
Dendritic Cells in Cancer Immunology and Immunotherapy
 , ,
, ,  , , ,
, , , 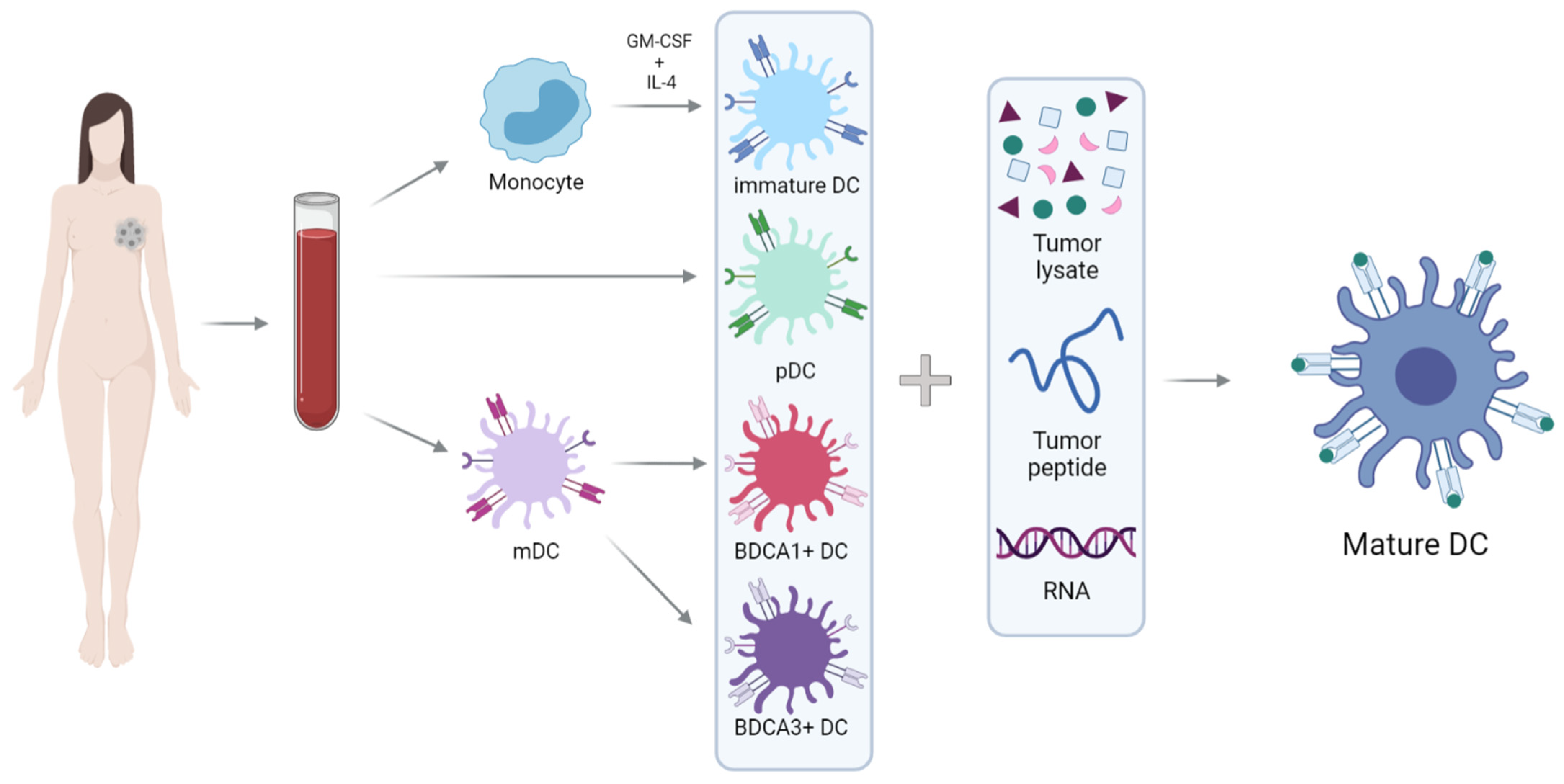{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
1.1. An Inadequate Formulation of the Vaccine
1.2. A Poor Choice of the Antigens
1.3. The Route of Administration of the Vaccine
1.4. Inappropriate Selection of Patients
2. DC Vaccines in Solid Tumors
2.1. Breast Cancer
2.2. Brain Tumors
2.3. Colorectal Cancer
2.4. Gynecological Cancers
2.5. Melanoma
2.6. Urologic Tumors
3. New Trends
DCV Pulsed with Neoantigens
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Colaprico, A.; Silva, T.C.; Chen, J.; An, H.; Ban, Y.; Huang, H.; Wang, L.; James, J.L.; Balko, J.M.; et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat. Commun. 2021, 12, 6276. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Berk, E.; Cintolo, J.A.; Xu, S.; Roses, R.E.; Czerniecki, B.J. Rationale for a Multimodality Strategy to Enhance the Efficacy of Dendritic Cell-Based Cancer Immunotherapy. Front. Immunol. 2015, 6, 271. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.S. Tumor vaccines for breast cancer. Cancer Investig. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Bucovec, R.; Meregalli, S.; Fumagalli, L.; Vigorè, L.; Ferrante, R.; Brivio, F. IL-2 Immunotherapy-Induced Increase in IL-12 Blood Concentrations May Depend on an Increase in Circulating Dendritic Cell Number. Int. J. Biol. Markers 1999, 14, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Diao, J.; Cattral, M.S. Molecular mechanisms involved in dendritic cell dysfunction in cancer. Cell. Mol. Life Sci. 2017, 74, 761–776. [Google Scholar] [CrossRef]
- Lurje, I.; Hammerich, L.; Tacke, F. Dendritic Cell and T Cell Crosstalk in Liver Fibrogenesis and Hepatocarcinogenesis: Implications for Prevention and Therapy of Liver Cancer. Int. J. Mol. Sci. 2020, 21, 7378. [Google Scholar] [CrossRef]
- Hsu, F.J.; Benike, C.; Fagnoni, F.; Liles, T.M.; Czerwinski, D.; Taidi, B.; Engleman, E.G.; Levy, R. Vaccination of patients with B–cell lymphoma using autologous antigen–pulsed dendritic cells. Nat. Med. 1996, 2, 52–58. [Google Scholar] [CrossRef]
- Nestle, F.O.; Tun Kyi, A.; Gilliet, M.; Burg, G. Vaccination therapy of malignant melanoma. Ther. Umsch. 1999, 56, 334–337. [Google Scholar] [CrossRef]
- Nestle, F.O.; Tun-Kyi, A.; Gilliet, M.F. Antigen-pulsed dendritic cell approach to melanoma. Methods Mol. Med. 2001, 61, 195–202. [Google Scholar]
- Raaijmakers, T.K.; Ansems, M. Microenvironmental derived factors modulating dendritic cell function and vaccine efficacy: The effect of prostanoid receptor and nuclear receptor ligands. Cancer Immunol. Immunother. 2018, 67, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.J.; Ning, Y.L.; Han, Y.S.; Min, H.Y.; Ye, H.; Zhu, Y.L.; Qian, K.Q. Autologous dendritic cell vaccine for estrogen receptor (ER)/progestin receptor (PR) double-negative breast cancer. Cancer Immunol. Immunother. 2012, 61, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Aarntzen, E.H.J.G.; Hout, F.E.M.I.; Schreibelt, G.; Creemers, J.H.A.; Lesterhuis, W.J.; Gerritsen, W.R.; Grunhagen, D.J.; Verhoef, C.; Punt, C.J.A.; et al. Favorable overall survival in stage III melanoma patients after adjuvant dendritic cell vaccination. OncoImmunology 2016, 5, e1057673. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; de Vries, I.J.M.; Figdor, C.G. Dendritic Cell–Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef]
- Gulley, J.L.; Madan, R.A.; Tsang, K.Y.; Jochems, C.; Marté, J.L.; Farsaci, B.; Tucker, J.A.; Hodge, J.W.; Liewehr, D.J.; Steinberg, S.M.; et al. Immune Impact Induced by PROSTVAC (PSA-TRICOM), a Therapeutic Vaccine for Prostate Cancer. Cancer Immunol. Res. 2014, 2, 133–141. [Google Scholar] [CrossRef]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2018, 9, 3176. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Niemi, J.V.L.; Sokolov, A.V.; Schiöth, H.B. Neoantigen Vaccines; Clinical Trials, Classes, Indications, Adjuvants and Combinatorial Treatments. Cancers 2022, 14, 5163. [Google Scholar] [CrossRef] [PubMed]
- González, F.E.; Gleisner, A.; Falcón-Beas, F.; Osorio, F.; López, M.N.; Salazar-Onfray, F. Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum. Vaccines Immunother. 2014, 10, 3261–3269. [Google Scholar] [CrossRef] [PubMed]
- de Vries, I.J.M.; Bernsen, M.R.; Lesterhuis, W.J.; Scharenborg, N.M.; Strijk, S.P.; Gerritsen, M.-J.P.; Ruiter, D.J.; Figdor, C.G.; Punt, C.J.; Adema, G.J. Immunomonitoring Tumor-Specific T Cells in Delayed-Type Hypersensitivity Skin Biopsies After Dendritic Cell Vaccination Correlates With Clinical Outcome. J. Clin. Oncol. 2005, 23, 5779–5787. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; de Vries, I.J.M.; Schreibelt, G.; Lambeck, A.J.; Aarntzen, E.H.; Jacobs, J.F.; Scharenborg, N.M.; van de Rakt, M.W.; de Boer, A.J.; Croockewit, S.; et al. Route of Administration Modulates the Induction of Dendritic Cell Vaccine–Induced Antigen-Specific T Cells in Advanced Melanoma Patients. Clin. Cancer Res. 2011, 17, 5725–5735. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Mailliard, R.B.; Wankowicz-Kalinska, A.; Cai, Q.; Wesa, A.; Hilkens, C.M.; Kapsenberg, M.L.; Kirkwood, J.M.; Storkus, W.J.; Kalinski, P. alpha-type-1 polarized dendritic cells: A novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004, 64, 5934–5937. [Google Scholar] [CrossRef] [PubMed]
