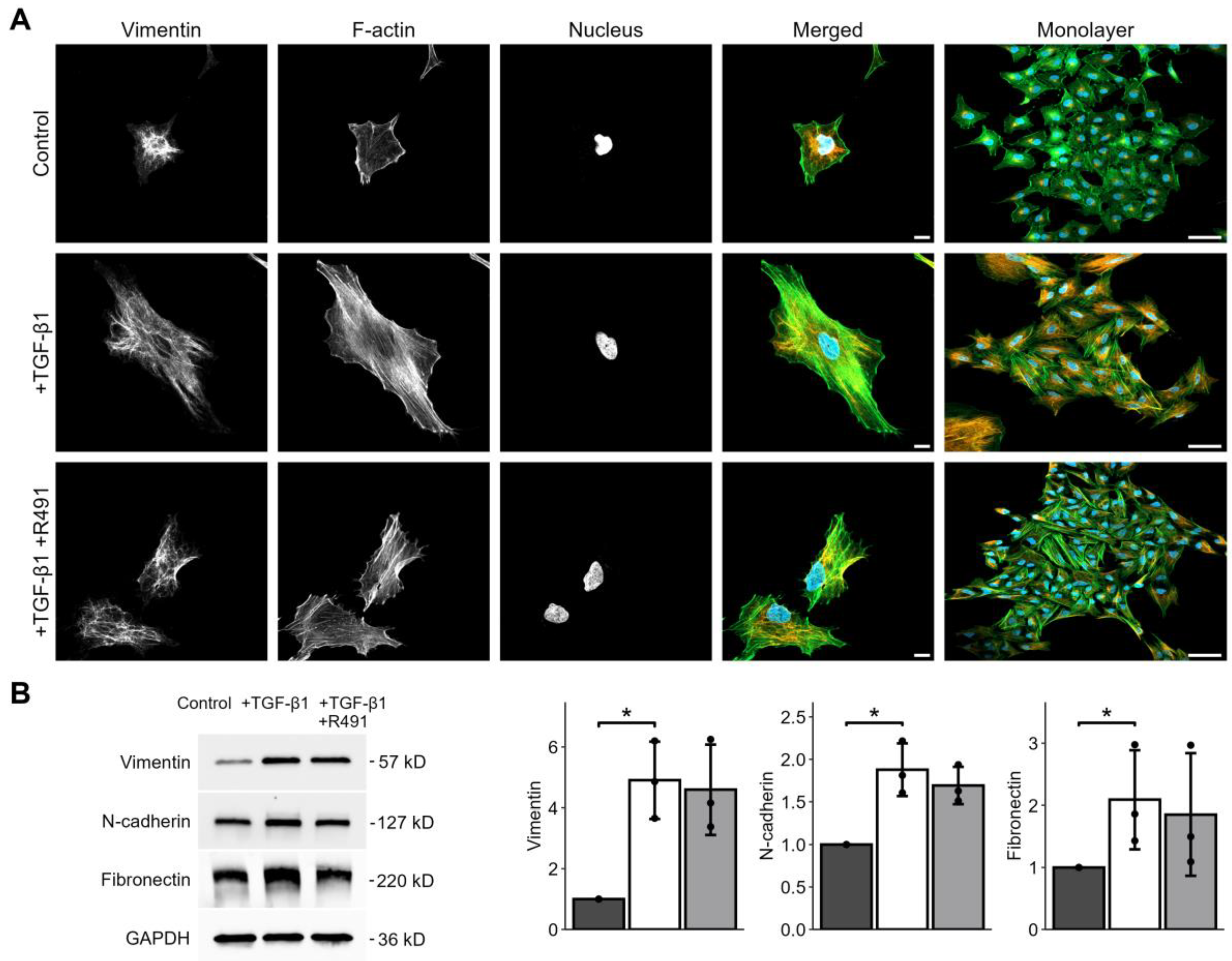
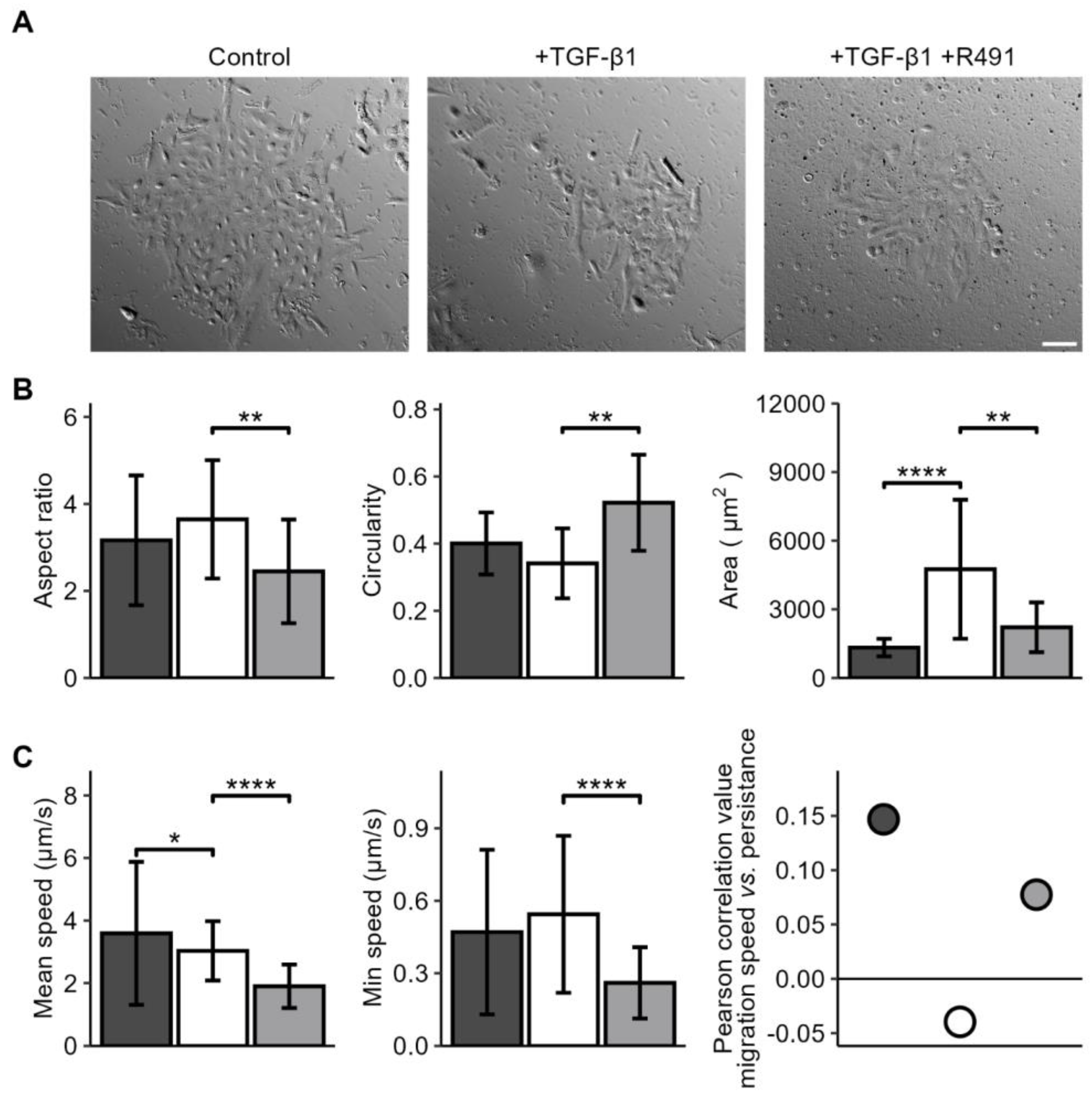
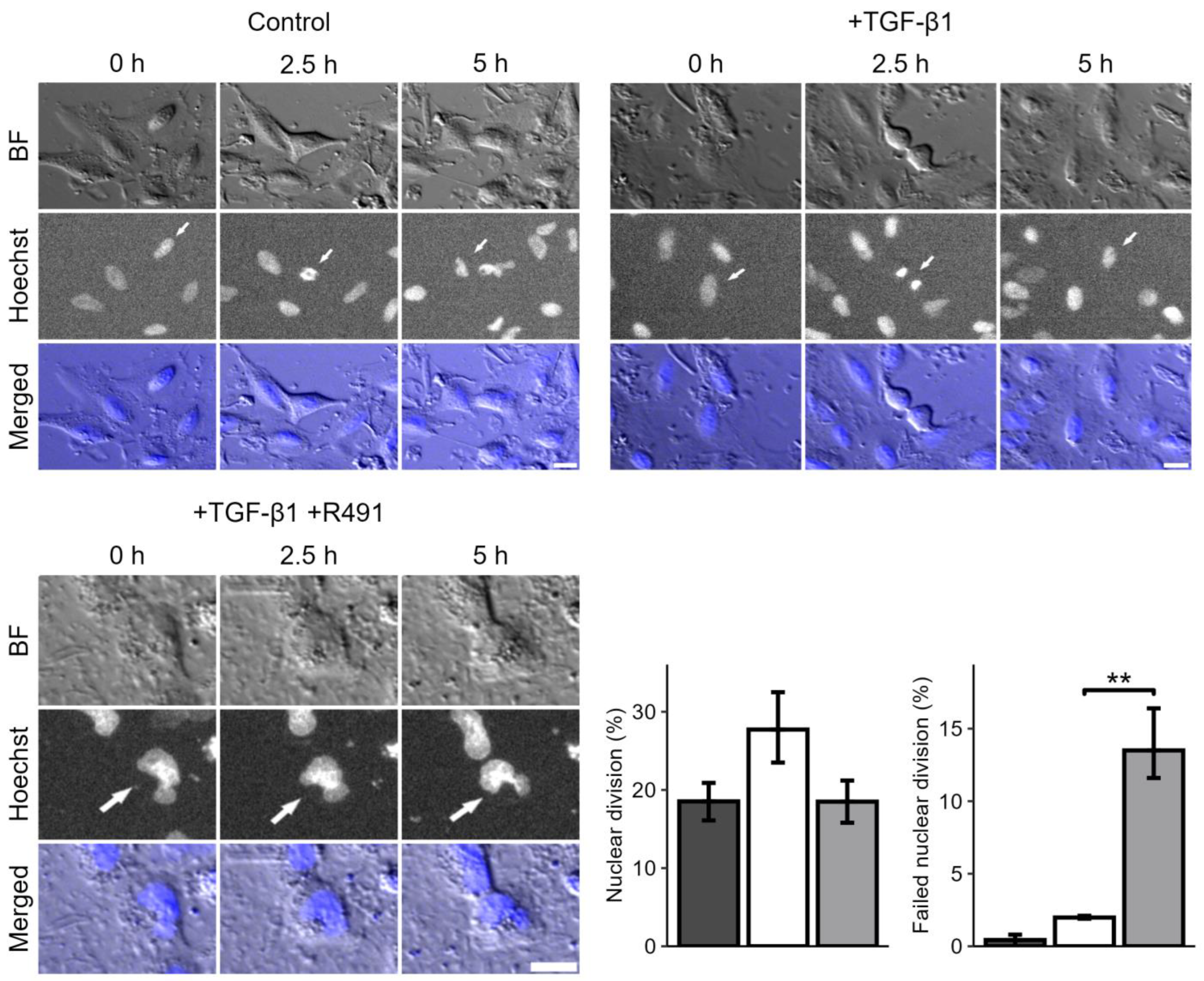
3.1. ALD-R491 Partially Reverses the Phenotypes of the Epithelial-to-Mesenchymal Transition
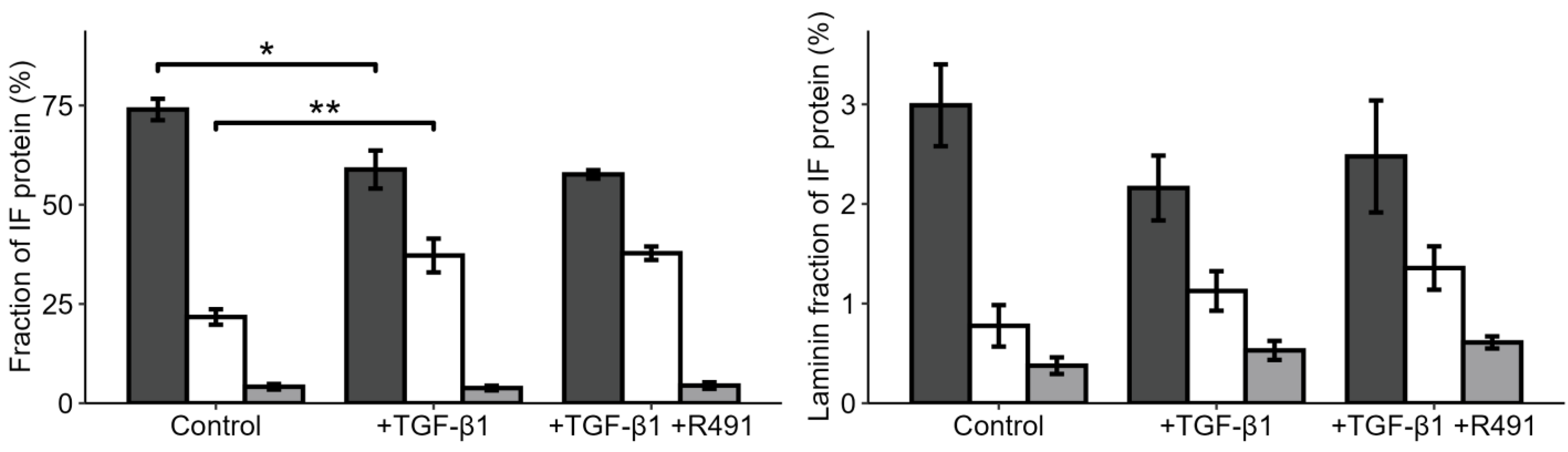
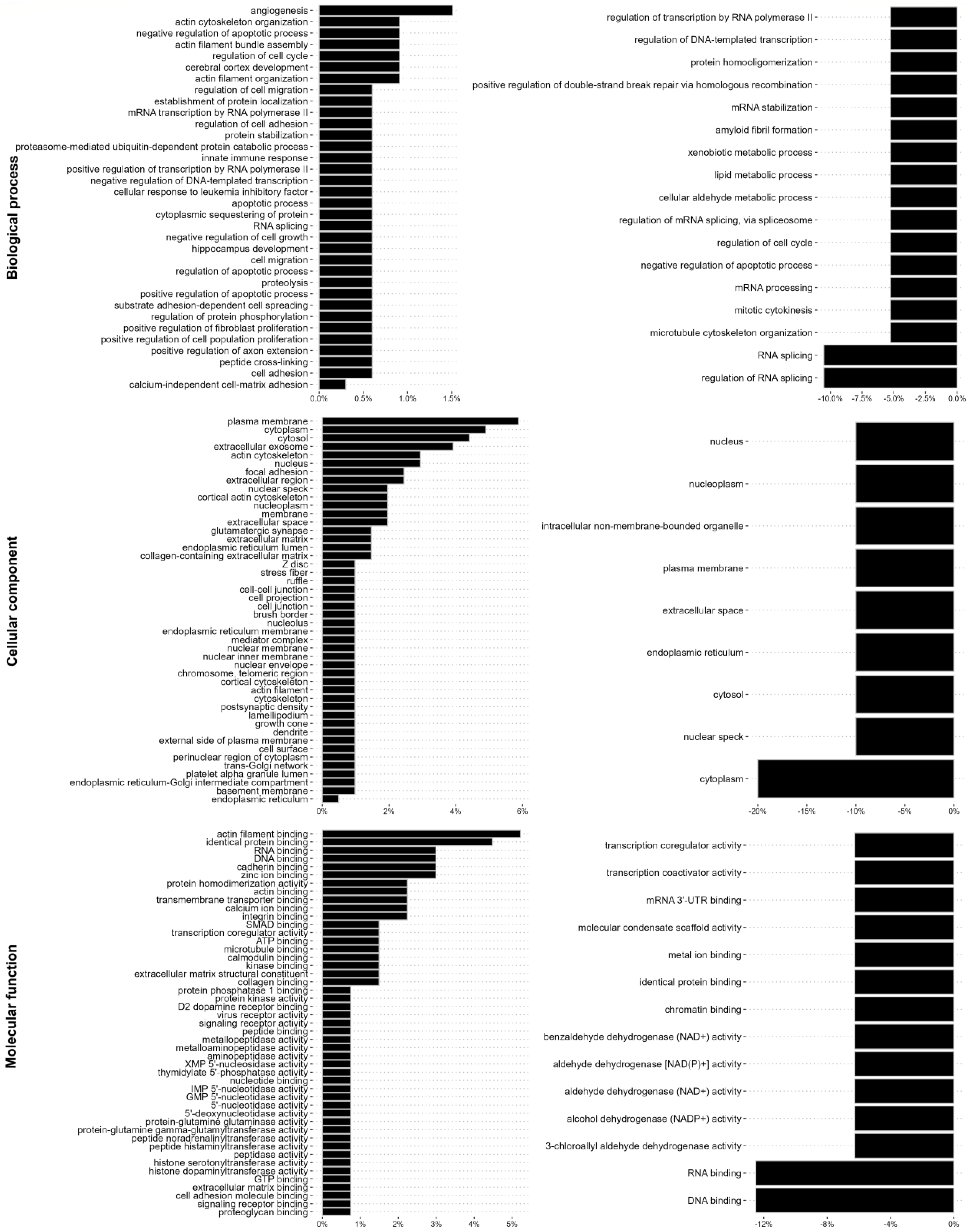
3.2. EMT-Increased Binding of Extracellular Matrix, Cell Motility, Cytokinesis, Cytoskeletal, and RNA-Binding Proteins to Vimentin, Is Partially Reversed by ALD-R491
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Menko, A.S.; Bleaken, B.M.; Libowitz, A.A.; Zhang, L.; Stepp, M.A.; Walker, J.L. A Central Role for Vimentin in Regulating Repair Function during Healing of the Lens Epithelium. Mol. Biol. Cell 2014, 25, 776–790. [Google Scholar] [CrossRef]
- Ridge, K.M.; Eriksson, J.E.; Pekny, M.; Goldman, R.D. Roles of Vimentin in Health and Disease. Genes Dev. 2022, 36, 391–407. [Google Scholar] [CrossRef]
- Danielsson, F.; Peterson, M.K.; Araújo, H.C.; Lautenschläger, F.; Gad, A.K.B. Vimentin Diversity in Health and Disease. Cells 2018, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin Induces Changes in Cell Shape, Motility, and Adhesion during the Epithelial to Mesenchymal Transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Baldovini, C.; Rossi, G.; Ciarrocchi, A. Approaches to Tumor Classification in Pulmonary Sarcomatoid Carcinoma. Lung Cancer 2019, 10, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, T.; Ishii, G.; Nagai, K.; Yoshida, J.; Nishimura, M.; Hishida, T.; Neri, S.; Hasegawa, S.; Ochiai, A. Characteristic Immunophenotype of Solid Subtype Component in Lung Adenocarcinoma. Ann. Surg. Oncol. 2012, 19, 3943–3952. [Google Scholar] [CrossRef]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The Role of Vimentin Intermediate Filaments in the Progression of Lung Cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Berr, A.L.; Wiese, K.; dos Santos, G.; Koch, C.M.; Anekalla, K.R.; Kidd, M.; Davis, J.M.; Cheng, Y.; Hu, Y.S.; Ridge, K.M. Vimentin Is Required for Tumor Progression and Metastasis in a Mouse Model of Non–Small Cell Lung Cancer. Oncogene 2023, 42, 2074–2087. [Google Scholar] [CrossRef]
