1. Introduction
This study investigated the immediate effect of low concentrations (0.001–1.0 µM) of erastin on the transporters of the blood–brain barrier (BBB) in rats. The blood-brain barrier (BBB) resides in the vast array of microvessels of the brain. Structurally, the BBB is arranged as a tri-laminate, consisting of luminal-facing endothelial cells with an outward layering of pericytes and astrocytic feet [
1]. Tight junctions between the luminal-facing endothelial cells provide a formidable physical barrier, while luminal-bound transporters discretely allow entry of needed nutrients and chemicals that serve the biological needs of the brain. Unwanted and potential harmful blood-born molecules are defended against by a host of luminal-bound ATP-binding cassette (ABC) transporters that selectively restrict xenobiotics, drugs, and harmful metabolites from entering the brain. ABC transporters hydrolyze ATP to ADP + Pi to derive sufficient energy to efflux harmful substrates back into the circulating blood. Arguably, the most important and most studied ABC transporter of the BBB is P-glycoprotein (ABCB1, MDR1) [
2]. P-glycoprotein is a 170 kD monomeric protein highly expressed on the luminal endothelial plasma membrane that is dynamically regulated at the activity and expression level through a vast array of signaling pathways. It is also highly expressed in cancer cells and is the primary contributor to chemotherapeutic drug resistance in the clinic [
3,
4]. It has an expansive and diverse array of substrates that include prescription drugs, xenobiotics, and metabolites [
5].
The BBB responds to a myriad of stress responses through the activation of biological signaling pathways. These pathways regulate BBB function by altering the expression and/or activity of ABC transporters at the barrier [
6]. Since previous work has shown that oxidative stress has the capacity to modulate transport activity at the BBB [
7], we investigated whether erastin could also modulate transport activity at the BBB. To accomplish this, we exposed freshly isolated rat brain capillaries ex vivo to low nanomolar concentrations of erastin and measured P-glycoprotein transport activity using a steady state confocal microscopy-based assay. Furthermore, we extended our findings to humans by exposing cancer cell lines in vitro to erastin and measuring the cytotoxic effect of a known cytotoxic P-glycoprotein substrate.
The chemotherapeutic, erastin (2-[1-[4-[2-(4-Chlorophenoxy)acetyl]-1-piperazinyl]ethyl]-3-(2-ethoxyphenyl)-4(3
H)-quinazolinone), was identified in a selective screen of small molecules capable of killing cancer cells that overexpress small-T antigen and oncogenic RAS [
8]. Cancer cells exposed to micromolar levels of erastin die from a non-apoptotic death that is associated with membrane lipid peroxidation, increased oxidative stress, reduced antioxidative capacity, and high levels of iron. This erastin-induced, iron-dependent form of programmed cell death was named “ferroptosis” [
9]. Studies show that erastin can induce ferroptosis through multiple pathways that include but are not limited to perturbations of the system X
C– (glutamate /cystine antiporter) [
10], the mitochondria-bound voltage-dependent anion channel (VDAC) [
11], and the tumor suppressor p53 gene [
12]. Mechanistic studies on ferroptosis found that cancer cells exposed to micromolar levels of erastin were depleted of the important antioxidant, glutathione (GSH) [
13]. The depletion of intracellular GSH was found to result from erastin’s ability to inhibit the plasma membrane-bound glutamate/cystine antiporter, system X
C−. The X
C− antiporter functions to import extracellular cystine in exchange for an abundant intracellular glutamate [
13]. Upon entry, intracellular cystine is rapidly reduced to cysteine and utilized in the synthesis and maintenance of intracellular GSH levels to help protect against oxidative stress. We chose erastin for our studies because it had been previously shown to induce iNOS, presumably through its interaction with the Xc- antiporter. We hypothesized that NO production from erastin mediated iNOS induction could affect the activity and/or expression of P-glycoprotein at the BBB.
2. Methods
2.1. Animals
All animal experiments presented in this work were approved by the Animal Care and Use Committee at the National Institute of Environmental Health Sciences in accordance with the National Institutes of Health. The data derived from these experiments are presented in compliance within the guidelines of the Animal Research Reporting In Vivo Experiments (ARRIVE). All male and female Sprague Dawley rats aged 12–50 weeks were purchased from Envigo and housed in humidity- and temperature-controlled rooms with a 12 h light/dark cycle. All were provided with access to both food and water ad libitum. Rats were killed by the approved method of CO2 inhalation and decapitation.
2.2. Materials
Ficoll, Bovine Serum Albumin (BSA), sodium pyruvate, L-Cysteine, and D-(+)-glucose, were all purchased from Sigma-Aldrich (Saint Louis, MO, USA). Thirty-micrometer pluriStrainer filters were purchased from pluriSelect, and 300 µm nylon mesh was purchased from Spectrum Labs. Erastin was purchased from Sigma-Aldrich. The P-glycoprotein fluorescent substrate N-ɛ(4-nitrobenzofurazan-7-yl)-D-Lys8 cyclosporin A (NBD-CSA) was provided as a gift from Dr. Bjoern Bauer from the University of Kentucky. The BCRP fluorescent substrate BODIPY® FL prazosin was purchased from Invitrogen (Waltham, MA, USA), and the MRP2 fluorescent substrate sulforhodamine 101 free acid (Texas Red, St. Louis, MO 68178, USA) was purchased from Sigma-Aldrich, St. Louis, MO, USA. PSC833 (valspodar), the P-glycoprotein-specific inhibitor, was purchased from Tocris Bioscience. Two-well 1.5 chamber slides were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The antioxidant N-acetylcystine (NAC) was gifted by Alex Merrick. Ten-well Invitrogen 4–12% Bis-Tri NuPAGE Gels NP0321, Novex Nitrocellulose membranes LC2001, the XCell SureLock® Mini-Cell and the XCell II™ Blot Module were all purchased from Thermo Fisher Scientific. Odyssey® Blocking Buffer was obtained from Li-Cor Biosciences (Lincoln, NE, USA). Immobilon-FL membranes were purchased from Sigma-Aldrich. Mouse monoclonal P-glycoprotein antibody C219 was purchased from Covance (Princeton, NJ, USA). Secondary goat anti-mouse IgG Alex Fluor 488 antibodies were purchased from Invitrogen. Mouse monoclonal β-actin antibody A1978 was purchased from Sigma-Aldrich. The iNOS antibody was purchased from Invitrogen. The iNOS inhibitor 1400 W was obtained from Abcam (Cambridge, UK), and the pan-NOS inhibitor L-NAME was obtained from Cayman (Ann Arbor, MI, USA). Thermo Scientific MES, 0.5 M buffer solution, pH 6.0 was purchased from Thermo Fisher Scientific. The reducing agent tris(2-carbosyethyl)phosphine hydrochloride, the cystine analog seleno-L-cystine, the fluorescent molecule fluorescein O,O’-diacrylate, and the system XC− inhibitor sulfasalazine were all purchased from Sigma-Aldrich.