- Trepiakas, R.; Pedersen, A.E.; Met, Ö.; Hansen, M.H.; Berntsen, A.; Svane, I.M. Comparison of α-Type-1 polarizing and standard dendritic cell cytokine cocktail for maturation of therapeutic monocyte-derived dendritic cell preparations from cancer patients. Vaccine 2008, 26, 2824–2832. [Google Scholar] [CrossRef]
- Lapenta, C.; Gabriele, L.; Santini, S.M. IFN-Alpha-Mediated Differentiation of Dendritic Cells for Cancer Immunotherapy: Advances and Perspectives. Vaccines 2020, 8, 617. [Google Scholar] [CrossRef]
- Colonna, M.; Trinchieri, G.; Liu, Y.-J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef]
- Schreibelt, G.; Tel, J.; Sliepen, K.H.E.W.J.; Benitez-Ribas, D.; Figdor, C.G.; Adema, G.J.; De Vries, I.J.M. Toll-like receptor expression and function in human dendritic cell subsets: Implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1573–1582. [Google Scholar] [CrossRef]
- Liu, Y.J.; Kanzler, H.; Soumelis, V.; Gilliet, M. Dendritic cell lineage, plasticity and cross-regulation. Nat. Immunol. 2001, 2, 585–589. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Piccioli, D.; Sammicheli, C.; Tavarini, S.; Nuti, S.; Frigimelica, E.; Manetti, A.G.; Nuccitelli, A.; Aprea, S.; Valentini, S.; Borgogni, E.; et al. Human plasmacytoid dendritic cells are unresponsive to bacterial stimulation and require a novel type of cooperation with myeloid dendritic cells for maturation. Blood 2009, 113, 4232–4239. [Google Scholar] [CrossRef] [PubMed]
- Mathan, T.S.M.M.; Figdor, C.G.; Buschow, S.I. Human Plasmacytoid Dendritic Cells: From Molecules to Intercellular Communication Network. Front. Immunol. 2013, 4, 372. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Schreibelt, G.; Sittig, S.P.; Mathan, T.S.M.; Buschow, S.I.; Cruz, L.J.; Lambeck, A.J.A.; Figdor, C.G.; de Vries, I.J.M. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood 2013, 121, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural Human Plasmacytoid Dendritic Cells Induce Antigen-Specific T-Cell Responses in Melanoma Patients. Cancer Res 2013, 73, 1063–1075. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J. Immunother. Cancer 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.P.A.; Munster, D.J.; Clark, G.J.; Dzionek, A.; Schmitz, J.; Hart, D.N.J. Characterization of human blood dendritic cell subsets. Blood 2002, 100, 4512–4520. [Google Scholar] [CrossRef] [PubMed]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef]
- Yan, Z.; Wu, Y.; Du, J.; Li, G.; Wang, S.; Cao, W.; Zhou, X.; Wu, C.; Zhang, D.; Jing, X.; et al. A novel peptide targeting Clec9a on dendritic cell for cancer immunotherapy. Oncotarget 2016, 7, 40437–40450. [Google Scholar] [CrossRef]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis E Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef]
- Iborra, S.; Izquierdo, H.M.; Martínez-López, M.; Blanco-Menéndez, N.; Reis E Sousa, C.; Sancho, D. The DC receptor DNGR-1 mediates cross-priming of CTLs during vaccinia virus infection in mice. J. Clin. Investig. 2012, 122, 1628–1643. [Google Scholar] [CrossRef] [PubMed]
- Iborra, S.; Martínez-López, M.; Khouili, S.C.; Enamorado, M.; Cueto, F.J.; Conde-Garrosa, R.; del Fresno, C.; Sancho, D. Optimal Generation of Tissue-Resident but Not Circulating Memory T Cells during Viral Infection Requires Crosspriming by DNGR-1 + Dendritic Cells. Immunity 2016, 45, 847–860. [Google Scholar] [CrossRef]
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martínez, D.; Hernanz-Falcón, P.; Rosewell, I.; Reis E Sousa, C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009, 458, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-G.; Czabotar, P.E.; Policheni, A.N.; Caminschi, I.; Wan, S.S.; Kitsoulis, S.; Tullett, K.M.; Robin, A.Y.; Brammananth, R.; van Delft, M.F.; et al. The Dendritic Cell Receptor Clec9A Binds Damaged Cells via Exposed Actin Filaments. Immunity 2012, 36, 646–657. [Google Scholar] [CrossRef]
- Bachem, A.; Güttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.-L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef]
- Fields, R.C.; Shimizu, K.; Mule, J.J. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 9482–9487. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Apostolopoulos, V.; Chen, D.; Chen, H.; Koido, S.; Gendler, S.J.; Mckenzie, I.F.; Kufe, D. Selection and characterization of MUC1-specific CD8+ T cells from MUC1 transgenic mice immunized with dendritic-carcinoma fusion cells. Immunology 2000, 101, 316–324. [Google Scholar] [CrossRef]
