
Immune System Disorder and Cancer-Associated Cachexia

Abstract
Simple Summary
Abstract

1. Introduction
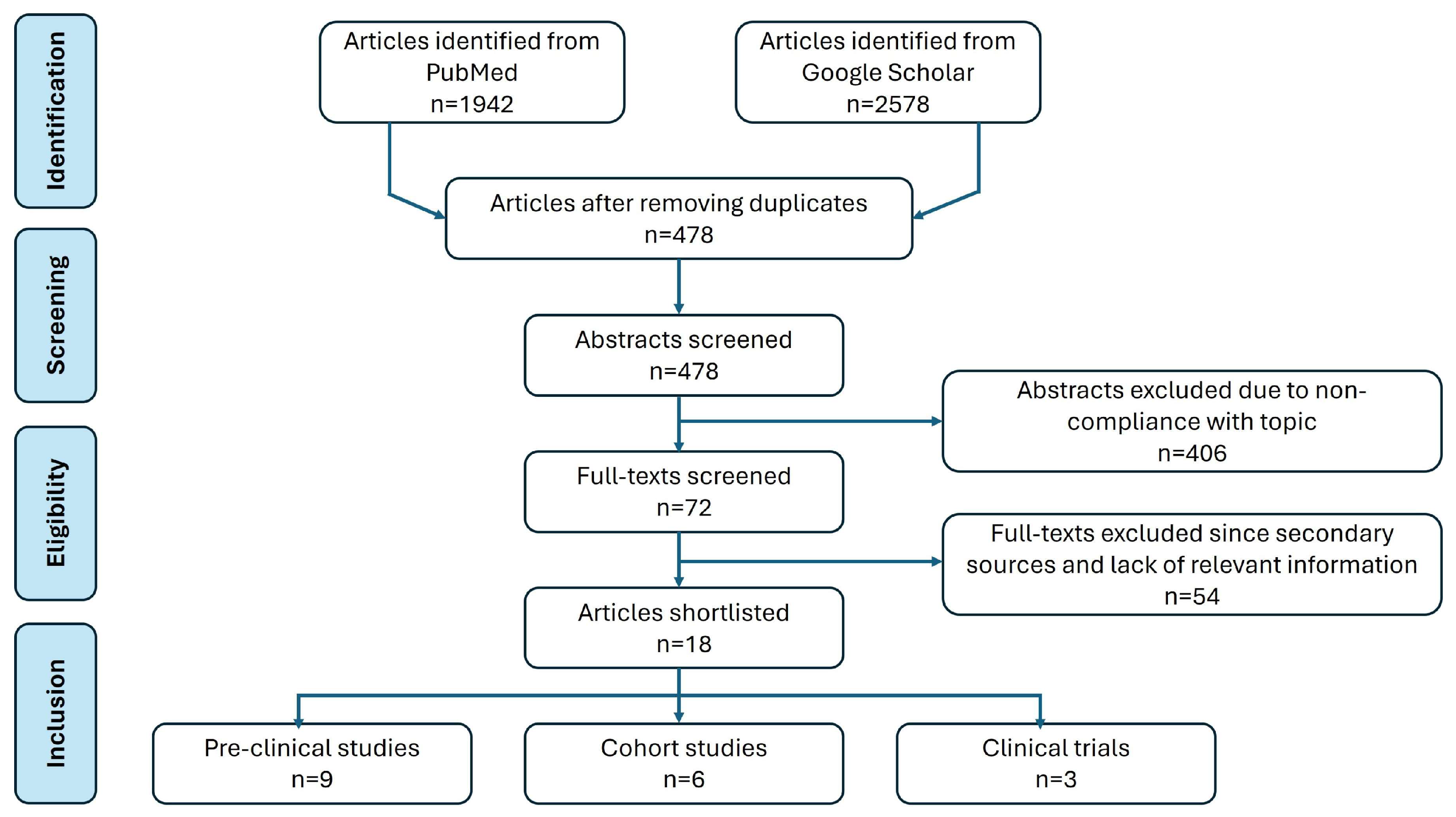
2. Methodology
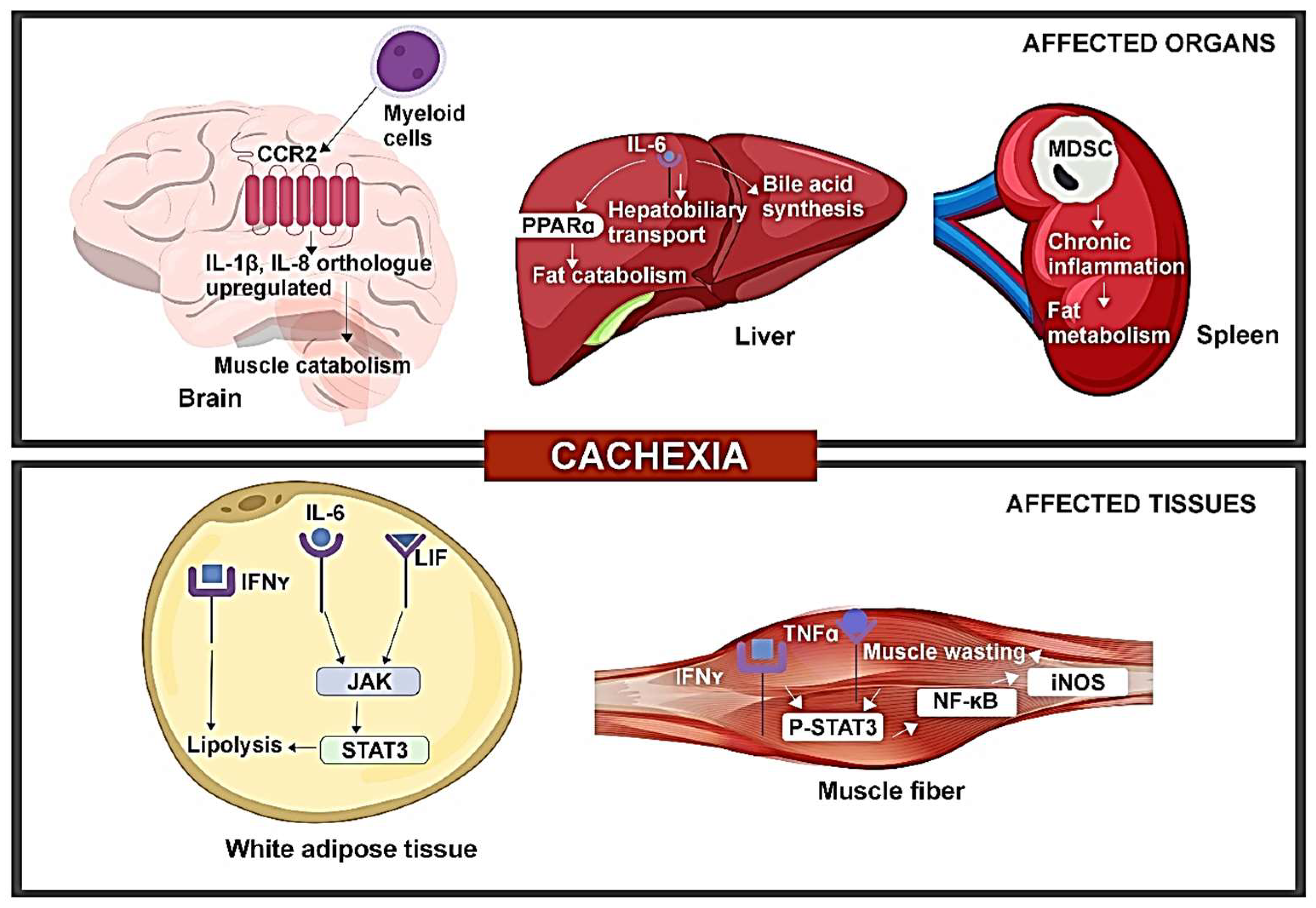
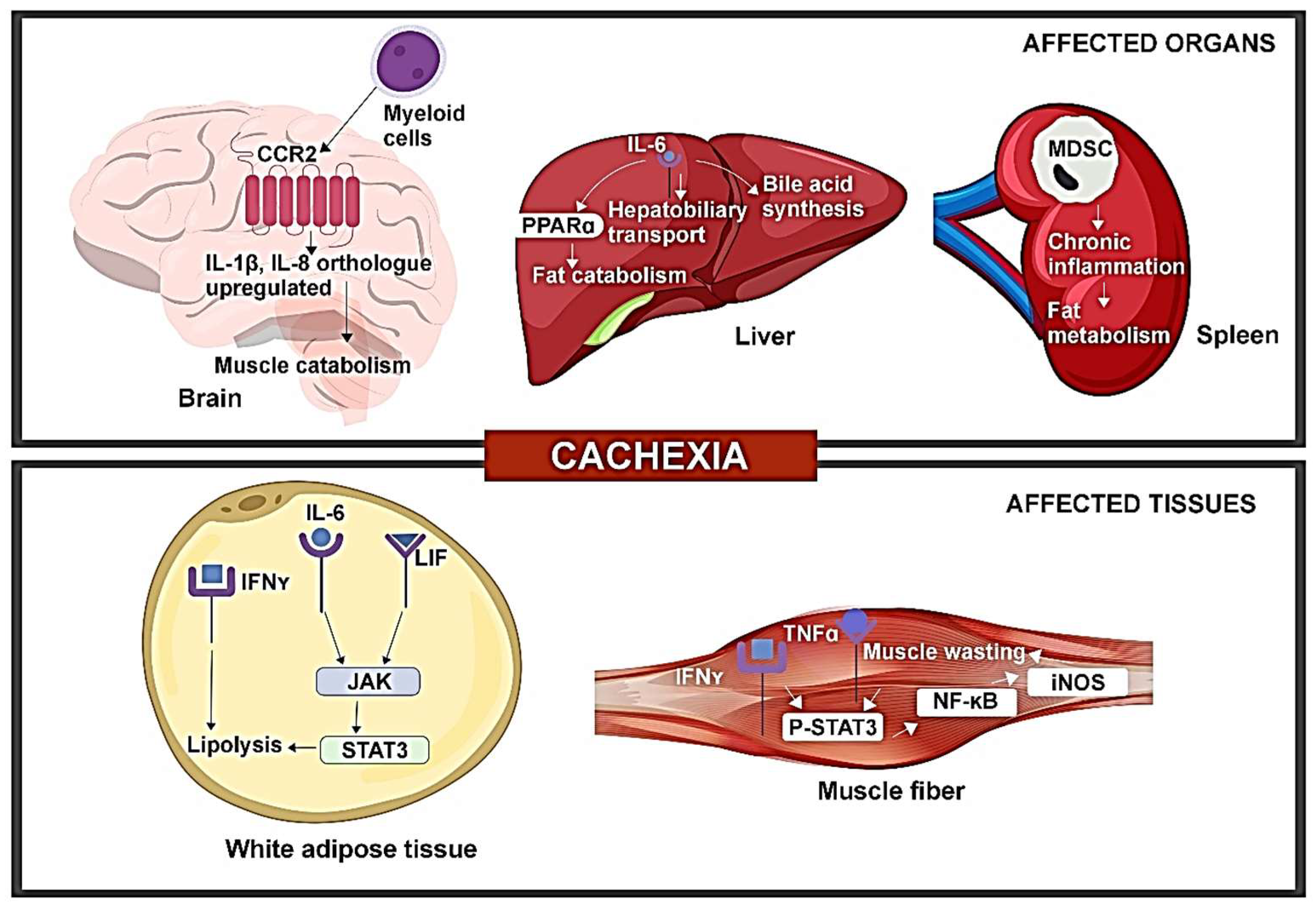
3. Pre-Clinical Evidence of Tumor-Induced Immune Disorder Driving CAC
4. Cellular and Molecular Immunomodulation Associated with CAC in Patients with Late-Stage Cancer
5. Clinical Implications and Future Directions
6. Strengths and Limitations of the Study
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, Q.; Liu, Z.; Li, B.; Liu, Y.E.; Wang, P. Immunoregulation in cancer-associated cachexia. J. Adv. Res. 2023, 58, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Khatami, M. Unresolved inflammation:‘immune tsunami’or erosion of integrity in immune-privileged and immune-responsive tissues and acute and chronic inflammatory diseases or cancer. Expert Opin. Biol. Ther. 2011, 11, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.; Samaden, M.; Ruggieri, E.; Venereau, E. Cancer cachexia as a multiorgan failure: Reconstruction of the crime scene. Front. Cell Dev. Biol. 2022, 10, 960341. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Marceca, G.P.; Londhe, P.; Calore, F. Management of Cancer Cachexia: Attempting to Develop New Pharmacological Agents for New Effective Therapeutic Options. Front. Oncol. 2020, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; An, Z.Y.; Lin, D.H.; Jin, W.L. Targeting cancer cachexia: Molecular mechanisms and clinical study. MedComm 2022, 3, e164. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Tsai, K.W.; Lu, K.C.; Shih, L.J.; Hu, W.C. Cancer as a Dysfunctional Immune Disorder: Pro-Tumor TH1-like Immune Response and Anti-Tumor THαβ Immune Response Based on the Complete Updated Framework of Host Immunological Pathways. Biomedicines 2022, 10, 2497. [Google Scholar] [CrossRef]