2.3. Cell Culture
Authenticated human ovarian tumor cells, OVCAR-8 and NCI/ADR-RES, were obtained from the National Cancer Institute (Bethesda, MD, USA) and were grown in Phenol Red-free RPMI 1640 media supplemented with 10% fetal bovine serum and antibiotics. Tumor cells were routinely used for 20–25 passages, after which the cells were discarded and a new cell culture was started from the frozen stock.
2.4. Capillary Isolation
Brain capillaries were isolated from male and female Sprague Dawley rats. Using an established protocol [
14], rats were killed with CO
2 inhalation and subsequently decapitated. Rat brains were extracted from the skull and placed in a cold PBS buffer solution containing KCl 2.7 mM, KH
2PO
4 1.5 mM, NaCl 136.9 mM, Na
2HPO
4 4.3 mM, CaCl
2 0.7 mM, MgCl
2 0.5 mM, augmented with D-glucose 5.0 mM, and sodium pyruvate 1.0 mM at pH 7.4. Dissection of the rat brains involved removal of meninges, midbrain, olfactory bulbs, choroid plexus, blood vessels, and cortical white matter. Remaining grey matter brain tissue was homogenized in cold PBS buffer solution using a tissue grinder followed by ten up and down strokes in a Dounce homogenizer. The resulting homogenate was centrifuged in an aliquot of 30% Ficoll for 20 min at 5800×
g at 4 °C, separating the lipid fraction from the brain parenchyma. The capillary pellet was resuspended in 1% BSA in PBS, passed through a 300 μm nylon mesh filter, and then passed through a series of 30 μm pluriSelect cell strainers. Capillaries were washed off the filters with PBS to remove them and resuspended, followed by a second centrifugation of 350×
g for 10 min at 4 °C. Following final centrifugation, the supernatant was removed, and capillaries were used immediately for transport or protein studies.
2.5. Transport Assay
Following capillary isolation, the pellet was resuspended in a cold PBS buffer solution and plated into 2-well chamber slides. Capillaries could then be treated with various antagonists, agonists, or inhibitors. For dosing experiments, serial dilutions of erastin were prepared immediately before use in PBS buffer solution using 10.0 mM erastin stock solution made in DMSO. Solutions containing the fluorescent substrate specific for P-glycoprotein (NBD-CSA) and appropriate dosing of erastin were then added to respective chamber slides at volumes of 2.0 mL. Time course studies were staggered such that each slide could be viewed consecutively on a confocal microscope at 15 min/slide. For studies involving BCRP and MRP2, fluorescent substrates BODIPY® FL prazosin and sulforhodamine 101 free acid (Texas Red) were used for the transporters, respectively. Transport activity was measured as a function of steady-state luminal fluorescence. In the P-gp studies, background fluorescence was measured using the P-gp-specific inhibitor, PSC833 10 μM (valspodar), and subtracted from raw treatment values to obtain specific luminal fluorescence as a result of inhibition or activation of the transporter.
2.6. Confocal Microscopy
Capillaries were imaged using the Zeiss LSM 710 multiphoton confocal microscope and the Zeiss LSM 880 confocal microscope through a 40× water-immersion objective with a numeric aperture of 1.5. A 488 nm laser line for both NBD-CSA and BODIPY
® FL prazosin and a 543 nm laser line for Texas Red were used. The resulting images were saved to a data storage drive, which were then analyzed and quantified using ImageJ software. This published method was described in detail by Chan and Cannon in 2017 [
14].
2.7. Western Blotting
The capillary isolation protocol was outlined previously. Following isolation, capillary pellets were treated with 1.0 nM erastin for 3 h, then centrifuged at 350× g for 10 min at 4 °C and placed in a −80 °C freezer until further use. Thawed samples were separated into cytosolic, membrane, and nuclear fractions using lysis buffer with a protease inhibitor. Membrane and cytosolic protein fractions were mixed with loading dye, sample reducing buffer, and water and loaded onto a 4–12% Bis-Tris NuPAGE gel. Samples were subsequently electrophoresed using the XCell SureLock® Mini-Cell and transferred to an Immobilon-FL membrane using the XCell II™ Blot Module according to the manufacturer’s instructions. Following transfer, blocking buffer was added to the membrane for 40 min. The membrane was rinsed with 0.1% Tween in PBS, treated with mouse or rabbit monoclonal primary antibodies, and allowed to hybridize overnight. After overnight hybridization, the membrane was then rinsed with 0.1% Tween in PBS and stained with goat anti-mouse or goat anti-rabbit secondary antibodies for 1 h. The membrane was rinsed in three 40-min intervals with 0.1% Tween in PBS and imaged using an Odyssey infrared imaging system from Li-Cor Biosciences. Images were analyzed, and values were normalized to the β-actin loading control.