- Kosibaty, Z.; Brustugun, O.T.; Zwicky Eide, I.J.; Tsakonas, G.; Grundberg, O.; De Petris, L.; McGowan, M.; Hydbring, P.; Ekman, S. Ras-Related Protein Rab-32 and Thrombospondin 1 Confer Resistance to the EGFR Tyrosine Kinase Inhibitor Osimertinib by Activating Focal Adhesion Kinase in Non-Small Cell Lung Cancer. Cancers 2022, 14, 3430. [Google Scholar] [CrossRef]
- Guo, M.; Ehrlicher, A.J.; Mahammad, S.; Fabich, H.; Jensen, M.H.; Moore, J.R.; Fredberg, J.J.; Goldman, R.D.; Weitz, D.A. The Role of Vimentin Intermediate Filaments in Cortical and Cytoplasmic Mechanics. Biophys. J. 2013, 105, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Jiu, Y.; Peränen, J.; Schaible, N.; Cheng, F.; Eriksson, J.E.; Krishnan, R.; Lappalainen, P. Vimentin Intermediate Filaments Control Actin Stress Fiber Assembly through GEF-H1 and RhoA. J. Cell Sci. 2017, 130, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Ding, L.; Burckhardt, C.J.; Lowery, J.; Zaritsky, A.; Sitterley, K.; Mota, A.; Costigliola, N.; Starker, C.G.; Voytas, D.F.; et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst. 2016, 3, 252–263.e8. [Google Scholar] [CrossRef] [PubMed]
- Coelho-Rato, L.S.; Parvanian, S.; Modi, M.K.; Eriksson, J.E. Vimentin at the Core of Wound Healing. Trends Cell Biol. 2024, 34, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Costigliola, N.; Ding, L.; Burckhardt, C.J.; Han, S.J.; Gutierrez, E.; Mota, A.; Groisman, A.; Mitchison, T.J.; Danuser, G. Vimentin Fibers Orient Traction Stress. Proc. Natl. Acad. Sci. USA 2017, 114, 5195–5200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Stefanovic, B. LARP6 Meets Collagen MRNA: Specific Regulation of Type I Collagen Expression. Int. J. Mol. Sci. 2016, 17, 419. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska-Podhorodecka, Z.; Ding, I.; Norouzi, M.; McCulloch, C.A. Impact of Vimentin on Regulation of Cell Signaling and Matrix Remodeling. Front. Cell Dev. Biol. 2022, 10, 869069. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Warrington, S.J.; López-Guajardo, A.; Al Hennawi, K.; Cook, S.L.; Griffith, Z.D.J.; Symmes, D.; Zhang, T.; Qu, Z.; Xu, Y.; et al. ALD-R491 Regulates Vimentin Filament Stability and Solubility, Cell Contractile Force, Cell Migration Speed and Directionality. Front. Cell Dev. Biol. 2022, 10, 926283. [Google Scholar] [CrossRef]
- Nöding, B.; Herrmann, H.; Köster, S. Direct Observation of Subunit Exchange along Mature Vimentin Intermediate Filaments. Biophys. J. 2014, 107, 2923–2931. [Google Scholar] [CrossRef]
