
Promises of Protein Kinase Inhibitors in Recalcitrant Small-Cell Lung Cancer: Recent Scenario and Future Possibilities
 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Origin and Progression of SCLC
3. Clinical Features of SCLC
4. Immunotherapy in SCLC
5. Role of Protein Kinases in Immunotherapy for SCLC
5.1. CDK4/6
5.2. WEE1
5.3. CHK1
5.4. Macrophage-Mediated Anti-Cancer Therapies
6. Repurposing Protein Kinase and Other Inhibitors against SCLC
7. Protein Kinases in Personalized Medicine for SCLC
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cancer Facts & Figures 2022|American Cancer Society. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2022.html (accessed on 16 August 2023).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Ridge, C.A.; McErlean, A.M.; Ginsberg, M.S. Epidemiology of Lung Cancer. Semin. Interv. Radiol. 2013, 30, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, M.; Ding, X.-J.; Cao, Y. Familial Risk for Lung Cancer. Oncol. Lett. 2017, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.H.; Abraham, S.; Callister, M.E.; Grice, A.; Hamilton, W.T.; Lopez, R.R.; Shinkins, B.; Neal, R.D. Sensitivity of Chest X-ray for Detecting Lung Cancer in People Presenting with Symptoms: A Systematic Review. Br. J. Gen. Pract. 2019, 69, e827–e835. [Google Scholar] [CrossRef] [PubMed]
- Gergen, A.K.; Scott, C.D.; Mitchell, J.D. Surgery for Limited Stage Small Cell Lung Cancer. J. Thorac. Dis. 2020, 12, 6291–6297. [Google Scholar] [CrossRef] [PubMed]
- Kalemkerian, G.P. Staging and Imaging of Small Cell Lung Cancer. Cancer Imaging 2012, 11, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic, I.S.; Stjepanovic, M.; Mitrovic, D. Distribution Patterns of the Metastases of the Lung Carcinoma in Relation to Histological Type of the Primary Tumor: An Autopsy Study. Ann. Thorac. Med. 2017, 12, 191–198. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.V.; Reck, M.; Mansfield, A.S.; Mok, T.; Scherpereel, A.; Reinmuth, N.; Garassino, M.C.; De Castro Carpeno, J.; Califano, R.; Nishio, M.; et al. Updated Overall Survival and PD-L1 Subgroup Analysis of Patients with Extensive-Stage Small-Cell Lung Cancer Treated with Atezolizumab, Carboplatin, and Etoposide (IMpower133). J. Clin. Oncol. 2021, 39, 619–630. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Każarnowicz, A.; Karaseva, N.; Sánchez, A.; De Boer, R.; Andric, Z.; Reck, M.; Atagi, S.; Lee, J.-S.; Garassino, M.; et al. Safety and Patient-Reported Outcomes of Atezolizumab, Carboplatin, and Etoposide in Extensive-Stage Small-Cell Lung Cancer (IMpower133): A Randomized Phase I/III Trial. Ann. Oncol. 2020, 31, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Sugawara, S.; Atagi, S.; Akamatsu, H.; Sakai, H.; Okamoto, I.; Takayama, K.; Hayashi, H.; Nakagawa, Y.; Kawakami, T. Subgroup Analysis of Japanese Patients in a Phase III Study of Atezolizumab in Extensive-Stage Small-Cell Lung Cancer (IMpower133). Clin. Lung Cancer 2019, 20, 469–476.e1. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and Cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Tsurutani, J.; West, K.A.; Sayyah, J.; Gills, J.J.; Dennis, P.A. Inhibition of the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin Pathway but Not the MEK/ERK Pathway Attenuates Laminin-Mediated Small Cell Lung Cancer Cellular Survival and Resistance to Imatinib Mesylate or Chemotherapy. Cancer Res. 2005, 65, 8423–8432. [Google Scholar] [CrossRef]