2.8. Cystine Uptake Assay via XC− Antiporter
The capillary isolation protocol was outlined previously, and the cystine uptake assay via XC− was adapted for brain capillaries. Once capillary isolation was completed, capillary pellets were transferred to a microfuge tube and washed with pre-warmed, cystine-free, serum-free media. Capillaries were centrifuged at 350× g for 10 min at 4 °C. The supernatant was removed, and capillaries were resuspended in 200 µL of pre-warmed cystine-free, serum-free media and divided into microfuge tubes for a variety of treatments. Capillaries were centrifuged again at 350× g for 10 min at 4 °C, and the supernatant was removed. A total of 200 µL of pre-warmed cystine-free, serum-free media was then added to each microfuge tube in addition to pretreatments of erastin and sulfasalazine, both known inhibitors of the XC−. Erastin was added at a dose of 1.0 nM, while sulfasalazine was added at a dose of 500 µM. Tubes were shielded from light and allowed to sit for 15–20 min. Following pretreatment, tubes were incubated at 37 °C for 5 min and then centrifuged at 350× g for 10 min at 4 °C. The supernatant was then removed, and 200 µL of pre-warmed cystine analog solution (200 µM selenocysteine) was added to each microfuge tube, except for the controls. To the respective tubes, 1.0 nM erastin and 500 µM sulfasalazine were added. To the controls, 200 µL of cystine-free, serum-free media was added. The tubes were incubated at 37 °C for 30 min. The supernatant was removed, and capillaries were rinsed three times with 200 µL of ice-cold PBS solution. Supernatant was removed and 60 µL of 100% methanol was added to each tube. A working solution was prepared immediately before use that consisted of 100 mM MES buffer (pH 6.0), 10.0 µM fluorescein O,O’-diacrylate, and 200 µM tris92-carboxyethyl)phosphine hydrochloride. A total of 240 µL of working solution was added to each microfuge tube, mixed by pipetting, and incubated a second time for 30 min at 37 °C. Following incubation, capillaries were centrifuged at 20,800× g for 5 min at 4 °C. The final supernatant was transferred to new microfuge tubes, then plated into a Greiner Fluotrac flat-bottomed 96-well black plate. Fluorescence was read using a BioTek Synergy microplate reader. Fluorescence intensity was measured at Ex/Em = 490/535 and samples were read in triplicate.
2.9. Cytotoxicity Assay
The cytotoxicity studies were carried out with both a cell growth inhibition assay and Trypan Blue. Briefly, 50,000–75,000 cells/well were seeded onto a 6-well plate (in triplicate) and allowed to attach for 18 h. Various concentrations of drugs (adriamycin or combinations of adriamycin, erastin or NOS inhibitors) were added to cells (NCI/ADR-RES) in fresh complete media (2 mL) and incubated for 72 h. When used, ER and NOS inhibitors were preincubated with cells for 2 h before the addition of Adriamycin. DMSO (0.01–0.1%) was included as the vehicle control when used. Following trypsinization, surviving cells were collected and counted in a cell counter (Beckman, Brea, CA, USA) or 15 μL of cell mixtures were combined with 15 µL of trypan blue and counted in a T20 automatic cell counter (Bio-Rad, Hercules, CA, USA).
2.10. Statistical Analysis
Data were graphed and analyzed using the GraphPad Prism (v 9.2.0) software and expressed as mean ± SEM. Statistical analyses and determination of significance were performed by one-way ANOVA and the Tukey multiple comparison test. Significance was defined as p < 0.05. Values deemed significant were so when compared to control, unless otherwise specified.
4. Discussion
Erastin was originally discovered in a screen of small molecules exhibiting the ability to selectively kill cancer cell lines expressing
SV40 small T antigen and oncogenic
ras while leaving normal cells unharmed [
8]. The unique erastin-induced cell death of cancer cells was called ferroptosis and was determined to involve membrane lipid peroxidation, glutathione depletion, and high levels of iron [
9]. Further studies have shown that erastin kills cancer cells in a three-pronged attack: (1) inhibiting the uptake of cystine through the system X
C− antiporter [
10], (2) disrupting mitochondria function and releasing oxidants from the voltage-dependent anion-selective channel (
VDAC) [
11], and (3) modulating the expression and activity of the tumor suppressor gene, TP53 [
12]. The cumulative effect of these attacks weakens cancer cells’ oxidative defenses by depleting GSH while simultaneously inducing higher levels of oxidants. These events occur in cancer cells that contain high levels of ionic iron (Fe
+2) that freely participate in Fenton reactions to produce oxidized membrane lipids [
22].
A mechanistic understanding of erastin-induced ferroptosis is important beyond the therapeutic needs of killing cancer cells in the clinic. Numerous pathophysiological disease states closely mimic aspects of ferroptosis [
23,
24]. These include, but are not limited to, blood diseases, central nervous system (CNS) diseases, ischemia and reperfusion injuries, and kidney injuries. A greater understanding of molecular mechanisms that induce, inhibit, and regulate ferroptosis will help to mitigate the deleterious effects of many diseases and cancers.