- Wu, J.; Xie, Q.; Liu, Y.; Gao, Y.; Qu, Z.; Mo, L.; Xu, Y.; Chen, R.; Shi, L. A Small Vimentin-Binding Molecule Blocks Cancer Exosome Release and Reduces Cancer Cell Mobility. Front. Pharmacol. 2021, 12, 627394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qu, Z.; Wu, J.; Yao, S.; Zhang, Q.; Zhang, T.; Mo, L.; Yao, Q.; Xu, Y.; Chen, R. SARs of a Novel Series of S-Triazine Compounds Targeting Vimentin to Induce Methuotic Phenotype. Eur. J. Med. Chem. 2021, 214, 113188. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Alexeyenko, A.; Hennig, K.; Danielsson, F.; Lebbink, R.J.; Fielden, M.; Turunen, S.P.; Lehti, K.; Kashuba, V.; Madapura, H.S.; et al. RhoA Knockout Fibroblasts Lose Tumor-Inhibitory Capacity in Vitro and Promote Tumor Growth in Vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E1413–E1421. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Tinevez, J.Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An Open and Extensible Platform for Single-Particle Tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Leech, S.H.; Evans, C.A.; Shaw, L.; Wong, C.H.; Connolly, J.; Griffiths, J.R.; Whetton, A.D.; Corfe, B.M. Proteomic Analyses of Intermediate Filaments Reveals Cytokeratin8 Is Highly Acetylated—Implications for Colorectal Epithelial Homeostasis. Proteomics 2008, 8, 279–288. [Google Scholar] [CrossRef]
- Evans, C.A.; Kim, H.R.; Macfarlane, S.C.; Nowicki, P.I.A.; Baltes, C.; Xu, L.; Widengren, J.; Lautenschläger, F.; Corfe, B.M.; Gad, A.K.B. Metastasising Fibroblasts Show an HDAC6-Dependent Increase in Migration Speed and Loss of Directionality Linked to Major Changes in the Vimentin Interactome. Int. J. Mol. Sci. 2022, 23, 1961. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Schwanhüusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.D.; Goode, R.J.A.; Huang, C.; Powell, D.R.; Schittenhelm, R.B. LFQ-Analyst: An Easy-To-Use Interactive Web Platform to Analyze and Visualize Label-Free Proteomics Data Preprocessed with MaxQuant. J. Proteome Res. 2020, 19, 204–211. [Google Scholar] [CrossRef]