- Guo, L.; Zhou, Y.; Sun, Y.; Zhang, F. Non-Receptor Tyrosine Kinase Etk Regulation of Drug Resistance in Small-Cell Lung Cancer. Eur. J. Cancer 2010, 46, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kim, I.-K.; Kallakury, B.; Chahine, J.J.; Iwama, E.; Pierobon, M.; Petricoin, E.; McCutcheon, J.N.; Zhang, Y.-W.; Umemura, S.; et al. Acquired Small Cell Lung Cancer Resistance to Chk1 Inhibitors Involves Wee1 Up-Regulation. Mol. Oncol. 2021, 15, 1130–1145. [Google Scholar] [CrossRef]
- Herzog, B.H.; Devarakonda, S.; Govindan, R. Overcoming Chemotherapy Resistance in SCLC. J. Thorac. Oncol. 2021, 16, 2002–2015. [Google Scholar] [CrossRef]
- Taniguchi, H.; Caeser, R.; Chavan, S.S.; Zhan, Y.A.; Chow, A.; Manoj, P.; Uddin, F.; Kitai, H.; Qu, R.; Hayatt, O.; et al. WEE1 Inhibition Enhances the Antitumor Immune Response to PD-L1 Blockade by the Concomitant Activation of STING and STAT1 Pathways in SCLC. Cell Rep. 2022, 39, 110814. [Google Scholar] [CrossRef]
- Sen, T.; Della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef]
- Ruff, M.R.; Pert, C.B. Small Cell Carcinoma of the Lung: Macrophage-Specific Antigens Suggest Hemopoietic Stem Cell Origin. Science 1984, 225, 1034–1036. [Google Scholar] [CrossRef]
- Bunn, P.A.; Linnoila, I.; Minna, J.D.; Carney, D.; Gazdar, A.F. Small Cell Lung Cancer, Endocrine Cells of the Fetal Bronchus, and Other Neuroendocrine Cells Express the Leu-7 Antigenic Determinant Present on Natural Killer Cells. Blood 1985, 65, 764–768. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.-Y.; Berns, A. Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of Small Cell Lung Cancer by Somatic Inactivation of Both Trp53 and Rb1 in a Conditional Mouse Model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef]
- Park, K.-S.; Liang, M.-C.; Raiser, D.M.; Zamponi, R.; Roach, R.R.; Curtis, S.J.; Walton, Z.; Schaffer, B.E.; Roake, C.M.; Zmoos, A.-F.; et al. Characterization of the Cell of Origin for Small Cell Lung Cancer. Cell Cycle 2011, 10, 2806–2815. [Google Scholar] [CrossRef]
- Chang, R.B.; Strochlic, D.E.; Williams, E.K.; Umans, B.D.; Liberles, S.D. Vagal Sensory Neuron Subtypes That Differentially Control Breathing. Cell 2015, 161, 622–633. [Google Scholar] [CrossRef]
- Sui, P.; Wiesner, D.L.; Xu, J.; Zhang, Y.; Lee, J.; Van Dyken, S.; Lashua, A.; Yu, C.; Klein, B.S.; Locksley, R.M.; et al. Pulmonary Neuroendocrine Cells Amplify Allergic Asthma Responses. Science 2018, 360, eaan8546. [Google Scholar] [CrossRef]
- Youngson, C.; Nurse, C.; Yeger, H.; Cutz, E. Oxygen Sensing in Airway Chemoreceptors. Nature 1993, 365, 153–155. [Google Scholar] [CrossRef]
- Stevens, T.P.; McBride, J.T.; Peake, J.L.; Pinkerton, K.E.; Stripp, B.R. Cell Proliferation Contributes to PNEC Hyperplasia after Acute Airway Injury. Am. J. Physiol. 1997, 272, L486–L493. [Google Scholar] [CrossRef] [PubMed]
- Ouadah, Y.; Rojas, E.R.; Riordan, D.P.; Capostagno, S.; Kuo, C.S.; Krasnow, M.A. Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, P53, and Notch. Cell 2019, 179, 403–416.e23. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive Genomic Profiles of Small Cell Lung Cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative Genome Analyses Identify Key Somatic Driver Mutations of Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Cai, H.; Murray, C.W.; Li, C.; Shue, Y.T.; Andrejka, L.; He, A.L.; Holzem, A.M.E.; Drainas, A.P.; Ko, J.H.; et al. A Multiplexed in Vivo Approach to Identify Driver Genes in Small Cell Lung Cancer. Cell Rep. 2023, 42, 111990. [Google Scholar] [CrossRef]