- Neidhardt-Berard, E.-M.; Berard, F.; Banchereau, J.; Palucka, A.K. Dendritic cells loaded with killed breast cancer cells induce differentiation of tumor-specific cytotoxic T lymphocytes. Breast Cancer Res. 2004, 6, R322–R328. [Google Scholar] [CrossRef]
- Brossart, P. Dendritic cells in vaccination therapies of malignant diseases. Transfus. Apher. Sci. 2002, 27, 183–186. [Google Scholar] [CrossRef]
- Zheng, J.; Liu, Q.; Yang, J.; Ren, Q.; Cao, W.; Yang, J.; Yu, Z.; Yu, F.; Wu, Y.; Shi, H.; et al. Co-culture of apoptotic breast cancer cells with immature dendritic cells: A novel approach for DC-based vaccination in breast cancer. Braz. J. Med. Biol. Res. 2012, 45, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Avigan, D. Dendritic cell/tumour fusions vaccine. Dev. Biol. 2004, 116, 161–168, discussion 179–186. [Google Scholar]
- Baek, S.; Lee, S.J.; Kim, M.J.; Lee, H. Dendritic Cell (DC) Vaccine in Mouse Lung Cancer Minimal Residual Model; Comparison of Monocyte-derived DC vs. Hematopoietic Stem Cell Derived-DC. Immune Netw. 2012, 12, 269–276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morse, M.A.; Hobeika, A.C.; Osada, T.; Serra, D.; Niedzwiecki, D.; Lyerly, H.K.; Clay, T.M. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood 2008, 112, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Maeng, H.M.; Moore, B.N.; Bagheri, H.; Steinberg, S.M.; Inglefield, J.; Dunham, K.; Wei, W.-Z.; Morris, J.C.; Terabe, M.; England, L.C.; et al. Phase I Clinical Trial of an Autologous Dendritic Cell Vaccine Against HER2 Shows Safety and Preliminary Clinical Efficacy. Front. Oncol. 2021, 11, 789078. [Google Scholar] [CrossRef] [PubMed]
- Lowenfeld, L.; Mick, R.; Datta, J.; Xu, S.; Fitzpatrick, E.; Fisher, C.S.; Fox, K.R.; DeMichele, A.; Zhang, P.J.; Weinstein, S.P.; et al. Dendritic Cell Vaccination Enhances Immune Responses and Induces Regression of HER2pos DCIS Independent of Route: Results of Randomized Selection Design Trial. Clin. Cancer Res. 2017, 23, 2961–2971. [Google Scholar] [CrossRef]
- Santisteban, M.; Solans, B.P.; Hato, L.; Urrizola, A.; Mejías, L.D.; Salgado, E.; Sánchez-Bayona, R.; Toledo, E.; Rodríguez-Spiteri, N.; Olartecoechea, B.; et al. Final results regarding the addition of dendritic cell vaccines to neoadjuvant chemotherapy in early HER2-negative breast cancer patients: Clinical and translational analysis. Ther. Adv. Med. Oncol. 2021, 13, 17588359211064653. [Google Scholar] [CrossRef]
- Mejías Sosa, L.; López-Janeiro, Á.; Córdoba Iturriagagoitia, A.; Sala, P.; Solans, B.P.; Hato, L.; Inogés, S.; López-Díaz de Cerio, A.; Guillén-Grima, F.; Espinós, J.; et al. Modification of Breast Cancer Milieu with Chemotherapy plus Dendritic Cell Vaccine: An Approach to Select Best Therapeutic Strategies. Biomedicines 2023, 11, 238. [Google Scholar] [CrossRef]
- Denkert, C.; Von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Ahmed, F.S.; Gaule, P.; McGuire, J.; Patel, K.; Blenman, K.; Pusztai, L.; Rimm, D.L. PD-L1 Protein Expression on Both Tumor Cells and Macrophages are Associated with Response to Neoadjuvant Durvalumab with Chemotherapy in Triple-negative Breast Cancer. Clin. Cancer Res. 2020, 26, 5456–5461. [Google Scholar] [CrossRef]
- Hammerl, D.; Martens, J.W.M.; Timmermans, M.; Smid, M.; Trapman-Jansen, A.M.; Foekens, R.; Isaeva, O.I.; Voorwerk, L.; Balcioglu, H.E.; Wijers, R.; et al. Spatial immunophenotypes predict response to anti-PD1 treatment and capture distinct paths of T cell evasion in triple negative breast cancer. Nat. Commun. 2021, 12, 5668. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.-U.; Grischke, E.-M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.; Patwardhan, G.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef]
- Nanda, R.; Liu, M.C.; Yau, C.; Shatsky, R.; Pusztai, L.; Wallace, A.; Chien, A.J.; Forero-Torres, A.; Ellis, E.; Han, H.; et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020, 6, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Gianni, L.; Huang, C.; Egle, D.; Bermejo, B.; Zamagni, C.; Thill, M.; Anton, A.; Zambelli, S.; Bianchini, G.; Russo, S.; et al. Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple-negative, early high-risk and locally advanced breast cancer: NeoTRIP Michelangelo randomized study. Ann. Oncol. 2022, 33, 534–543. [Google Scholar] [CrossRef]
- Schmid, P.; Salgado, R.; Park, Y.; Muñoz-Couselo, E.; Kim, S.; Sohn, J.; Im, S.-A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef]
- Le Gall, C.M.; Weiden, J.; Eggermont, L.J.; Figdor, C.G. Dendritic cells in cancer immunotherapy. Nat. Mater. 2018, 17, 474–475. [Google Scholar] [CrossRef]
- Sadeghzadeh, M.; Bornehdeli, S.; Mohahammadrezakhani, H.; Abolghasemi, M.; Poursaei, E.; Asadi, M.; Zafari, V.; Aghebati-Maleki, L.; Shanehbandi, D. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci. 2020, 254, 117580. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Song, B.; Koido, S.; Calderwood, S.K.; Gong, J. Immunotherapy of Radioresistant Mammary Tumors with Early Metastasis Using Molecular Chaperone Vaccines Combined with Ionizing Radiation. J. Immunol. 2013, 191, 755–763. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol. 2022, 24 (Suppl. S5), v1–v95. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Datsi, A.; Sorg, R.V. Dendritic Cell Vaccination of Glioblastoma: Road to Success or Dead End. Front. Immunol. 2021, 12, 770390. [Google Scholar] [CrossRef]