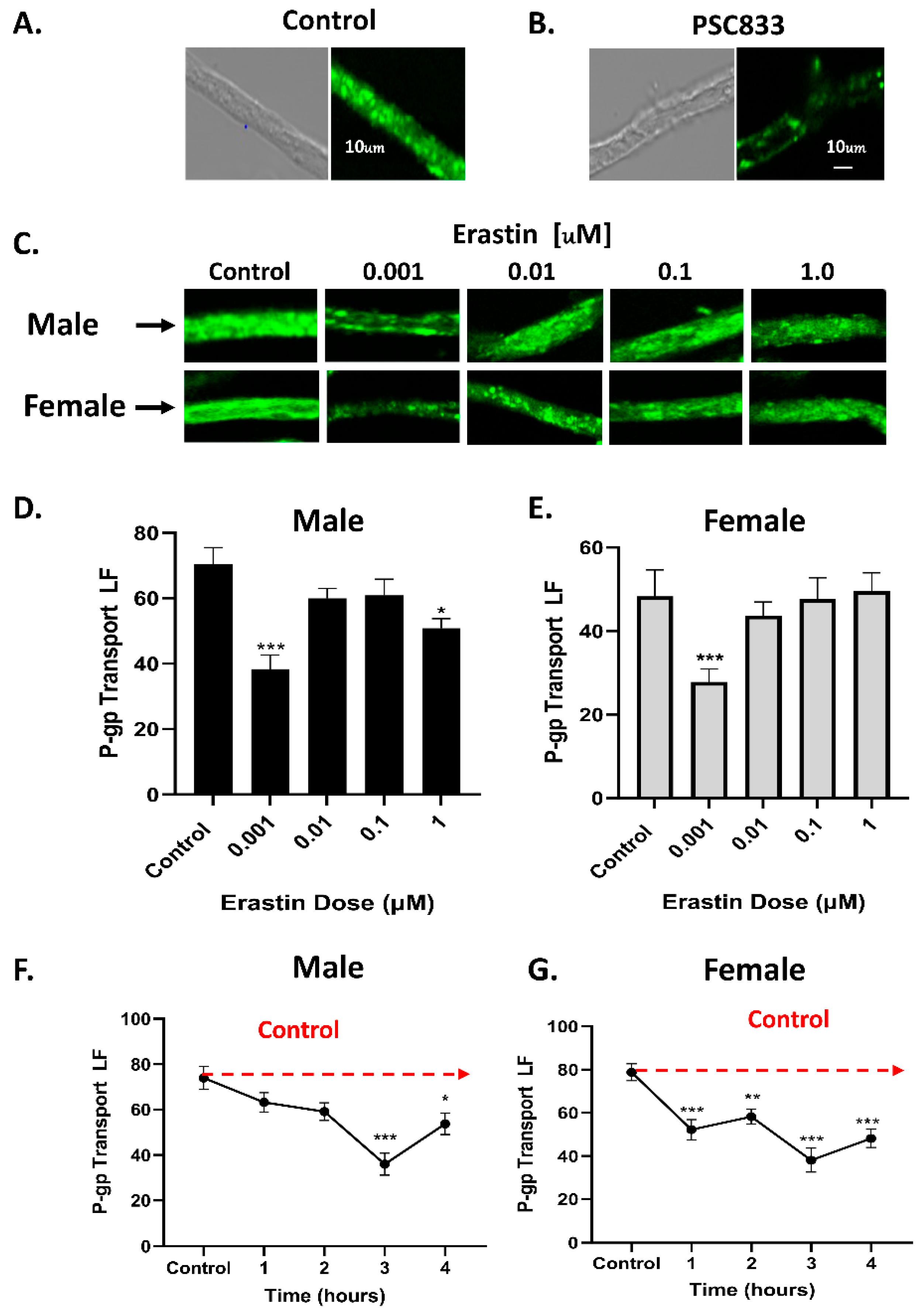
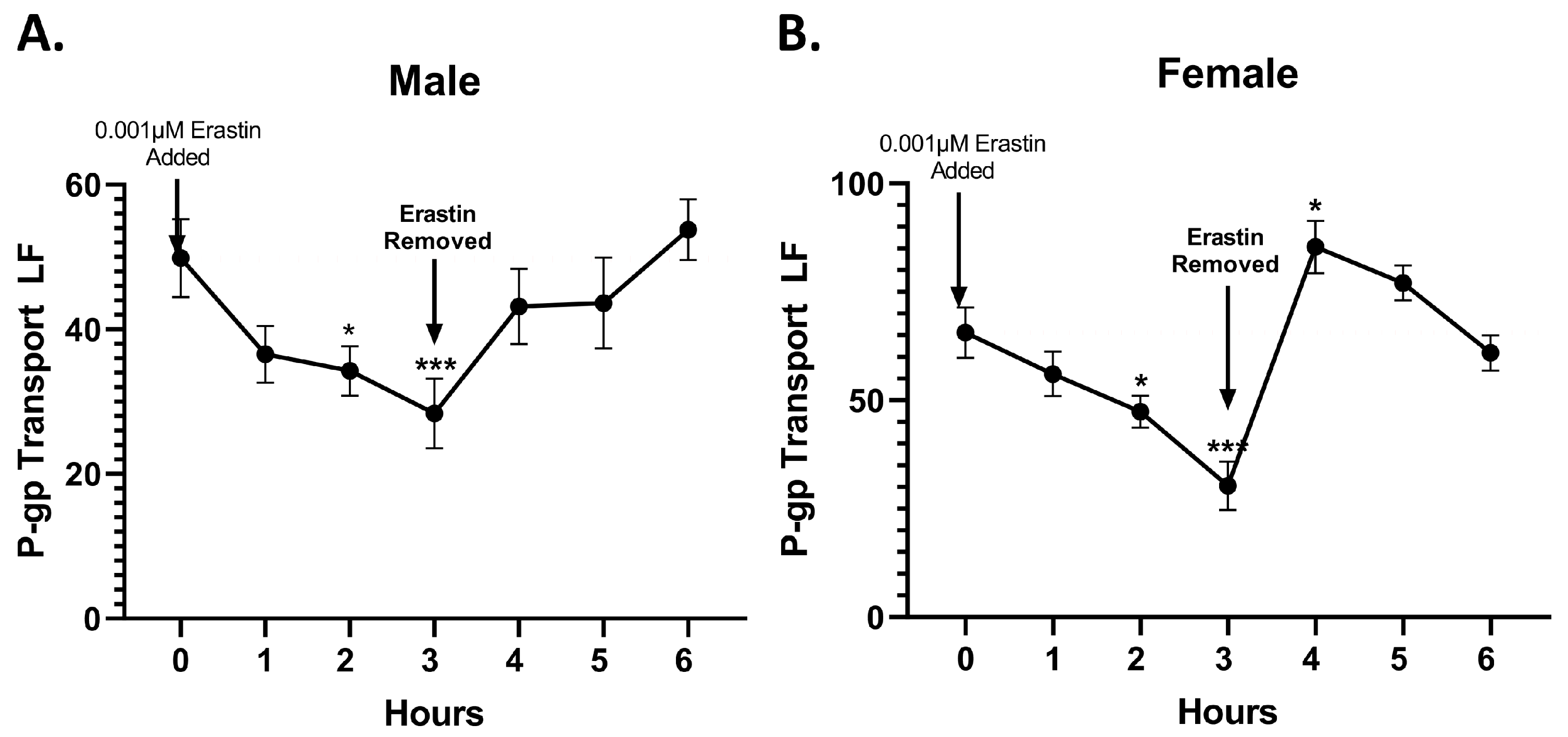
In this study, we investigated the rapid effects of low nM concentrations of erastin on the BBB of rats and the tumor cell lines of humans. Specifically, using freshly isolated rat brain capillaries, we measured the effects of nM concentrations of erastin on P-glycoprotein transport activity and expression at the BBB. We found that erastin caused a dose- and time-dependent reduction in P-glycoprotein transport activity in both male and female rats. The maximal reductions in transport occurred in 3 h in capillaries that were exposed to 1.0 nM erastin. One interesting aspect of the erastin reduction in P-glycoprotein transport is that males were not significantly reduced until 3 h of exposure to 1.0 nM erastin, while P-glycoprotein transport in females was significantly reduced at 1 h of exposure and sustained thereafter. The reason for this early and sustained response to erastin in females is unknown. Additionally, it can be noted that in both male and female rats, P-glycoprotein transport activity appears to slightly increase following the 3-h mark, suggesting that when treated with nM concentrations of erastin, P-gp transport activity could reverse without removal of erastin. These observed sex differences of erastin on P-glycoprotein activity could be related to the intrinsic differences in the basal and/or inducible NO levels in males compared to females. Multiple studies have shown that systemic NO is higher in females than males [
25,