- Jia, D.; Augert, A.; Kim, D.-W.; Eastwood, E.; Wu, N.; Ibrahim, A.H.; Kim, K.-B.; Dunn, C.T.; Pillai, S.P.S.; Gazdar, A.F.; et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov. 2018, 8, 1422–1437. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Z.; Li, Y.; Peng, H.; Liu, J.; Zhang, J.; Xiao, X. The Role of CREBBP/EP300 and Its Therapeutic Implications in Hematological Malignancies. Cancers 2023, 15, 1219. [Google Scholar] [CrossRef]
- Kim, K.-B.; Kabra, A.; Kim, D.-W.; Xue, Y.; Huang, Y.; Hou, P.-C.; Zhou, Y.; Miranda, L.J.; Park, J.-I.; Shi, X.; et al. KIX Domain Determines a Selective Tumor-Promoting Role for EP300 and Its Vulnerability in Small Cell Lung Cancer. Sci. Adv. 2022, 8, eabl4618. [Google Scholar] [CrossRef]
- Schaffer, B.E.; Park, K.-S.; Yiu, G.; Conklin, J.F.; Lin, C.; Burkhart, D.L.; Karnezis, A.N.; Sweet-Cordero, E.A.; Sage, J. Loss of P130 Accelerates Tumor Development in a Mouse Model for Human Small-Cell Lung Carcinoma. Cancer Res. 2010, 70, 3877–3883. [Google Scholar] [CrossRef]
- Rogers, Z.N.; McFarland, C.D.; Winters, I.P.; Naranjo, S.; Chuang, C.-H.; Petrov, D.; Winslow, M.M. A Quantitative and Multiplexed Approach to Uncover the Fitness Landscape of Tumor Suppression in Vivo. Nat. Methods 2017, 14, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Brady, J.J.; Tsai, M.K.; Li, C.; Winters, I.P.; Tang, R.; Andrejka, L.; Ma, R.K.; Kunder, C.A.; Chu, P.; et al. An LKB1-SIK Axis Suppresses Lung Tumor Growth and Controls Differentiation. Cancer Discov. 2019, 9, 1590–1605. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.T.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin Activation of Rheb, a Mediator of mTOR/S6K/4E-BP Signaling, Is Inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR Function in Response to Hypoxia by REDD1 and the TSC1/TSC2 Tumor Suppressor Complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef]
- Brennan, J.; O’Connor, T.; Makuch, R.W.; Simmons, A.M.; Russell, E.; Linnoila, R.I.; Phelps, R.M.; Gazdar, A.F.; Ihde, D.C.; Johnson, B.E. Myc Family DNA Amplification in 107 Tumors and Tumor Cell Lines from Patients with Small Cell Lung Cancer Treated with Different Combination Chemotherapy Regimens. Cancer Res. 1991, 51, 1708–1712. [Google Scholar]
- Kim, K.-B.; Kim, Y.; Rivard, C.J.; Kim, D.-W.; Park, K.-S. FGFR1 Is Critical for RBL2 Loss-Driven Tumor Development and Requires PLCG1 Activation for Continued Growth of Small Cell Lung Cancer. Cancer Res. 2020, 80, 5051–5062. [Google Scholar] [CrossRef]
- Pardo, O.E.; Latigo, J.; Jeffery, R.E.; Nye, E.; Poulsom, R.; Spencer-Dene, B.; Lemoine, N.R.; Stamp, G.W.; Aboagye, E.O.; Seckl, M.J. The Fibroblast Growth Factor Receptor Inhibitor PD173074 Blocks Small Cell Lung Cancer Growth in Vitro and in Vivo. Cancer Res. 2009, 69, 8645–8651. [Google Scholar] [CrossRef]
- Ferone, G.; Song, J.-Y.; Krijgsman, O.; van der Vliet, J.; Cozijnsen, M.; Semenova, E.A.; Adams, D.J.; Peeper, D.; Berns, A. FGFR1 Oncogenic Activation Reveals an Alternative Cell of Origin of SCLC in Rb1/P53 Mice. Cell Rep. 2020, 30, 3837–3850.e3. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Augert, A.; Rongione, M.; Conkrite, K.; Parazzoli, S.; Nikitin, A.Y.; Ingolia, N.; MacPherson, D. PTEN Is a Potent Suppressor of Small Cell Lung Cancer. Mol. Cancer Res. 2014, 12, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Quintanal-Villalonga, Á.; Gao, V.R.; Xie, Y.; Allaj, V.; Chaudhary, O.; Masilionis, I.; Egger, J.; Chow, A.; Walle, T.; et al. Signatures of Plasticity, Metastasis, and Immunosuppression in an Atlas of Human Small Cell Lung Cancer. Cancer Cell 2021, 39, 1479–1496.e18. [Google Scholar] [CrossRef]