- Sakowska, J.; Arcimowicz, Ł.; Jankowiak, M.; Papak, I.; Markiewicz, A.; Dziubek, K.; Kurkowiak, M.; Kote, S.; Kaźmierczak-Siedlecka, K.; Połom, K.; et al. Autoimmunity and Cancer—Two Sides of the Same Coin. Front. Immunol. 2022, 13, 793234. [Google Scholar] [CrossRef] [PubMed]
- Valencia, J.C.; Egbukichi, N.; Erwin-Cohen, R.A. Autoimmunity and Cancer, the Paradox Comorbidities Challenging Therapy in the Context of Preexisting Autoimmunity. J. Interferon Cytokine Res. 2019, 39, 72–84. [Google Scholar] [CrossRef]
- Han, J.; Lu, C.; Meng, Q.; Halim, A.; Yean, T.J.; Wu, G. Plasma concentration of interleukin-6 was upregulated in cancer cachexia patients and was positively correlated with plasma free fatty acid in female patients. Nutr. Metab. 2019, 16, 80. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Huisman, C.; Buenafe, A.C.; Olson, B.; Michaelis, K.A.; Torres, E.R.; Jeng, S.; et al. Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. Elife 2020, 9, e54095. [Google Scholar] [CrossRef]
- Ma, J.F.; Sanchez, B.J.; Hall, D.T.; Tremblay, A.K.; Di Marco, S.; Gallouzi, I.E. STAT3 promotes IFNγ/TNFα-induced muscle wasting in an NF-κB-dependent and IL-6-independent manner. EMBO Mol. Med. 2017, 9, 622–637. [Google Scholar] [CrossRef]
- Lima, J.; Simoes, E.; de Castro, G.; Morais, M.; de Matos-Neto, E.M.; Alves, M.J.; Pinto, N.I.; Figueredo, R.G.; Zorn, T.M.T.; Felipe-Silva, A.S.; et al. Tumour-derived transforming growth factor-β signalling contributes to fibrosis in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 1045–1059. [Google Scholar] [CrossRef]
- Yamashita, A.S.; das Neves, R.X.; Rosa-Neto, J.C.; Lira, F.D.; Batista, M.L., Jr.; Alcantara, P.S.; Otoch, J.P.; Seelaender, M. White adipose tissue IFN-γ expression and signalling along the progression of rodent cancer cachexia. Cytokine 2016, 89, 122–126. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, C.; Moya, R.; Davies, J.D. A novel role for CD4+ T cells in the control of cachexia. J. Immunol. 2008, 181, 4676–4684. [Google Scholar] [CrossRef]