26]. The observed biphasic response of P-glycoprotein as erastin concentrations approach micromolar levels may also be related to the multiple biological downstream effectors that are known targets of erastin at higher concentrations. These are known to produce cellular stressors related to oxidative stress, mitochondrial dysfunction and dysregulation of important stress response genes like P53 [
9,
12]. Similar stressors have been associated with signaling pathways that increase P-glycoprotein transport activity over longer periods of time [
6].
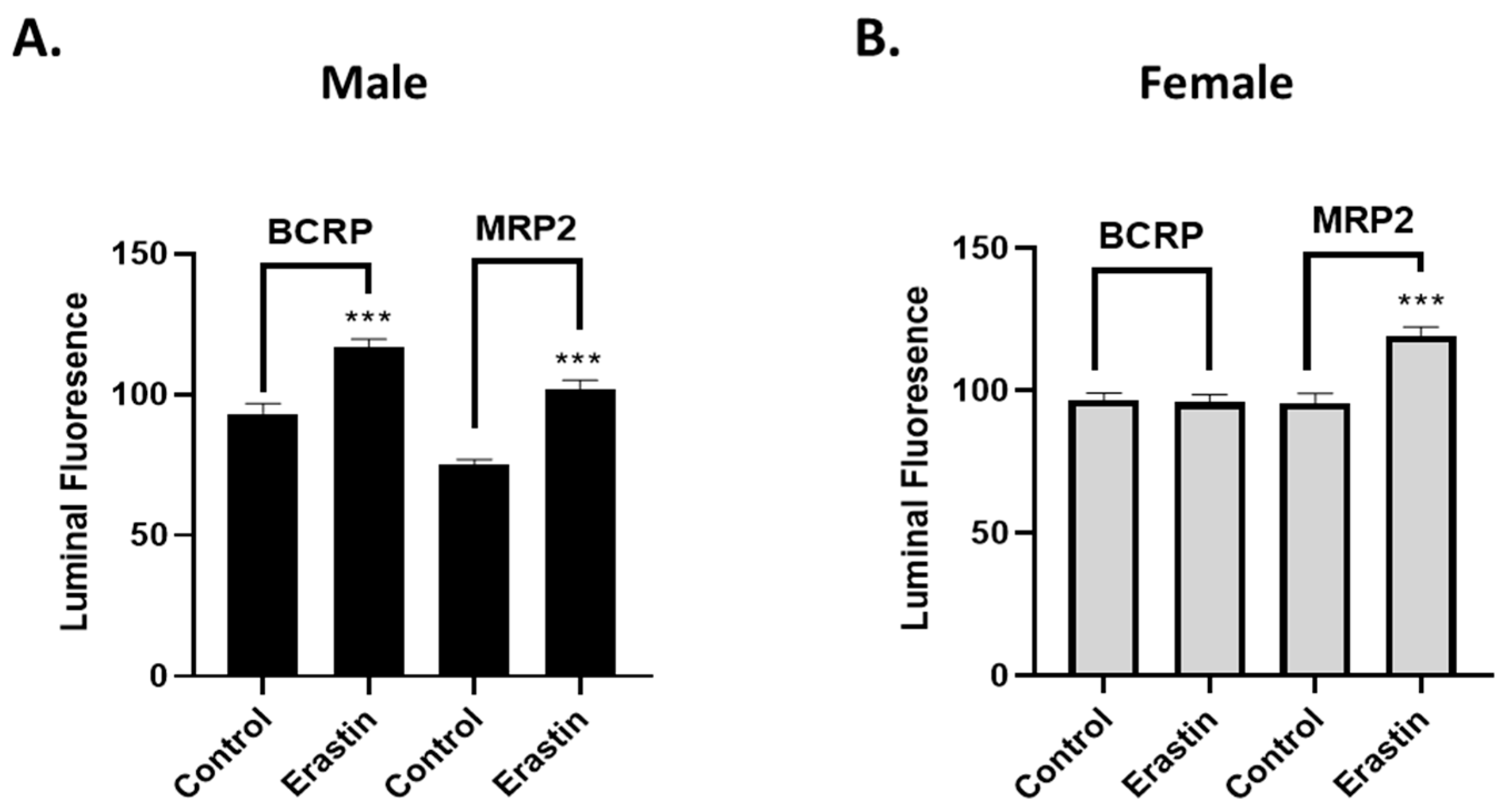
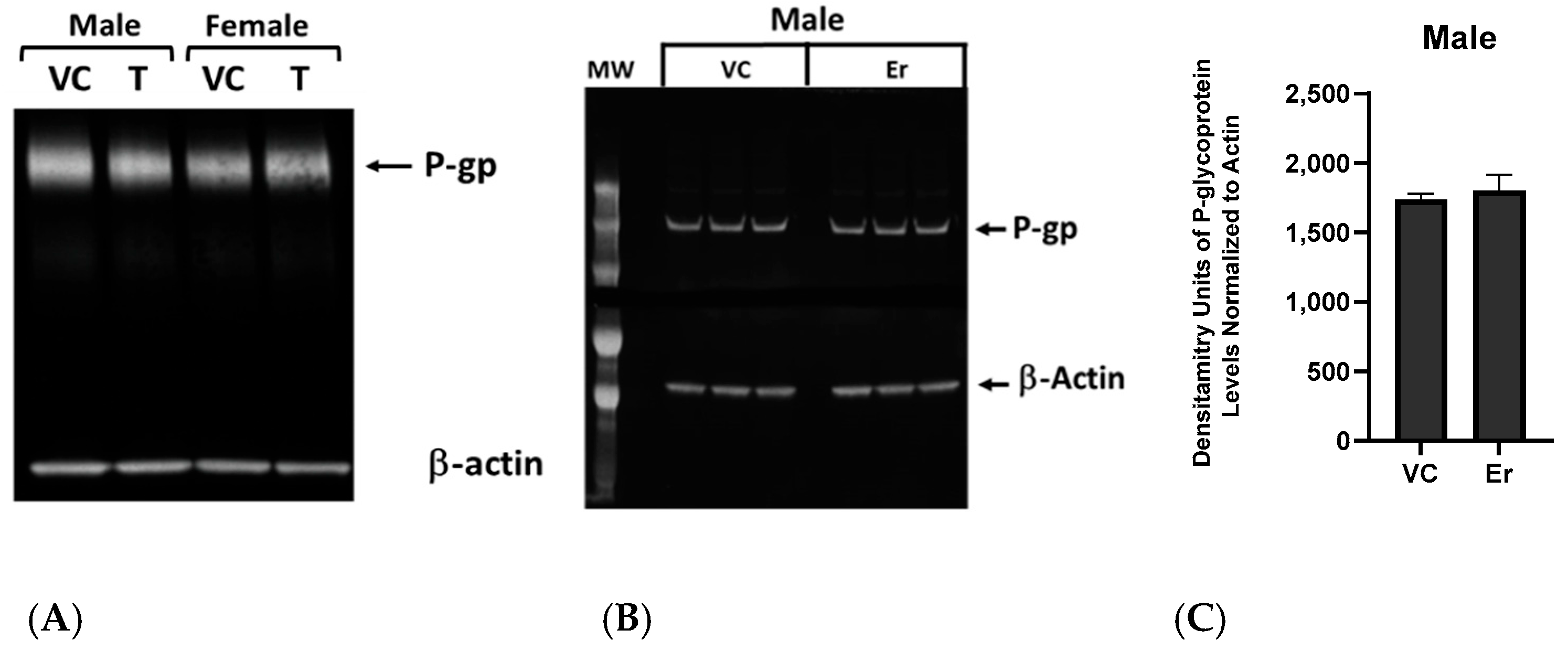
Mechanistically, our experiments indicate that the erastin-induced reductions in P-glycoprotein at the BBB are not due to physical damage or loss of barrier integrity. Our work also shows that erastin is transporter-specific. For example, under identical exposure conditions that rapidly reduce P-glycoprotein activity in males, we observed erastin-induced increases in BCRP and MRP2 transport activities. However, in females, only MRP2 transport activity was increased by 1.0 nM erastin, indicating that the effects of erastin on the transporters of BBB are transporter- and sex-specific. Our experiments also show that erastin-dependent reductions in P-glycoprotein transport activity are rapidly reversed upon erastin removal. This rapid reversal back to basal levels suggests the involvement of signaling pathways that affect transport activity without changing protein levels of expression. Our Western blots, measuring protein levels, confirmed that erastin