- Mpakali, A.; Stratikos, E. The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy. Cancers 2021, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus Platinum-Etoposide versus Platinum-Etoposide in First-Line Treatment of Extensive-Stage Small-Cell Lung Cancer (CASPIAN): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.W.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab, with or without Tremelimumab, plus Platinum-Etoposide versus Platinum-Etoposide Alone in First-Line Treatment of Extensive-Stage Small-Cell Lung Cancer (CASPIAN): Updated Results from a Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 51–65. [Google Scholar] [CrossRef]
- Rudin, C.M.; Awad, M.M.; Navarro, A.; Gottfried, M.; Peters, S.; Csőszi, T.; Cheema, P.K.; Rodriguez-Abreu, D.; Wollner, M.; Yang, J.C.-H.; et al. Pembrolizumab or Placebo Plus Etoposide and Platinum as First-Line Therapy for Extensive-Stage Small-Cell Lung Cancer: Randomized, Double-Blind, Phase III KEYNOTE-604 Study. J. Clin. Oncol. 2020, 38, 2369–2379. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, C.; Yao, W.; Wang, Q.; Min, X.; Chen, G.; Xu, X.; Li, X.; Xu, F.; Fang, Y.; et al. Adebrelimab or Placebo plus Carboplatin and Etoposide as First-Line Treatment for Extensive-Stage Small-Cell Lung Cancer (CAPSTONE-1): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2022, 23, 739–747. [Google Scholar] [CrossRef]
- Cheng, Y.; Han, L.; Wu, L.; Chen, J.; Sun, H.; Wen, G.; Ji, Y.; Dvorkin, M.; Shi, J.; Pan, Z.; et al. Effect of First Line Serplulimab vs Placebo Added to Chemotherapy on Survival in Patients with Extensive-Stage Small Cell Lung Cancer: The ASTRUM-005 Randomized Clinical Trial. JAMA 2022, 328, 1223–1232. [Google Scholar] [CrossRef]
- Acheampong, E.; Abed, A.; Morici, M.; Bowyer, S.; Amanuel, B.; Lin, W.; Millward, M.; Gray, E.S. Tumour PD-L1 Expression in Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis. Cells 2020, 9, 2393. [Google Scholar] [CrossRef]
- Yu, S.; Jia, M.; Li, Y.; Sun, P.-L.; Gao, H. Differential Expression of PD-L1 in Central and Peripheral and TTF1-Positive and -Negative Small-Cell Lung Cancer. Front. Med. 2020, 7, 621838. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, Y. Immunotherapy for Extensive-Stage Small-Cell Lung Cancer: Current Landscape and Future Perspectives. Front. Oncol. 2023, 13, 1142081. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, F.; Zhao, S.; Shi, P.; Wang, S.; Cui, D. Correlation between PD-1/PD-L1 Expression and Polarization in Tumor-Associated Macrophages: A Key Player in Tumor Immunotherapy. Cytokine Growth Factor Rev. 2022, 67, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Bisi, J.E.; Sorrentino, J.A.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-Induced Myelosuppression. Mol. Cancer Ther. 2016, 15, 783–793. [Google Scholar] [CrossRef]
- Lai, A.Y.; Sorrentino, J.A.; Dragnev, K.H.; Weiss, J.M.; Owonikoko, T.K.; Rytlewski, J.A.; Hood, J.; Yang, Z.; Malik, R.K.; Strum, J.C.; et al. CDK4/6 Inhibition Enhances Antitumor Efficacy of Chemotherapy and Immune Checkpoint Inhibitor Combinations in Preclinical Models and Enhances T-Cell Activation in Patients with SCLC Receiving Chemotherapy. J. Immunother. Cancer 2020, 8, e000847. [Google Scholar] [CrossRef] [PubMed]