- Danielsson, F.; Skogs, M.; Huss, M.; Rexhepaj, E.; O’Hurley, G.; Klevebring, D.; Pontén, F.; Gad, A.K.B.; Uhlén, M.; Lundberg, E. Majority of Differentially Expressed Genes Are Down-Regulated during Malignant Transformation in a Four-Stage Model. Proc. Natl. Acad. Sci. USA 2013, 110, 6853–6858. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Pallari, H.M.; Nevo, J.; Eriksson, J.E. Novel Functions of Vimentin in Cell Adhesion, Migration, and Signaling. Exp. Cell Res. 2007, 313, 2050–2062. [Google Scholar] [CrossRef]
- De Oliveira Ramos, G.; Bernardi, L.; Lauxen, I.; Filho, M.S.A.; Horwitz, A.R.; Lamers, M.L. Fibronectin Modulates Cell Adhesion and Signaling to Promote Single Cell Migration of Highly Invasive Oral Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0151338. [Google Scholar] [CrossRef]
- Missirlis, D.; Haraszti, T.; Kessler, H.; Spatz, J.P. Fibronectin Promotes Directional Persistence in Fibroblast Migration through Interactions with Both Its Cell-Binding and Heparin-Binding Domains. Sci. Rep. 2017, 7, 3711. [Google Scholar] [CrossRef]
- Odii, B.O.; Coussons, P. Biological Functionalities of Transglutaminase 2 and the Possibility of Its Compensation by Other Members of the Transglutaminase Family. Sci. World J. 2014, 2014, 714561. [Google Scholar] [CrossRef]
- Jones, R.A.; Kotsakis, P.; Johnson, T.S.; Chau, D.Y.S.; Ali, S.; Melino, G.; Griffin, M. Matrix Changes Induced by Transglutaminase 2 Lead to Inhibition of Angiogenesis and Tumor Growth. Cell Death Differ 2006, 13, 1442–1453. [Google Scholar] [CrossRef]
- Tatsukawa, H.; Furutani, Y.; Hitomi, K.; Kojima, S. Transglutaminase 2 Has Opposing Roles in the Regulation of Cellular Functions as Well as Cell Growth and Death. Cell Death Dis. 2016, 7, e2244. [Google Scholar] [CrossRef]
- Crowley, J.L.; Smith, T.C.; Fang, Z.; Takizawa, N.; Luna, E.J. Supervillin Reorganizes the Actin Cytoskeleton and Increases Invadopodial Efficiency. Mol. Biol. Cell 2009, 20, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Hleihel, R.; El Hajj, H.; Wu, H.C.; Berthier, C.; Zhu, H.H.; Massoud, R.; Chakhachiro, Z.; El Sabban, M.; de The, H.; Bazarbachi, A. A Pin1/PML/P53 Axis Activated by Retinoic Acid in NPM-1c Acute Myeloid Leukemia. Haematologica 2021, 106, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Shinde, D.N.N.; Reijnders, M.R.R.F.; Hauser, N.S.S.; Belmonte, R.L.L.; Wilson, G.R.R.; Bosch, D.G.G.M.; Bubulya, P.A.A.; Shashi, V.; Petrovski, S.; et al. De Novo Mutations in SON Disrupt RNA Splicing of Genes Essential for Brain Development and Metabolism, Causing an Intellectual-Disability Syndrome. Am. Soc. Hum. Genet. 