- Arora, G.; Gupta, A.; Guo, T.; Gandhi, A.; Laine, A.; Williams, D.; Ahn, C.; Iyengar, P.; Infante, R. JAK Inhibitors Suppress Cancer Cachexia-Associated Anorexia and Adipose Wasting in Mice. JCSM Rapid Commun. 2020, 3, 115–128. [Google Scholar] [CrossRef]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef]
- Rounis, K.; Makrakis, D.; Tsigkas, A.-P.; Georgiou, A.; Galanakis, N.; Papadaki, C.; Monastirioti, A.; Vamvakas, L.; Kalbakis, K.; Vardakis, N.; et al. Cancer cachexia syndrome and clinical outcome in patients with metastatic non-small cell lung cancer treated with PD-1/PD-L1 inhibitors: Results from a prospective, observational study. Transl. Lung Cancer Res. 2021, 10, 3538–3549. [Google Scholar] [CrossRef]
- Fujii, H.; Araki, A.; Iihara, H.; Kaito, D.; Hirose, C.; Kinomura, M.; Yamazaki, M.; Endo, J.; Inui, T.; Yanase, K.; et al. Cancer cachexia as a determinant of efficacy of first-line pembrolizumab in patients with advanced non-small cell lung cancer. Mol. Clin. Oncol. 2022, 16, 91. [Google Scholar] [CrossRef]
- Daas, S.I.; Rizeq, B.R.; Nasrallah, G.K. Adipose tissue dysfunction in cancer cachexia. J. Cell. Physiol. 2018, 234, 13–22. [Google Scholar] [CrossRef]
- Cuenca, A.G.; Cuenca, A.L.; Winfield, R.D.; Joiner, D.N.; Gentile, L.; Delano, M.J.; Kelly-Scumpia, K.M.; Scumpia, P.O.; Matheny, M.K.; Scarpace, P.J.; et al. Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J. Immunol. 2014, 192, 6111–6119. [Google Scholar] [CrossRef]
- Thibaut, M.M.; Sboarina, M.; Roumain, M.; Potgens, S.A.; Neyrinck, A.M.; Destree, F.; Gillard, J.; Leclercq, I.A.; Dachy, G.; Demoulin, J.B.; et al. Inflammation-induced cholestasis in cancer cachexia. J. Cachexia Sarcopenia Muscle 2021, 12, 70–90. [Google Scholar] [CrossRef]