- Daniel, D.; Kuchava, V.; Bondarenko, I.; Ivashchuk, O.; Reddy, S.; Jaal, J.; Kudaba, I.; Hart, L.; Matitashvili, A.; Pritchett, Y.; et al. Trilaciclib Prior to Chemotherapy and Atezolizumab in Patients with Newly Diagnosed Extensive-Stage Small Cell Lung Cancer: A Multicentre, Randomised, Double-Blind, Placebo-Controlled Phase II Trial. Int. J. Cancer 2020, 148, 2557–2570. [Google Scholar] [CrossRef]
- Weiss, J.; Goldschmidt, J.; Andric, Z.; Dragnev, K.H.; Gwaltney, C.; Skaltsa, K.; Pritchett, Y.; Antal, J.M.; Morris, S.R.; Daniel, D. Effects of Trilaciclib on Chemotherapy-Induced Myelosuppression and Patient-Reported Outcomes in Patients with Extensive-Stage Small Cell Lung Cancer: Pooled Results from Three Phase II Randomized, Double-Blind, Placebo-Controlled Studies. Clin. Lung Cancer 2021, 22, 449–460. [Google Scholar] [CrossRef] [PubMed]
- PosthumaDeBoer, J.; Würdinger, T.; Graat, H.C.A.; van Beusechem, V.W.; Helder, M.N.; van Royen, B.J.; Kaspers, G.J.L. WEE1 Inhibition Sensitizes Osteosarcoma to Radiotherapy. BMC Cancer 2011, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Booher, R.N.; Kraker, A.; Lawrence, T.; Leopold, W.R.; Sun, Y. Radiosensitization of P53 Mutant Cells by PD0166285, a Novel G(2) Checkpoint Abrogator. Cancer Res. 2001, 61, 8211–8217. [Google Scholar] [PubMed]
- Wang, Y.; Decker, S.J.; Sebolt-Leopold, J. Knockdown of Chk1, Wee1 and Myt1 by RNA Interference Abrogates G2 Checkpoint and Induces Apoptosis. Cancer Biol. Ther. 2004, 3, 305–313. [Google Scholar] [CrossRef]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-Molecule Inhibition of Wee1 Kinase by MK-1775 Selectively Sensitizes P53-Deficient Tumor Cells to DNA-Damaging Agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef]
- SCLC: Combining WEE1 Inhibition with PD-L1 Blockade. Available online: https://dailyreporter.esmo.org/esmo-targeted-anticancer-therapies-congress-2022/congress-highlights/combining-wee1-inhibition-with-pd-l1-blockade-shows-promise-in-sclc-models (accessed on 16 August 2023).
- Sen, T.; Tong, P.; Diao, L.; Li, L.; Fan, Y.; Hoff, J.; Heymach, J.V.; Wang, J.; Byers, L.A. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 6239–6253. [Google Scholar] [CrossRef]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients with Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef]
- Cole, K.P.; Groh, J.M.; Johnson, M.D.; Burcham, C.L.; Campbell, B.M.; Diseroad, W.D.; Heller, M.R.; Howell, J.R.; Kallman, N.J.; Koenig, T.M.; et al. Kilogram-Scale Prexasertib Monolactate Monohydrate Synthesis under Continuous-Flow CGMP Conditions. Science 2017, 356, 1144–1150. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Mouw, K.W.; Konstantinopoulos, P.A. From Checkpoint to Checkpoint: DNA Damage ATR/Chk1 Checkpoint Signalling Elicits PD-L1 Immune Checkpoint Activation. Br. J. Cancer 2018, 118, 933–935. [Google Scholar] [CrossRef]
- Attili, I.; Karachaliou, N.; Bonanno, L.; Berenguer, J.; Bracht, J.; Codony-Servat, J.; Codony-Servat, C.; Ito, M.; Rosell, R. STAT3 as a Potential Immunotherapy Biomarker in Oncogene-Addicted Non-Small Cell Lung Cancer. Ther. Adv. Med. Oncol. 2018, 10, 1758835918763744. [Google Scholar] [CrossRef]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.B.; Fan, Y.H.; Li, L.; Glisson, B.S.; et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017, 77, 3870–3884. [Google Scholar] [CrossRef]