treatments did not alter P-glycoprotein expression levels. These data taken together suggest that erastin-induced reductions in P-glycoprotein transport activity were not due to protein degradation. The data are more consistent with the notion that erastin is modulating important signaling pathways that regulate transport activity at the BBB.
To understand the effect of nM concentrations of erastin in BBB endothelial cells, we used a novel fluorescent-based assay that measured the uptake of selenocystine through the system X
C− antiporter [
15]. To our knowledge, this is the first time it has been used on brain microvessels. All previous reports exposed cells to high µM concentrations of erastin to cause inhibition of cystine uptake. Using freshly isolated rat brain capillaries, we found that low nM amounts of erastin rapidly inhibited cystine uptake. We logically reasoned that lower cysteine levels could contribute to GSH depletion. Furthermore, we wondered if reduced GSH levels were also responsible for the rapid decreases in P-glycoprotein transport activity we observed in our rat brain capillaries. To address this, we co-treated capillaries with 1.0 nM erastin plus NAC or cysteine, two known precursors to GSH. We found that co-treating with either alone blocked the repressive effects of erastin on P-glycoprotein transport activity. These experiments suggested that oxidative stress in the absence of sufficient levels of GSH could activate signaling pathways that altered the pump activities of important transporters at the BBB (e.g., P-glycoprotein).