- Chao, T.; Wang, X.; Zhiqi, L.; Qi, Y.; Yang, Z.; Kun, F.; Hoon, D.; Wei, H. A systemic review of clinical trials on dendritic-cells based vaccines against maligang glioma. J. Carcinog. Mutagen. 2015, 6, 1–7. [Google Scholar]
- Inogés, S.; Tejada, S.; de Cerio, A.L.-D.; Pérez-Larraya, J.G.; Espinós, J.; Idoate, M.A.; Domínguez, P.D.; de Eulate, R.G.; Aristu, J.; Bendandi, M.; et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J. Transl. Med. 2017, 15, 104. [Google Scholar] [CrossRef]
- Wheeler, C.J.; Black, K.L.; Liu, G.; Mazer, M.; Zhang, X.-X.; Pepkowitz, S.; Goldfinger, D.; Ng, H.; Irvin, D.; Yu, J.S. Vaccination Elicits Correlated Immune and Clinical Responses in Glioblastoma Multiforme Patients. Cancer Res 2008, 68, 5955–5964. [Google Scholar] [CrossRef] [PubMed]
- Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Preusser, M.; Stockhammer, G.; Nowosielski, M.; Iglseder, S.; Freyschlag, C.F.; Oberndorfer, S.; et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers 2018, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-Y.; Yang, W.-K.; Lee, H.-C.; Hsu, D.-M.; Lin, H.-L.; Lin, S.-Z.; Chen, C.-C.; Harn, H.-J.; Liu, C.-L.; Lee, W.-Y.; et al. Adjuvant Immunotherapy with Whole-Cell Lysate Dendritic Cells Vaccine for Glioblastoma Multiforme: A Phase II Clinical Trial. World Neurosurg. 2012, 77, 736–744. [Google Scholar] [CrossRef]
- Jie, X.; Hua, L.; Jiang, W.; Feng, F.; Feng, G.; Hua, Z. Clinical Application of a Dendritic Cell Vaccine Raised Against Heat-Shocked Glioblastoma. Cell Biochem. Biophys. 2012, 62, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Luo, F.; Tang, C.; Chen, D.; Qin, Z.; Hua, W.; Xu, M.; Zhong, P.; Yu, S.; Chen, D.; et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: An exploratory randomized phase II clinical trial. Cancer Immunol. Immunother. 2018, 67, 1777–1788. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.-J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Preusser, M.; van den Bent, M.J. Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination (DCVax-L) in glioblastoma: Breakthrough or fata morgana? Neuro Oncol. 2022, 25, 631–634. [Google Scholar] [CrossRef]
- Parney, I.F.; Anderson, S.K.; Gustafson, M.P.; Steinmetz, S.; Peterson, T.E.; Kroneman, T.N.; Raghunathan, A.; O’neill, B.P.; Buckner, J.C.; Solseth, M.; et al. Phase I trial of adjuvant mature autologous dendritic cell/allogeneic tumor lysate vaccines in combination with temozolomide in newly diagnosed glioblastoma. Neuro Oncol. Adv. 2022, 4, vdac089. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F.; Bsc, M.F.B.; Me, J.F.; Soerjomataram, M.I.; et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Gato, M.; Martínez-Aguillo, M.; Morilla, I.; Fernández, G.; Hernández, B.; López, P.; Alberdi, N.; et al. Early Detection of Hyperprogressive Disease in Non-Small Cell Lung Cancer by Monitoring of Systemic T Cell Dynamics. Cancers 2020, 12, 344. [Google Scholar] [CrossRef]
- Bakarurraini, N.A.A.R.; Ab Mutalib, N.S.; Jamal, R.; Abu, N. The Landscape of Tumor-Specific Antigens in Colorectal Cancer. Vaccines 2020, 8, 371. [Google Scholar] [CrossRef]
- Barth, R.J.; Fisher, D.A.; Wallace, P.K.; Channon, J.Y.; Noelle, R.J.; Gui, J.; Ernstoff, M.S. A Randomized Trial of Ex vivo CD40L Activation of a Dendritic Cell Vaccine in Colorectal Cancer Patients: Tumor-Specific Immune Responses Are Associated with Improved Survival. Clin. Cancer Res. 2010, 16, 5548–5556. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; de Vries, I.J.M.; Aarntzen, E.A.; de Boer, A.; Scharenborg, N.M.; van de Rakt, M.; van Spronsen, D.-J.; Preijers, F.W.; Figdor, C.G.; Adema, G.J.; et al. A pilot study on the immunogenicity of dendritic cell vaccination during adjuvant oxaliplatin/capecitabine chemotherapy in colon cancer patients. Br. J. Cancer 2010, 103, 1415–1421. [Google Scholar] [CrossRef]
- Caballero-Baños, M.; Benitez-Ribas, D.; Tabera, J.; Varea, S.; Vilana, R.; Bianchi, L.; Ayuso, J.R.; Pagés, M.; Carrera, G.; Cuatrecasas, M.; et al. Phase II randomised trial of autologous tumour lysate dendritic cell plus best supportive care compared with best supportive care in pre-treated advanced colorectal cancer patients. Eur. J. Cancer 2016, 64, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Español-Rego, M.; Fernández-Martos, C.; Elez, E.; Foguet, C.; Pedrosa, L.; Rodríguez, N.; Ruiz-Casado, A.; Pineda, E.; Cid, J.; Cabezón, R.; et al. A Phase I-II multicenter trial with Avelumab plus autologous dendritic cell vaccine in pre-treated mismatch repair-proficient (MSS) metastatic colorectal cancer patients; GEMCAD 1602 study. Cancer Immunol. Immunother. 