- Byers, L.A.; Navarro, A.; Schaefer, E.; Johnson, M.; Özgüroğlu, M.; Han, J.-Y.; Bondarenko, I.; Cicin, I.; Dragnev, K.H.; Abel, A.; et al. A Phase II Trial of Prexasertib (LY2606368) in Patients with Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in Cell Biology and Cancer Therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic Profiling Identifies Dysregulated Pathways in Small Cell Lung Cancer and Novel Therapeutic Targets Including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef]
- Barayan, R.; Ran, X.; Lok, B.H. PARP Inhibitors for Small Cell Lung Cancer and Their Potential for Integration into Current Treatment Approaches. J. Thorac. Dis. 2020, 12, 6240–6252. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J. Thorac. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.; Ross, K.; Kim, S.; De Jonge, M.; Barlesi, F.; Postel-Vinay, S.; Domchek, S.; Lee, J.; Angell, H.; Bui, K.; et al. P1.15-004 An Open-Label, Multitumor Phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): Results in Patients with Relapsed SCLC. J. Thorac. Oncol. 2017, 12, S2044–S2045. [Google Scholar] [CrossRef]
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.; Norris, C.; Mu, S.; et al. Pamiparib in Combination with Tislelizumab in Patients with Advanced Solid Tumours: Results from the Dose-Escalation Stage of a Multicentre, Open-Label, Phase 1a/b Trial. Lancet Oncol. 2019, 20, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, A.M.; Scheel, A.H.; Ozretić, L.; George, J.; Thomas, R.K.; Hagemann, T.; Zander, T.; Wolf, J.; Buettner, R. PD-L1 Expression in Small Cell Neuroendocrine Carcinomas. Eur. J. Cancer 2015, 51, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Maharaj, K.; Powers, J.J.; Achille, A.; Mediavilla-Varela, M.; Gamal, W.; Burger, K.L.; Fonseca, R.; Jiang, K.; Miskin, H.P.; Maryanski, D.; et al. The Dual PI3Kδ/CK1ε Inhibitor Umbralisib Exhibits Unique Immunomodulatory Effects on CLL T Cells. Blood Adv. 2020, 4, 3072–3084. [Google Scholar] [CrossRef] [PubMed]
- Janovská, P.; Normant, E.; Miskin, H.; Bryja, V. Targeting Casein Kinase 1 (CK1) in Hematological Cancers. Int. J. Mol. Sci. 2020, 21, 9026. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J. Mining the Microenvironment for Therapeutic Targets in Chronic Lymphocytic Leukemia. Cancer J. 2021, 27, 306–313. [Google Scholar] [CrossRef]
- Ahirwar, D. Casein Kinase-1 Epsilon as a Novel Therapeutic Target against Small Cell Lung Cancer; U.S. Army Medical Research and Development Command: Fort Detrick, MD, USA, 2023. [Google Scholar]
- Brägelmann, J.; Böhm, S.; Guthrie, M.R.; Mollaoglu, G.; Oliver, T.G.; Sos, M.L. Family Matters: How MYC Family Oncogenes Impact Small Cell Lung Cancer. Cell Cycle 2017, 16, 1489–1498. [Google Scholar] [CrossRef]
- Sos, M.L.; Dietlein, F.; Peifer, M.; Schöttle, J.; Balke-Want, H.; Müller, C.; Koker, M.; Richters, A.; Heynck, S.; Malchers, F.; et al. A Framework for Identification of Actionable Cancer Genome Dependencies in Small Cell Lung Cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 17034–17039. [Google Scholar] [CrossRef]
- Fiorentino, F.P.; Tokgün, E.; Solé-Sánchez, S.; Giampaolo, S.; Tokgün, O.; Jauset, T.; Kohno, T.; Perucho, M.; Soucek, L.; Yokota, J. Growth Suppression by MYC Inhibition in Small Cell Lung Cancer Cells with TP53 and RB1 Inactivation. Oncotarget 2016, 7, 31014–31028. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Reversible Tumorigenesis by MYC in Hematopoietic Lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained Loss of a Neoplastic Phenotype by Brief Inactivation of MYC. Science 2002, 297, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Tam, A.; Pomerantz, J.; Wong, M.; Holash, J.; Bardeesy, N.; Shen, Q.; O’Hagan, R.; Pantginis, J.; Zhou, H.; et al. Essential Role for Oncogenic Ras in Tumour Maintenance. Nature 1999, 400, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Huettner, C.S.; Zhang, P.; Van Etten, R.A.; Tenen, D.G. Reversibility of Acute B-Cell Leukaemia Induced by BCR-ABL1. Nat. Genet. 2000, 24, 57–60. [Google Scholar] [CrossRef]