In endothelial cells, oxidative stress can lead to increases in NO production. This is especially true during high and rapid states of cytokine release from inflammatory conditions such as rheumatoid arthritis (RA) [
27] or chronic brain disorders such as multiple sclerosis (MS) [
28]. This associative link between oxidative stress and NO (NOS activity) extends to previously described signaling pathways that regulate P-glycoprotein in rat brain capillaries [
7]. We found that human cancer cells treated with NO donor compounds become more sensitive to killing by P-glycoprotein substrates [
29]. These findings are consistent with the notion that NO is involved (required) in the inhibition of P-glycoprotein transport activity. Lastly, it has been recently reported that human cancer cells exposed to erastin exhibit increases in iNOS activity and expression [
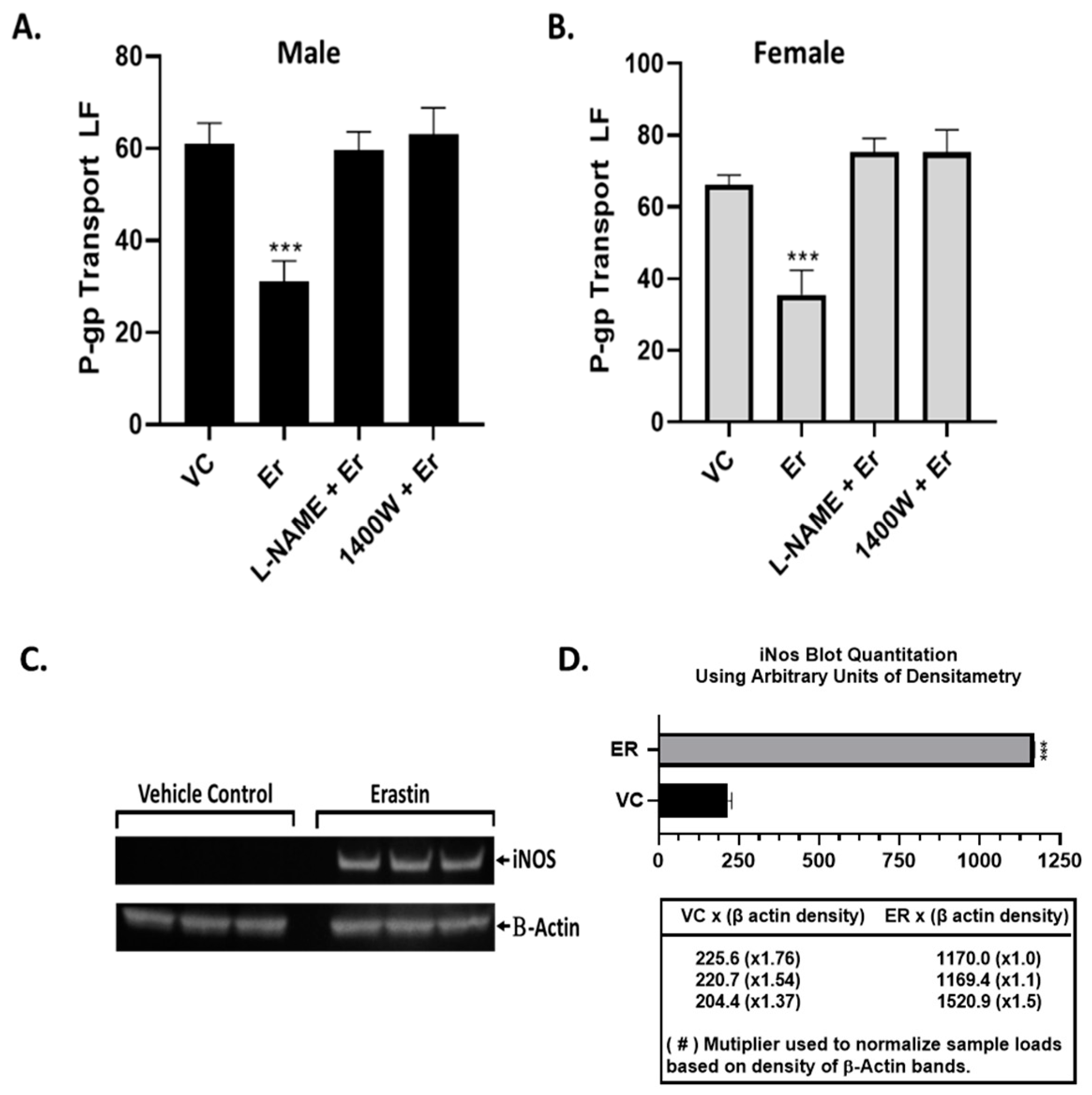
21]. Our Western blots measuring iNOS protein levels clearly show that iNOS is rapidly induced by nM levels of erastin. Furthermore, we found either the pan-NOS inhibitor (L-NAME) or the selective iNOS inhibitor (1400 W) blocked the erastin-induced inhibition of P-glycoprotein transport activity. These observations are consistent with previous studies involving iNOS-containing signaling pathways that rapidly inhibit P-glycoprotein transport activity.