2022, 72, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ruiz, M.E.; Perez-Gracia, J.L.; Rodríguez, I.; Alfaro, C.; Oñate, C.; Pérez, G.; Gil-Bazo, I.; Benito, A.; Inogés, S.; López-Diaz de Cerio, A.; et al. Combined immunotherapy encompassing intratumoral poly-ICLC, dendritic-cell vaccination and radiotherapy in advanced cancer patients. Ann. Oncol. 2018, 29, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Castañón, E.; Perez-Gracia, J.L.; Rodriguez, I.; Viudez, A.; Alfaro, C.; Oñate, C.; Perez, G.; Rotellar, F.; Inogés, S.; et al. A randomized phase II clinical trial of dendritic cell vaccination following complete resection of colon cancer liver metastasis. J. Immunother. Cancer 2018, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Rufo, N.; Garg, A.D.; Agostinis, P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Wu, Y.; Zhang, X.-S.; Yang, J.-L.; Li, H.-L.; Mao, Y.-Q.; Wang, Y.; Cheng, X.; Li, Y.-Q.; Xia, J.-C.; et al. Single administration of low dose cyclophosphamide augments the antitumor effect of dendritic cell vaccine. Cancer Immunol. Immunother. 2007, 56, 1597–1604. [Google Scholar] [CrossRef]
- Obeid, M.; Panaretakis, T.; Joza, N.; Tufi, R.; Tesniere, A.; van Endert, P.; Zitvogel, L.; Kroemer, G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007, 14, 1848–1850. [Google Scholar] [CrossRef]
- Bose, A.; Lowe, D.B.; Rao, A.; Storkus, W.J. Combined vaccine+axitinib therapy yields superior antitumor efficacy in a murine melanoma model. Melanoma Res. 2012, 22, 236–243. [Google Scholar] [CrossRef]
- Long, J.; Hu, Z.; Xue, H.; Wang, Y.; Chen, J.; Tang, F.; Zhou, J.; Liu, L.; Qiu, W.; Zhang, S.; et al. Vascular endothelial growth factor (VEGF) impairs the motility and immune function of human mature dendritic cells through the VEGF receptor 2-RhoA-cofilin1 pathway. Cancer Sci. 2019, 110, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Mpekris, F.; Voutouri, C.; Baish, J.W.; Duda, D.G.; Munn, L.L.; Stylianopoulos, T.; Jain, R.K. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3728–3737. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Wentzensen, N.; Mackie, A.; Berkhof, J.; Brotherton, J.; Giorgi-Rossi, P.; Kupets, R.; Smith, R.; Arrossi, S.; Bendahhou, K.; et al. The IARC Perspective on Cervical Cancer Screening. N. Engl. J. Med. 2021, 385, 1908–1918. [Google Scholar] [CrossRef]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Usta, E.H.; Yañez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. N. Engl. J. Med. 2021, 385, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Makker, V.; Colombo, N.; Herráez, A.C.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Baron-Hay, S.; Ray-Coquard, I.; et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N. Engl. J. Med. 2022, 386, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Berton, D.; Floquet, A.; Lescaut, W.; Baron, G.; Kaminsky, M.-C.; Toussaint, P.; Largillier, R.; Savoye, A.-M.; Alexandre, J.; Delbaldo, C.; et al. Real-World Experience of Bevacizumab as First-Line Treatment for Ovarian Cancer: The GINECO ENCOURAGE Cohort of 468 French Patients. Front. Pharmacol. 2021, 12, 711813. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Jin, F.; Norwood, K.; Aurora-Garg, D. Tumour mutational burden in treatment-resistant tumours—Authors’ reply. Lancet Oncol. 2020, 21, e552. [Google Scholar] [CrossRef] [PubMed]
- Naumann, R.W.; Hollebecque, A.; Meyer, T.; Devlin, M.-J.; Oaknin, A.; Kerger, J.; López-Picazo, J.M.; Machiels, J.-P.; Delord, J.-P.; Evans, T.R.J.; et al. Safety and Efficacy of Nivolumab Monotherapy in Recurrent or Metastatic Cervical, Vaginal, or Vulvar Carcinoma: Results From the Phase I/II CheckMate 358 Trial. J. Clin. Oncol. 2019, 37, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Zhang, X.; He, T.; Li, Y.; Chen, L.; Liu, H.; Wu, Y.; Guo, H. Dendritic Cell Vaccines in Ovarian Cancer. Front. Immunol. 2020, 11, 613773. [Google Scholar] [CrossRef]
- Bispo, S.; Farias, T.D.J.; de Araujo-Souza, P.S.; Cintra, R.; dos Santos, H.G.; Jorge, N.A.N.; Castro, M.A.A.; Wajnberg, G.; Scherer, N.d.M.; Genta, M.L.N.D.; et al. Dysregulation of Transcription Factor Networks Unveils Different Pathways in Human Papillomavirus 16-Positive Squamous Cell Carcinoma and Adenocarcinoma of the Uterine Cervix. Front. Oncol. 2021, 11, 626187. [Google Scholar] [CrossRef]
- Cao, G.; Cui, R.; Liu, C.; Zhang, G.; Zhang, Z. MTBHsp70-exFPR1-pulsed Dendritic Cells Enhance the Immune Response against Cervical Cancer. J. Cancer 2019, 10, 6364–6373. [Google Scholar] [CrossRef]
- Strickler, H.D.; Martinson, J.; Desai, S.; Xie, X.; Burk, R.D.; Anastos, K.; Massad, L.S.; Minkoff, H.; Xue, X.; D’Souza, G.; et al. The Relation of Plasmacytoid Dendritic Cells (pDCs) and Regulatory T-Cells (Tregs) with HPV Persistence in HIV-Infected and HIV-Uninfected Women. Viral Immunol. 2014, 27, 20–25. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef]