- Restifo, N.P. Can Antitumor Immunity Help to Explain “Oncogene Addiction”? Cancer Cell 2010, 18, 403–405. [Google Scholar] [CrossRef][Green Version]
- Felsher, D.W. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes. Cancer 2010, 1, 597–604. [Google Scholar] [CrossRef]
- Casey, S.C.; Bellovin, D.I.; Felsher, D.W. Noncanonical Roles of the Immune System in Eliciting Oncogene Addiction. Curr. Opin. Immunol. 2013, 25, 246–258. [Google Scholar] [CrossRef]
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E.; et al. CD4+ T Cells Contribute to the Remodeling of the Microenvironment Required for Sustained Tumor Regression upon Oncogene Inactivation. Cancer Cell 2010, 18, 485–498. [Google Scholar] [CrossRef]
- Dabir, S.; Babakoohi, S.; Kluge, A.; Morrow, J.J.; Kresak, A.; Yang, M.; MacPherson, D.; Wildey, G.; Dowlati, A. RET Mutation and Expression in Small-Cell Lung Cancer. J. Thorac. Oncol. 2014, 9, 1316–1323. [Google Scholar] [CrossRef]
- Rusmini, M.; Griseri, P.; Matera, I.; Pontarini, E.; Ravazzolo, R.; Mavilio, D.; Ceccherini, I. Expression Variability and Function of the RET Gene in Adult Peripheral Blood Mononuclear Cells. J. Cell Physiol. 2014, 229, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.M.; Seymour, L.; Smylie, M.; Ding, K.; Ung, Y.; Findlay, B.; Lee, C.W.; Djurfeldt, M.; Whitehead, M.; Ellis, P.; et al. Phase II Study of Vandetanib or Placebo in Small-Cell Lung Cancer Patients after Complete or Partial Response to Induction Chemotherapy with or without Radiation Therapy: National Cancer Institute of Canada Clinical Trials Group Study BR.20. J. Clin. Oncol. 2007, 25, 4278–4284. [Google Scholar] [CrossRef]
- Ocak, S.; Yamashita, H.; Udyavar, A.R.; Miller, A.N.; Gonzalez, A.L.; Zou, Y.; Jiang, A.; Yi, Y.; Shyr, Y.; Estrada, L.; et al. DNA Copy Number Aberrations in Small-Cell Lung Cancer Reveal Activation of the Focal Adhesion Pathway. Oncogene 2010, 29, 6331–6342. [Google Scholar] [CrossRef]
- Ocak, S.; Chen, H.; Callison, C.; Gonzalez, A.L.; Massion, P.P. Expression of Focal Adhesion Kinase in Small-Cell Lung Carcinoma. Cancer 2012, 118, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Aboubakar Nana, F.; Hoton, D.; Ambroise, J.; Lecocq, M.; Vanderputten, M.; Sibille, Y.; Vanaudenaerde, B.; Pilette, C.; Bouzin, C.; Ocak, S. Increased Expression and Activation of FAK in Small-Cell Lung Cancer Compared to Non-Small-Cell Lung Cancer. Cancers 2019, 11, 1526. [Google Scholar] [CrossRef]
- Aboubakar Nana, F.; Vanderputten, M.; Ocak, S. Role of Focal Adhesion Kinase in Small-Cell Lung Cancer and Its Potential as a Therapeutic Target. Cancers 2019, 11, 1683. [Google Scholar] [CrossRef]
- Aboubakar Nana, F.; Lecocq, M.; Ladjemi, M.Z.; Detry, B.; Dupasquier, S.; Feron, O.; Massion, P.P.; Sibille, Y.; Pilette, C.; Ocak, S. Therapeutic Potential of Focal Adhesion Kinase Inhibition in Small Cell Lung Cancer. Mol. Cancer Ther. 2019, 18, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.C.; Serrels, A.; Stupack, D.G.; Schlaepfer, D.D.; Frame, M.C. Targeting FAK in Anticancer Combination Therapies. Nat. Rev. Cancer 2021, 21, 313–324. [Google Scholar] [CrossRef]