2016, 99, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.F.; Jiang, T.; Chen, F.; Shi, P.C.; Li, H.Q.; Bai, J.; Song, J. KIF4A Facilitates Cell Proliferation via Induction of P21-Mediated Cell Cycle Progression and Promotes Metastasis in Colorectal Cancer. Cell Death Dis. 2018, 9, 477. [Google Scholar] [CrossRef]
- Lendeckel, U.; Karimi, F.; Al Abdulla, R.; Wolke, C. The Role of the Ectopeptidase APN/CD13 in Cancer. Biomedicines 2023, 11, 724. [Google Scholar] [CrossRef]
- Shirao, T.; Sekino, Y. General Introduction to Drebrin. In Drebrin; Advances in Experimental Medicine and Biology; Springer: Tokyo, Japan, 2017; Volume 1006, pp. 3–22. [Google Scholar] [CrossRef]
- Koizumi, H.; Gleeson, J.G. Sun Proteins Enlighten Nuclear Movement in Development. Neuron 2009, 64, 147–149. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, F.; Yang, X.; Wang, Q.; Chang, R.; Zhu, L.; Feitelson, M.A.; Chen, Z. TMEM43 Promotes the Development of Hepatocellular Carcinoma by Activating VDAC1 through USP7 Deubiquitination. Transl. Gastroenterol. Hepatol. 2024, 9, 9. [Google Scholar] [CrossRef]
- Ahn, E.Y.; DeKelver, R.C.; Lo, M.C.; Nguyen, T.A.; Matsuura, S.; Boyapati, A.; Pandit, S.; Fu, X.D.; Zhang, D.E. SON Controls Cell-Cycle Progression by Coordinated Regulation of RNA Splicing. Mol. Cell 2011, 42, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, R.; Izquierdo, L.; van der Heijden, A.G.; Lozano, J.J.; Franco, M.; Ingelmo-Torres, M.; Roldan, F.L.; Llorens, M.; Ribal, M.J.; Mengual, L.; et al. Differential Gene Expression Profile between Progressive and de Novo Muscle Invasive Bladder Cancer and Its Prognostic Implication. Sci. Rep. 2021, 11, 6132. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.; Schrecker, C.; Bon, D.; Friedrichs, N.; Bankov, K.; Wild, P.; Plotz, G.; Zeuzem, S.; Herrmann, E.; Hansmann, M.L.; et al. Downregulation of SPTAN1 Is Related to MLH1 Deficiency and Metastasis in Colorectal Cancer. PLoS ONE 2019, 14, e0213411. [Google Scholar] [CrossRef]