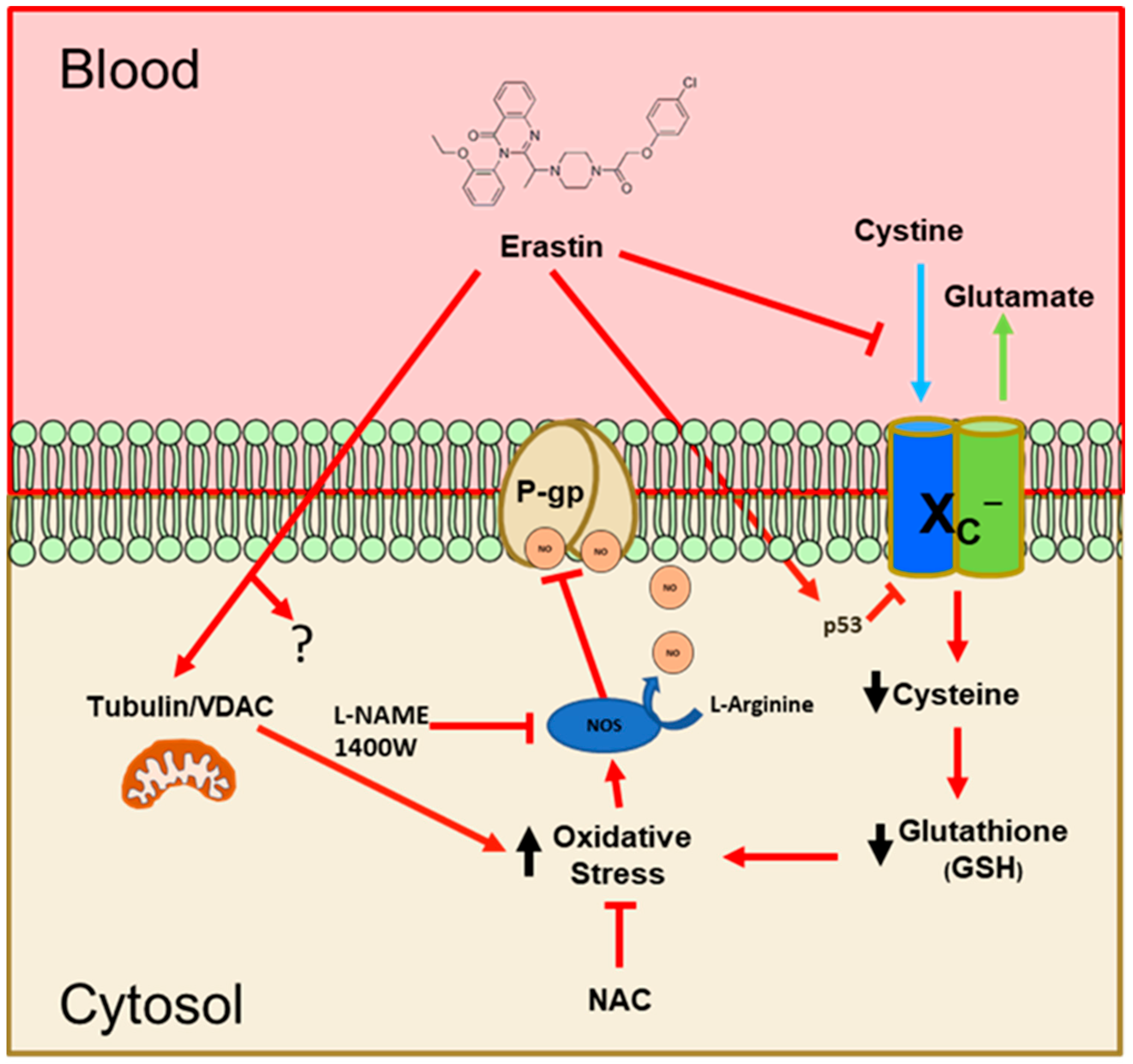
To summarize our work, we have proposed a model that integrates our findings and helps to understand how erastin rapidly reduces P-glycoprotein activity at the BBB. As shown in
Figure 8, our findings indicate that nM levels of erastin reduce the uptake of cystine through the system X
C− antiporter. This has been previously reported to occur in other cell types, but at concentrations that are 10–100 times higher [
30]. Inhibition of P-glycoprotein (ABCB1) by erastin has also been previously reported in human ovarian cancer cells [
31]. The suggested mechanism in these studies involved substrate competition. Their work also used erastin exposures of 48 h at 1.0–30.0 µM. We have not evaluated erastin at high µM concentrations in brain capillaries, but our work using low nM concentrations of erastin does not support a competitive substrate model of inhibition. Also, antioxidants and NOS inhibitors blocked the effects of erastin on P-glycoprotein in our experiments, eliminating a competitive model and supporting the notion that erastin is activating a signaling pathway to inhibit P-glycoprotein transport activity.
Two other possible erastin-mediated perturbations we have not examined are TP53 and/or mitochondrial VDAC function. We cannot eliminate the possibility that either or both may be involved in our observed effects of erastin. However, we found that exogenous NAC, or cysteine, completely blocks the repressive effects of erastin on P-glycoprotein transport, suggesting that the loss of cystine uptake is the primary and causal event. Loss of intracellular cystine and subsequent decreases in GSH are known to lead to oxidative stress. We show that in rat brain capillaries exposed to erastin, there is an increase in NO production via iNOS. Based on previous and present work, we believe that NO inhibits the ATPase activity of P-glycoprotein [
19]. Further work will be needed to confirm the identity of the nitrosylation sites on P-glycoprotein. However, we have previously identified putative sites within the ATPase domains that could be involved [
29]. This work is important because of the essential function the BBB plays in protecting the brain. Clinically, it suggests that erastin could rapidly lower P-glycoprotein activity at the BBB to facilitate the entry of drugs to reach targets within the brain/CNS. This work also suggests that erastin could be useful as an adjunct in cancer therapies by lowering chemoresistance (MDR), rendering cancer cells more sensitive to certain chemotherapeutic drugs. This could be clinically important given that the majority of chemotherapeutics are P-glycoprotein substrates.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}