- Chrysostomou, S.E.; Eder, S.; Pototschnig, I.; Mayer, A.L.; Derler, M.; Mussbacher, M.; Schauer, S.; Zhang, D.; Yan, D.; Liu, G.; et al. R-ketorolac ameliorates cancer-associated cachexia and prolongs survival of tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2024, 15, 562–574. [Google Scholar] [CrossRef]
- Zhang, Q.; Song, M.M.; Zhang, X.; Ding, J.S.; Ruan, G.T.; Zhang, X.W.; Liu, T.; Yang, M.; Ge, Y.Z.; Tang, M.; et al. Association of systemic inflammation with survival in patients with cancer cachexia: Results from a multicentre cohort study. J. Cachexia Sarcopenia Muscle 2021, 12, 1466–1476. [Google Scholar] [CrossRef]
- Alves, M.J.; Figueredo, R.G.; Azevedo, F.F.; Cavallaro, D.A.; Neto, N.I.; Lima, J.D.; Matos-Neto, E.; Radloff, K.; Riccardi, D.M.; Camargo, R.G.; et al. Adipose tissue fibrosis in human cancer cachexia: The role of TGFβ pathway. BMC Cancer 2017, 17, 190. [Google Scholar] [CrossRef]
- Hou, Y.C.; Wang, C.J.; Chao, Y.J.; Chen, H.Y.; Wang, H.C.; Tung, H.L.; Lin, J.T.; Shan, Y.S. Elevated Serum Interleukin-8 Level Correlates with Cancer-Related Cachexia and Sarcopenia: An Indicator for Pancreatic Cancer Outcomes. J. Clin. Med. 2018, 7, 502. [Google Scholar] [CrossRef]
- de Jesus, J.C.R.; Murari, A.S.P.; Radloff, K.; de Moraes, R.C.M.; Figueredo, R.G.; Pessoa, A.F.M.; Rosa-Neto, J.C.; Matos-Neto, E.M.; Alcantara, P.S.M.; Tokeshi, F.; et al. Activation of the Adipose Tissue NLRP3 Inflammasome Pathway in Cancer Cachexia. Front. Immunol. 2021, 12, 729182. [Google Scholar] [CrossRef]
- Suleyman, H.; Demircan, B.; Karagoz, Y. Anti-inflammatory and side effects of cyclo-oxygenase inhibitors. Pharmacol. Rep. 2007, 59, 247–258. [Google Scholar]