- Shan, Y.; Farmer, S.M.; Wray, S. Drebrin Regulates Cytoskeleton Dynamics in Migrating Neurons through Interaction with CXCR4. Proc. Natl. Acad. Sci. USA 2021, 118, e2009493118. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, V.; Paunu, K.; Kukkula, A.; Niva, S.; Junila, Y.; Toriseva, M.; Jokilehto, T.; Mäkelä, S.; Huhtaniemi, R.; Poutanen, M.; et al. Glucocorticoid Receptor-Induced Non-Muscle Caldesmon Regulates Metastasis in Castration-Resistant Prostate Cancer. Oncogenesis 2023, 12, 42. [Google Scholar] [CrossRef]
- Xie, G.F.; Zhao, L.D.; Chen, Q.; Tang, D.X.; Chen, Q.Y.; Lu, H.F.; Cai, J.R.; Chen, Z. High ACTN1 Is Associated with Poor Prognosis, and ACTN1 Silencing Suppresses Cell Proliferation and Metastasis in Oral Squamous Cell Carcinoma. Drug Des. Dev. Ther. 2020, 14, 1717–1727. [Google Scholar] [CrossRef]
- Wieczorek, K.; Wiktorska, M.; Sacewicz-Hofman, I.; Boncela, J.; Lewiński, A.; Kowalska, M.A.; Niewiarowska, J. Filamin A Upregulation Correlates with Snail-Induced Epithelial to Mesenchymal Transition (EMT) and Cell Adhesion but Its Inhibition Increases the Migration of Colon Adenocarcinoma HT29 Cells. Exp. Cell Res. 2017, 359, 163–170. [Google Scholar] [CrossRef]
- Huen, M.S.Y.; Sy, S.M.H.; Leung, K.M.; Ching, Y.P.; Tipoe, G.L.; Man, C.; Dong, S.; Chen, J. SON Is a Spliceosome-Associated Factor Required for Mitotic Progression. Cell Cycle 2010, 9, 2679–2685. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Liu, G.; Öztürk, A.; Hicks, G.G. ALS-Associated FUS Mutations Result in Compromised FUS Alternative Splicing and Autoregulation. PLoS Genet. 2013, 9, e1003895. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Reed, R. FUS Functions in Coupling Transcription to Splicing by Mediating an Interaction between RNAP II and U1 SnRNP. Proc. Natl. Acad. Sci. USA 2015, 112, 8608–8613. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Takanashi, K. FUS Interacts with Nuclear Matrix-Associated Protein SAFB1 as Well as Matrin3 to Regulate Splicing and Ligand-Mediated Transcription. Sci. Rep. 2016, 6, 35195. [Google Scholar] [CrossRef] [PubMed]
- Baechtold, H.; Kuroda, M.; Sok, J.; Ron, D.; Lopez, B.S.; Akhmedov, A.T. Human 75-KDa DNA-Pairing Protein Is Identical to the pro-Oncoprotein TLS/FUS and Is Able to Promote D-Loop Formation. J. Biol. Chem. 1999, 274, 34337–34342. [Google Scholar] [CrossRef]