- Sioud, M. Releasing the Immune System Brakes Using siRNAs Enhances Cancer Immunotherapy. Cancers 2019, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Ferrall, L.; Lin, K.Y.; Roden, R.B.; Hung, C.-F.; Wu, T.-C. Cervical Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2021, 27, 4953–4973. [Google Scholar] [CrossRef] [PubMed]
- Mastelic-Gavillet, B.; Sarivalasis, A.; Lozano, L.E.; Wyss, T.; Inoges, S.; de Vries, I.J.M.; Dartiguenave, F.; Jichlinski, P.; Derrè, L.; Coukos, G.; et al. Quantitative and qualitative impairments in dendritic cell subsets of patients with ovarian or prostate cancer. Eur. J. Cancer 2020, 135, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Caro, A.A.; Deschoemaeker, S.; Allonsius, L.; Coosemans, A.; Laoui, D. Dendritic Cell Vaccines: A Promising Approach in the Fight against Ovarian Cancer. Cancers 2022, 14, 4037. [Google Scholar] [CrossRef] [PubMed]
- van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic Cell Cancer Therapy: Vaccinating the Right Patient at the Right Time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef] [PubMed]
- Rob, L.; Cibula, D.; Knapp, P.; Mallmann, P.; Klat, J.; Minar, L.; Bartos, P.; Chovanec, J.; Valha, P.; Pluta, M.; et al. Safety and efficacy of dendritic cell-based immunotherapy DCVAC/OvCa added to first-line chemotherapy (carboplatin plus paclitaxel) for epithelial ovarian cancer: A phase 2, open-label, multicenter, randomized trial. J. Immunother. Cancer 2022, 10, e003190. [Google Scholar] [CrossRef] [PubMed]
- Gray, H.J.; Benigno, B.; Berek, J.; Chang, J.; Mason, J.; Mileshkin, L.; Mitchell, P.; Moradi, M.; Recio, F.O.; Michener, C.M.; et al. Progression-free and overall survival in ovarian cancer patients treated with CVac, a mucin 1 dendritic cell therapy in a randomized phase 2 trial. J. Immunother. Cancer 2016, 4, 34. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Laureano, R.S.; Sprooten, J.; Vanmeerbeerk, I.; Borras, D.M.; Govaerts, J.; Naulaerts, S.; Berneman, Z.N.; Beuselinck, B.; Bol, K.F.; Borst, J.; et al. Trial watch: Dendritic cell (DC)-based immunotherapy for cancer. OncoImmunology 2022, 11, 2096363. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Thurner, B.; Haendle, I.; Röder, C.; Dieckmann, D.; Keikavoussi, P.; Jonuleit, H.; Bender, A.; Maczek, C.; Schreiner, D.; Driesch, P.v.D.; et al. Vaccination with Mage-3a1 Peptide–Pulsed Mature, Monocyte-Derived Dendritic Cells Expands Specific Cytotoxic T Cells and Induces Regression of Some Metastases in Advanced Stage IV Melanoma. J. Exp. Med. 1999, 190, 1669–1678. [Google Scholar] [CrossRef]
- Slingluff, C.L.; Petroni, G.R.; Yamshchikov, G.V.; Barnd, D.L.; Eastham, S.; Galavotti, H.; Patterson, J.W.; Deacon, D.H.; Hibbitts, S.; Teates, D.; et al. Clinical and Immunologic Results of a Randomized Phase II Trial of Vaccination Using Four Melanoma Peptides Either Administered in Granulocyte-Macrophage Colony-Stimulating Factor in Adjuvant or Pulsed on Dendritic Cells. J. Clin. Oncol. 2003, 21, 4016–4026. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.J.G.; Duiveman-de Boer, T.; Van De Rakt, M.W.M.M.; Scharenborg, N.M.; De Boer, A.J.; Pots, J.M.; et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef]
- Bloemendal, M.; Bol, K.F.; Boudewijns, S.; Gorris, M.A.; de Wilt, J.H.; Croockewit, S.A.; van Rossum, M.M.; de Goede, A.L.; Petry, K.; Koornstra, R.H.; et al. Immunological responses to adjuvant vaccination with combined CD1c+ myeloid and plasmacytoid dendritic cells in stage III melanoma patients. OncoImmunology 2022, 11, 2015113. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.J.; Carvajal, R.D.; Postow, M.A.; Sharma, S.; Pronschinske, K.B.; Shyer, J.A.; Singh-Kandah, S.; Dickson, M.A.; D’Angelo, S.P.; Wolchok, J.D.; et al. Langerhans-type dendritic cells electroporated with TRP-2 mRNA stimulate cellular immunity against melanoma: Results of a phase I vaccine trial. OncoImmunology 2017, 7, e1372081. [Google Scholar] [CrossRef]
- Fukuda, K.; Funakoshi, T.; Sakurai, T.; Nakamura, Y.; Mori, M.; Tanese, K.; Tanikawa, A.; Taguchi, J.; Fujita, T.; Okamoto, M.; et al. Peptide-pulsed dendritic cell vaccine in combination with carboplatin and paclitaxel chemotherapy for stage IV melanoma. Melanoma Res. 2017, 27, 326–334. [Google Scholar] [CrossRef]
- Boudewijns, S.; Bloemendal, M.; de Haas, N.; Westdorp, H.; Bol, K.F.; Schreibelt, G.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Gorris, M.A.J.; Croockewit, A.; et al. Autologous monocyte-derived DC vaccination combined with cisplatin in stage III and IV melanoma patients: A prospective, randomized phase 2 trial. Cancer Immunol. Immunother. 2020, 69, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Gasser, O.; Sharples, K.J.; Barrow, C.; Williams, G.M.; Bauer, E.; Wood, C.E.; Mester, B.; Dzhelali, M.; Caygill, G.; Jones, J.; et al. A phase I vaccination study with dendritic cells loaded with NY-ESO-1 and α-galactosylceramide: Induction of polyfunctional T cells in high-risk melanoma patients. Cancer Immunol. Immunother. 2018, 67, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D.; Chen, Q.; Morris, L.; Quirk, J.; Stanley, M.; Tavarnesi, M.L.; Parente, P.; Cavicchiolo, T.; Hopkins, W.; Jackson, H.; et al. Blood Dendritic Cells Generated With Flt3 Ligand and CD40 Ligand Prime CD8+ T Cells Efficiently in Cancer Patients. J. Immunother. 2006, 29, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Geskin, L.J.; Damiano, J.J.; Patrone, C.C.; Butterfield, L.H.; Kirkwood, J.M.; Falo, L.D. Three antigen-loading methods in dendritic cell vaccines for metastatic melanoma. Melanoma Res. 2018, 28, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H.; Ribas, A.; Dissette, V.B.; Amarnani, S.N.; Vu, H.T.; Oseguera, D.; Wang, H.-J.; Elashoff, R.M.; McBride, W.H.; Mukherji, B.; et al. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin. Cancer. Res. 2003, 9, 998–1008. [Google Scholar] [PubMed]