- Bonomi, P.; Fidler, M.J.; Shah, P.; Borgia, J. Theoretical and Practical Implications of Treating Cachexia in Advanced Lung Cancer Patients. Cancers 2019, 11, 1619. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Ascierto, P.A.; Pigozzo, J.; Del Vecchio, M.; Maio, M.; Antonini Cappellini, G.C.; Guidoboni, M.; Queirolo, P.; Savoia, P.; Mandala, M.; et al. Baseline neutrophils and derived neutrophil-to-lymphocyte ratio: Prognostic relevance in metastatic melanoma patients receiving ipilimumab. Ann. Oncol. 2016, 27, 732–738. [Google Scholar] [CrossRef]
- Mezquita, L.; Auclin, E.; Ferrara, R.; Charrier, M.; Remon, J.; Planchard, D.; Ponce, S.; Ares, L.P.; Leroy, L.; Audigier-Valette, C.; et al. Association of the Lung Immune Prognostic Index with Immune Checkpoint Inhibitor Outcomes in Patients With Advanced Non-Small Cell Lung Cancer. JAMA Oncol. 2018, 4, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Monk, J.P.; Phillips, G.; Waite, R.; Kuhn, J.; Schaaf, L.J.; Otterson, G.A.; Guttridge, D.; Rhoades, C.; Shah, M.; Criswell, T.; et al. Assessment of tumor necrosis factor alpha blockade as an intervention to improve tolerability of dose-intensive chemotherapy in cancer patients. J. Clin. Oncol. 2006, 24, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, T.; Smith, J.; Schuste, r.M.; Dragnev, K.; Rigas, J. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin. Biol. Ther. 2011, 11, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Chasen, M.; Hirschman, S.Z.; Bhargava, R. Phase II study of the novel peptide-nucleic acid OHR118 in the management of cancer-related anorexia/cachexia. J. Am. Med. Dir. Assoc. 2011, 12, 62–67. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrial. A Feasibility Study of Ketorolac Treatment for Cachexia in Patients with Advanced Pancreatic Ductal Adenocarcinoma (KetoROCX). Available online: https://clinicaltrials.gov/study/NCT05336266 (accessed on 1 February 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study and Reference | Cancer Type | Sample Size | Objective | Outcome |
|---|---|---|---|---|