- Lynch, C.D.; Lazar, A.M.; Iskratsch, T.; Zhang, X.; Sheetz, M.P. Endoplasmic Spreading Requires Coalescence of Vimentin Intermediate Filaments at Force-Bearing Adhesions. Mol. Biol. Cell 2013, 24, 21–30. [Google Scholar] [CrossRef]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.; David Martin, W.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell-Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef]
- Tang, D.D.; Bai, Y.; Gunst, S.J. Silencing of P21-Activated Kinase Attenuates Vimentin Phosphorylation on Ser-56 and Reorientation of the Vimentin Network during Stimulation of Smooth Muscle Cells by 5-Hydroxytryptamine. Biochem. J. 2005, 388, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Rathje, L.S.Z.; Nordgren, N.; Pettersson, T.; Rönnlund, D.; Widengren, J.; Aspenström, P.; Gad, A.K.B. Oncogenes Induce a Vimentin Filament Collapse Mediated by HDAC6 That Is Linked to Cell Stiffness. Proc. Natl. Acad. Sci. USA 2014, 111, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska-Podhorodecka, Z.; Ding, I.; Lee, W.; Tanic, J.; Abbasi, S.; Arora, P.D.; Liu, R.S.; Patteson, A.E.; Janmey, P.A.; McCulloch, C.A. Vimentin Tunes Cell Migration on Collagen by Controlling Β1 Integrin Activation and Clustering. J. Cell Sci. 2021, 134, jcs254359. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Park, H.J.; Seong, W.; Kim, J.; Kim, C. Vimentin-Mediated Buffering of Internal Integrin Β1 Pool Increases Survival of Cells from Anoikis. BMC Biol. 2024, 22, 139. [Google Scholar] [CrossRef]
- Li, B.; Shen, W.; Peng, H.; Li, Y.; Chen, F.; Zheng, L.; Xu, J.; Jia, L. Fibronectin 1 Promotes Melanoma Proliferation and Metastasis by Inhibiting Apoptosis and Regulating EMT. OncoTargets Ther. 2019, 12, 3207–3221. [Google Scholar] [CrossRef] [PubMed]
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin Protects Cells against Nuclear Rupture and DNA Damage during Migration. J. Cell Biol. 2019, 218, 4079–4092. [Google Scholar] [CrossRef]
- Duarte, S.; Viedma-Poyatos, Á.; Navarro-Carrasco, E.; Martínez, A.E.; Pajares, M.A.; Pérez-Sala, D. Vimentin Filaments Interact with the Actin Cortex in Mitosis Allowing Normal Cell Division. Nat. Commun. 2019, 10, 4200. [Google Scholar] [CrossRef]
- Expression of SON in Lung Cancer—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000159140-SON/cancer/lung+cancer#cptac_lung_ac (accessed on 18 December 2024).