- Ribas, A.; Glaspy, J.A.; Lee, Y.; Dissette, V.B.; Seja, E.; Vu, H.T.; Tchekmedyian, N.S.; Oseguera, D.; Comin-Anduix, B.; Wargo, J.A.; et al. Role of Dendritic Cell Phenotype, Determinant Spreading, and Negative Costimulatory Blockade in Dendritic Cell-Based Melanoma Immunotherapy. J. Immunother. 2004, 27, 354–367. [Google Scholar] [CrossRef]
- Butterfield, L.H.; Gooding, W.; Whiteside, T.L. Development of a Potency Assay for Human Dendritic Cells: IL-12p70 Production. J. Immunother. 2008, 31, 89–100. [Google Scholar] [CrossRef]
- Butterfield, L.H.; Vujanovic, L.; Santos, P.M.; Maurer, D.M.; Gambotto, A.; Lohr, J.; Li, C.; Waldman, J.; Chandran, U.; Lin, Y.; et al. Multiple antigen-engineered DC vaccines with or without IFNalpha to promote antitumor immunity in melanoma. J. Immunother. Cancer 2019, 7, 113. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- van Willigen, W.W.; Bloemendal, M.; Boers-Sonderen, M.J.; de Groot, J.W.B.; Koornstra, R.H.; van der Veldt, A.A.; Haanen, J.B.A.G.; Boudewijns, S.; Schreibelt, G.; Gerritsen, W.R.; et al. Response and survival of metastatic melanoma patients treated with immune checkpoint inhibition for recurrent disease on adjuvant dendritic cell vaccination. OncoImmunology 2020, 9, 1738814. [Google Scholar] [CrossRef] [PubMed]
- Tijtgat, J.; Schwarze, J.K.; Vander Mijnsbrugge, A.S.; Raeymaeckers, S.; Van Riet, I.; Geeraerts, X.; Stevens, L.; Tuyaerts, S.; Neyns, B. Phase I Clinical Trial on Intratumoral Administration of Autologous Cd1c (Bdca-1)(+)/ Cd141 (Bdca-3)(+) Myeloid Dendritic Cells Plus Ipilimumab and As01(B) in Combination with Intravenously Administered Nivolumab. J. Immunother. Cancer 2022, 10, A823. [Google Scholar]
- Schwarze, J.K.; Tijtgat, J.; Awada, G.; Cras, L.; Vasaturo, A.; Bagnall, C.; Forsyth, R.; Dufait, I.; Tuyaerts, S.; Van Riet, I.; et al. Intratumoral administration of CD1c (BDCA-1)+and CD141 (BDCA-3)+myeloid dendritic cells in combination with talimogene laherparepvec in immune checkpoint blockade refractory advanced melanoma patients: A phase I clinical trial. J. Immunother. Cancer 2022, 10, e005141. [Google Scholar] [CrossRef] [PubMed]
- Vreeland, T.J.; Clifton, G.T.; Hale, D.F.; Chick, R.C.; Hickerson, A.T.; Cindass, J.L.; Adams, A.M.; Bohan, P.M.K.; Andtbacka, R.H.I.; Berger, A.C.; et al. A Phase IIb Randomized Controlled Trial of the TLPLDC Vaccine as Adjuvant Therapy After Surgical Resection of Stage III/IV Melanoma: A Primary Analysis. Ann. Surg. Oncol. 2021, 28, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.; Bloemendal, M.; van Willigen, W.; Schreibelt, G.; Bree, S.H.-D.; de Goede, A.; Van der Veldt, A.; Figdor, C.; de Groot, J.; de Wilt, J.; et al. 1078MO MIND-DC: A randomized phase III trial to assess the efficacy of adjuvant dendritic cell vaccination in comparison to placebo in stage IIIB and IIIC melanoma patients. Ann. Oncol. 2020, 31, S732. [Google Scholar] [CrossRef]
- Fong, L.; Carroll, P.; Weinberg, V.; Chan, S.; Lewis, J.; Corman, J.; Amling, C.L.; Stephenson, R.A.; Simko, J.; Sheikh, N.A.; et al. Activated Lymphocyte Recruitment Into the Tumor Microenvironment Following Preoperative Sipuleucel-T for Localized Prostate Cancer. JNCI J. Natl. Cancer Inst. 2014, 106, dju268. [Google Scholar] [CrossRef]
- Maiorano, B.A.; Schinzari, G.; Ciardiello, D.; Rodriquenz, M.G.; Cisternino, A.; Tortora, G.; Maiello, E. Cancer Vaccines for Genitourinary Tumors: Recent Progresses and Future Possibilities. Vaccines 2021, 9, 623. [Google Scholar] [CrossRef]
- Xi, H.-B.; Wang, G.-X.; Fu, B.; Liu, W.-P.; Li, Y. Survivin and PSMA Loaded Dendritic Cell Vaccine for the Treatment of Prostate Cancer. Biol. Pharm. Bull. 2015, 38, 827–835. [Google Scholar] [CrossRef]
- Ogasawara, M.; Miyashita, M.; Ota, S. Vaccination of Urological Cancer Patients With WT1 Peptide-Pulsed Dendritic Cells in Combination With Molecular Targeted Therapy or Conventional Chemotherapy Induces Immunological and Clinical Responses. Ther. Apher. Dial. 2018, 22, 266–277. [Google Scholar] [CrossRef]
- Figlin, R.A.; Tannir, N.M.; Uzzo, R.G.; Tykodi, S.S.; Chen, D.Y.; Master, V.; Kapoor, A.; Vaena, D.; Lowrance, W.T.; Bratslavsky, G.; et al. Results of the ADAPT Phase 3 Study of Rocapuldencel-T in Combination with Sunitinib as First-Line Therapy in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2327–2336. [Google Scholar] [CrossRef]