| Zhang et al., 2021 [24] | Solid malignant tumors | N = 2612 | Evaluation of NLR as a biomarker in CAC | NLR increases in CAC NLR > 3.5 can predict survival |
| Alves et al., 2017 [25] | Gastrointestinal cancer | N = 59 | Assessment of changes in extracellular matrix in CAC | Fibrosis of adipose tissue and alterations in TGFβ level observed |
| CAC: 21 | ||||
| WSC: 17 | ||||
| Control: 21 | ||||
| Hou et al., 2018 [26] | Pancreatic cancer | N = 146 | Relation between CAC status and inflammatory cytokines | Serum IL-8 level can predict survival IL-8 can be an indicator of pancreatic cancer |
| LA group: 55 (CAC: 51, Non-CAC: 4) | ||||
| Resected group: 91 (CAC: 59, Non-CAC: 32) | ||||
| Han et al., 2019 [10] | Gastric cancer or colorectal cancer | N = 311 | Clinical characteristics of patients (CAC and non-CAC) regarding the level of inflammation factors and lipid metabolism parameters | IL-6 levels varied between patient groups; females with CAC exhibited elevated IL-6 and FFA levels |
| CAC: 74 | ||||
| Non-CAC: 237 | ||||
| de Jesus et al., 2021 [27] | Colorectal tumors | N = 28 CAC: 10 WSC: 10 Control: 8 | To confirm that NLRP3 inflammasome pathways are triggered in adipose tissue of CAC | Adipose tissue in CAC is involved in inflammation and its regulation is depot-specific |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Bonomi, P.D. Immune System Disorder and Cancer-Associated Cachexia. Cancers 2024, 16, 1709. https://doi.org/10.3390/cancers16091709
Zhang L, Bonomi PD. Immune System Disorder and Cancer-Associated Cachexia. Cancers. 2024; 16(9):1709. https://doi.org/10.3390/cancers16091709
Chicago/Turabian StyleZhang, Lingbing, and Philip D. Bonomi. 2024. "Immune System Disorder and Cancer-Associated Cachexia" Cancers 16, no. 9: 1709. https://doi.org/10.3390/cancers16091709
APA StyleZhang, L., & Bonomi, P. D. (2024). Immune System Disorder and Cancer-Associated Cachexia. Cancers, 16(9), 1709. https://doi.org/10.3390/cancers16091709