- Xiong, D.; Wu, Y.B.; Jin, C.; Li, J.J.; Gu, J.; Liao, Y.F.; Long, X.; Zhu, S.Q.; Wu, H.B.; Xu, J.J.; et al. Elevated FUS/TLS Expression Is Negatively Associated with E-Cadherin Expression and Prognosis of Patients with Non-Small Cell Lung Cancer. Oncol. Lett. 2018, 16, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Yin, R.; Wang, L.; Zhao, S.; Lv, D.; Yang, K.; Geng, S.; Yang, N.; Zhang, X.; Wang, H. ALDH3A1 Driving Tumor Metastasis Is Mediated by P53/BAG1 in Lung Adenocarcinoma. J. Cancer 2021, 12, 4780–4790. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Espinoza, M.; Kim, M.; Ow, C.; Hutchins, E.J. Beyond Transcription: How Post-Transcriptional Mechanisms Drive Neural Crest EMT. Genesis 2024, 62, e23553. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene | Protein ID | Signalling Pathway | TGFβ1 vs. A549 Foldchange (p-Value) |
|---|---|---|---|---|
| Fibronectin;Anastellin;Ugl-Y1;Ugl-Y2;Ugl-Y3 | FN1 | P02751 | Integrin, Collagen, Fibrin | 137.19 (3.09 × 10−13) |
| Transforming growth factor-β1-induced protein ig-h3 | TGF-Β1 | Q15582 | TGF-Β1, MAPK | 28.05 (1.33 × 10−10) |
| Protein-glutamine gamma-glutamyltransferase 2 | TGM2 | P21980 | FN1, SPFN1, HSPB6 | 17.63 (2.09 × 10−18) |
| 5-nucleotidase | NT5E | P21589 | AMP- NAD- nucleotides | 6.92 (4.91 × 10−12) |
| Aminopeptidase N | ANPEP | P15144 | an aminopeptidase | 5.58 (1.06 × 10−7) |
| Neurabin-2 | PPP1R9B | Q96SB3 | F-actin, Rac, Dopamine D1 | 4.92 (0.000159) |
| Zinc finger protein 185 | ZNF185 | O15231 | * Wnt | 4.86 (4.95 × 10−10) |
| Drebrin 1 | DBN1 | Q16643 | * F-actin | 4.32 (1.88 × 10−10) |
| Caldesmon | CALD1 | E9PGZ1 | F-actin,, myosin, calmodulin | 4.08 (5.49 × 10−10) |
| SUN-domain-containing protein 2 | SUN2 | Q9UH99 | LINC complex | 3.92 (0.000905) |
| SON | SON | P18583 | TUBG1, KATNB1, AURKB | 3.78 (0.0034) |
| CTP synthase 1 | CTPS1 | A0A3B3IRI2 | CTP | 3.68 (3.42 × 10−5) |
| Bcl-2-associated transcription factor 1 | BCLAF1 | A0A3B3ITZ9 | CCND1 mRNA | 3.48 (0.00372) |
| Supervillin | SVIL | A0A6I8PIX7 | F-actin | 3.23 (0.000905) |
| Protein PML | PML | P29590 | PML-NBs | 3.18 (0.000565) |
| Protein disulfide-isomerase A4 | PDIA4 | A0A499FI48 | * HSP, ERO1 | 2.85 (0.000398) |
| Transmembrane protein 43 | TMEM43 | Q9BTV4 | RNF26 | 2.62 (1.04 × 10−6) |
| Uncharacterised protein C17orf85 | C17orf85 | Q53F19 | mRNA | 2.58 (0.00418) |
| Thyroid-hormone-receptor-associated protein 3 | THRAP3 | A0A3B3ITZ9 | mRNA, DNA | 2.36 (0.00372) |
| Alpha-actinin-1 | ACTN1 | P12814 | F-actin | 2.25 (3.66 × 10−10) |
| LIM domain and actin-binding protein 1 | LIMA1 | Q9UHB6 | F-actin | 2.22 (0.000905) |
| Kinesin-like protein 14 | KIF14 | Q15058 | Tubulin, CDKN1B | 2.11 (0.00102) |
| Spectrin alpha chain, non-erythrocytic 1 | SPTAN1 | Q13813 | F-actin, Calmodulin | 2.06 (0.00151) |
| Filamin-A | FLNA | P21333 | F-actin, SEMA3A | 2.01 (9.05 × 10−6) |
| Protein | Gene | Protein ID | Signaling Pathway | TGFβ1 vs. A549 Fold Change (p-Value) |
|---|---|---|---|---|
| Protein SON | SON | P18583 | TUBG1, KATNB1, AURKB | −4.89 (0.00156) |
| Aldehyde dehydrogenase, dimeric NADP-preferring | ALDH3A1 | P30838 | Aldehyde substrates | −2.62 (0.00257) |
| RNA-binding protein FUS | FUS | P35637 | mRNA, DNA | −2.62 (0.00354) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosier, M.; Krstulović, A.; Kim, H.R.; Kaur, N.; Enakireru, E.M.; Symmes, D.; Dobra, K.; Chen, R.; Evans, C.A.; Gad, A.K.B. The Vimentin-Targeting Drug ALD-R491 Partially Reverts the Epithelial-to-Mesenchymal Transition and Vimentin Interactome of Lung Cancer Cells. Cancers 2025, 17, 81. https://doi.org/10.3390/cancers17010081
Rosier M, Krstulović A, Kim HR, Kaur N, Enakireru EM, Symmes D, Dobra K, Chen R, Evans CA, Gad AKB. The Vimentin-Targeting Drug ALD-R491 Partially Reverts the Epithelial-to-Mesenchymal Transition and Vimentin Interactome of Lung Cancer Cells. Cancers. 2025; 17(1):81. https://doi.org/10.3390/cancers17010081
Chicago/Turabian StyleRosier, Marieke, Anja Krstulović, Hyejeong Rosemary Kim, Nihardeep Kaur, Erhumuoghene Mary Enakireru, Deebie Symmes, Katalin Dobra, Ruihuan Chen, Caroline A. Evans, and Annica K. B. Gad. 2025. "The Vimentin-Targeting Drug ALD-R491 Partially Reverts the Epithelial-to-Mesenchymal Transition and Vimentin Interactome of Lung Cancer Cells" Cancers 17, no. 1: 81. https://doi.org/10.3390/cancers17010081
APA StyleRosier, M., Krstulović, A., Kim, H. R., Kaur, N., Enakireru, E. M., Symmes, D., Dobra, K., Chen, R., Evans, C. A., & Gad, A. K. B. (2025). The Vimentin-Targeting Drug ALD-R491 Partially Reverts the Epithelial-to-Mesenchymal Transition and Vimentin Interactome of Lung Cancer Cells. Cancers, 17(1), 81. https://doi.org/10.3390/cancers17010081