- Mapara, M.Y.; Sykes, M. Tolerance and Cancer: Mechanisms of Tumor Evasion and Strategies for Breaking Tolerance. J. Clin. Oncol. 2004, 22, 1136–1151. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef]
- Holm, J.S.; Funt, S.A.; Borch, A.; Munk, K.K.; Bjerregaard, A.-M.; Reading, J.L.; Maher, C.; Regazzi, A.; Wong, P.; Al-Ahmadie, H.; et al. Neoantigen-specific CD8 T cell responses in the peripheral blood following PD-L1 blockade might predict therapy outcome in metastatic urothelial carcinoma. Nat. Commun. 2022, 13, 1935. [Google Scholar] [CrossRef]
- Kim, S.P.; Vale, N.R.; Zacharakis, N.; Krishna, S.; Yu, Z.; Gasmi, B.; Gartner, J.J.; Sindiri, S.; Malekzadeh, P.; Deniger, D.C.; et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor–Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol. Res. 2022, 10, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Li, Q.; Zhang, R.; Xie, L.; Shu, Y.; Gao, S.; Wang, P.; Su, X.; Qin, Y.; Wang, Y.; et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct. Target. Ther. 2021, 6, 26. [Google Scholar] [CrossRef]
- Guo, Z.; Yuan, Y.; Chen, C.; Lin, J.; Ma, Q.; Liu, G.; Gao, Y.; Huang, Y.; Chen, L.; Chen, L.-Z.; et al. Durable complete response to neoantigen-loaded dendritic-cell vaccine following anti-PD-1 therapy in metastatic gastric cancer. npj Precis. Oncol. 2022, 6, 34. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.-C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma—A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol. 2019, 10, 1832. [Google Scholar]
- Leoni, G.; D’Alise, A.M.; Cotugno, G.; Langone, F.; Garzia, I.; De Lucia, M.; Fichera, I.; Vitale, R.; Bignone, V.; Tucci, F.G.; et al. A Genetic Vaccine Encoding Shared Cancer Neoantigens to Treat Tumors with Microsatellite Instability. Cancer Res 2020, 80, 3972–3982. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Stopfer, L.E.; Ahn, R.; Sanders, E.A.; Sandel, D.A.; Freed-Pastor, W.A.; Rideout, W.M.; Naranjo, S.; Fessenden, T.; Nguyen, K.B.; et al. Deciphering the immunopeptidome in vivo reveals new tumour antigens. Nature 2022, 607, 149–155. [Google Scholar] [CrossRef]
- Haen, S.P.; Löffler, M.W.; Rammensee, H.-G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Ouspenskaia, T.; Law, T.; Clauser, K.R.; Klaeger, S.; Sarkizova, S.; Aguet, F.; Li, B.; Christian, E.; Knisbacher, B.A.; Le, P.M.; et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol. 2022, 40, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fritsche, J.; Roszik, J.; Williams, L.J.; Peng, X.; Chiu, Y.; Tsou, C.-C.; Hoffgaard, F.; Goldfinger, V.; Schoor, O.; et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat. Commun. 2018, 9, 3919. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Lv, D.; Li, D.; Yin, J.; Ma, Y.; Luo, Y.; Fu, L.; Ding, N.; Li, Y.; Pan, Z.; et al. IEAtlas: An atlas of HLA-presented immune epitopes derived from non-coding regions. Nucleic Acids Res. 2023, 51, D409–D417. [Google Scholar] [CrossRef] [PubMed]
- Ingles Garces, A.H.; Au, L.; Mason, R.; Thomas, J.; Larkin, J. Building on the anti-PD1/PD-L1 backbone: Combination immunotherapy for cancer. Expert Opin. Investig. Drugs 2019, 28, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Thomas, J.; Larkin, J. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Yang, L.-J.; Cui, H. Recent Advances in Nanotechnology for Dendritic Cell-Based Immunotherapy. Front. Pharmacol. 2020, 11, 960. [Google Scholar] [CrossRef]
- Davodabadi, F.; Sarhadi, M.; Arabpour, J.; Sargazi, S.; Rahdar, A.; Díez-Pascual, A.M. Breast cancer vaccines: New insights into immunomodulatory and nano-therapeutic approaches. J. Control. Release 2022, 349, 844–875. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hato, L.; Vizcay, A.; Eguren, I.; Pérez-Gracia, J.L.; Rodríguez, J.; Gállego Pérez-Larraya, J.; Sarobe, P.; Inogés, S.; Díaz de Cerio, A.L.; Santisteban, M. Dendritic Cells in Cancer Immunology and Immunotherapy. Cancers 2024, 16, 981. https://doi.org/10.3390/cancers16050981
Hato L, Vizcay A, Eguren I, Pérez-Gracia JL, Rodríguez J, Gállego Pérez-Larraya J, Sarobe P, Inogés S, Díaz de Cerio AL, Santisteban M. Dendritic Cells in Cancer Immunology and Immunotherapy. Cancers. 2024; 16(5):981. https://doi.org/10.3390/cancers16050981
Chicago/Turabian StyleHato, Laura, Angel Vizcay, Iñaki Eguren, José L. Pérez-Gracia, Javier Rodríguez, Jaime Gállego Pérez-Larraya, Pablo Sarobe, Susana Inogés, Ascensión López Díaz de Cerio, and Marta Santisteban. 2024. "Dendritic Cells in Cancer Immunology and Immunotherapy" Cancers 16, no. 5: 981. https://doi.org/10.3390/cancers16050981
APA StyleHato, L., Vizcay, A., Eguren, I., Pérez-Gracia, J. L., Rodríguez, J., Gállego Pérez-Larraya, J., Sarobe, P., Inogés, S., Díaz de Cerio, A. L., & Santisteban, M. (2024). Dendritic Cells in Cancer Immunology and Immunotherapy. Cancers, 16(5), 981. https://doi.org/10.3390/cancers16050981