- Ott, P.A.; Elez, E.; Hiret, S.; Kim, D.-W.; Morosky, A.; Saraf, S.; Piperdi, B.; Mehnert, J.M. Pembrolizumab in Patients with Extensive-Stage Small-Cell Lung Cancer: Results from the Phase Ib KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 3823–3829. [Google Scholar] [CrossRef]
- Wang, W.; Hodkinson, P.; McLaren, F.; MacKinnon, A.; Wallace, W.; Howie, S.; Sethi, T. Small Cell Lung Cancer Tumour Cells Induce Regulatory T Lymphocytes, and Patient Survival Correlates Negatively with FOXP3+ Cells in Tumour Infiltrate. Int. J. Cancer 2012, 131, E928–E937. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated Regulation of Myeloid Cells by Tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Tian, T.; Gu, X.; Zhang, B.; Liu, Y.; Yuan, C.; Shao, L.; Guo, Y.; Fan, K. Increased Circulating CD14(+) HLA-DR-/Low Myeloid-Derived Suppressor Cells Are Associated with Poor Prognosis in Patients with Small-Cell Lung Cancer. Cancer Biomark. 2015, 15, 425–432. [Google Scholar] [CrossRef]
- Jean, C.; Chen, X.L.; Nam, J.-O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.G.; Ghassemian, M.; et al. Inhibition of Endothelial FAK Activity Prevents Tumor Metastasis by Enhancing Barrier Function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Lim, K.-H.; McWilliams, R.; Suresh, R.; Lockhart, A.C.; Brown, A.; Breden, M.; Belle, J.I.; Herndon, J.; Bogner, S.J.; et al. Defactinib, Pembrolizumab, and Gemcitabine in Patients with Advanced Treatment Refractory Pancreatic Cancer: A Phase I Dose Escalation and Expansion Study. Clin. Cancer Res. 2022, 28, 5254–5262. [Google Scholar] [CrossRef]
- Udyavar, A.R.; Hoeksema, M.D.; Clark, J.E.; Zou, Y.; Tang, Z.; Li, Z.; Li, M.; Chen, H.; Statnikov, A.; Shyr, Y.; et al. Co-Expression Network Analysis Identifies Spleen Tyrosine Kinase (SYK) as a Candidate Oncogenic Driver in a Subset of Small-Cell Lung Cancer. BMC Syst. Biol. 2013, 7 (Suppl. S5), S1. [Google Scholar] [CrossRef] [PubMed]
- Buchner, M.; Fuchs, S.; Prinz, G.; Pfeifer, D.; Bartholomé, K.; Burger, M.; Chevalier, N.; Vallat, L.; Timmer, J.; Gribben, J.G.; et al. Spleen Tyrosine Kinase Is Overexpressed and Represents a Potential Therapeutic Target in Chronic Lymphocytic Leukemia. Cancer Res. 2009, 69, 5424–5432. [Google Scholar] [CrossRef]
- Hahn, C.K.; Berchuck, J.E.; Ross, K.N.; Kakoza, R.M.; Clauser, K.; Schinzel, A.C.; Ross, L.; Galinsky, I.; Davis, T.N.; Silver, S.J.; et al. Proteomic and Genetic Approaches Identify Syk as an AML Target. Cancer Cell 2009, 16, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Johnson, A.J.; Byrd, J.C. The B-Cell Receptor Signaling Pathway as a Therapeutic Target in CLL. Blood 2012, 120, 1175–1184. [Google Scholar] [CrossRef]
- Chen, L.; Monti, S.; Juszczynski, P.; Daley, J.; Chen, W.; Witzig, T.E.; Habermann, T.M.; Kutok, J.L.; Shipp, M.A. SYK-Dependent Tonic B-Cell Receptor Signaling Is a Rational Treatment Target in Diffuse Large B-Cell Lymphoma. Blood 2008, 111, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Mócsai, A.; Ruland, J.; Tybulewicz, V.L.J. The SYK Tyrosine Kinase: A Crucial Player in Diverse Biological Functions. Nat. Rev. Immunol. 2010, 10, 387–402. [Google Scholar] [CrossRef]
- Cheng, S.; Coffey, G.; Zhang, X.H.; Shaknovich, R.; Song, Z.; Lu, P.; Pandey, A.; Melnick, A.M.; Sinha, U.; Wang, Y.L. SYK Inhibition and Response Prediction in Diffuse Large B-Cell Lymphoma. Blood 2011, 118, 6342–6352. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, J.W.; Sharman, J.; Sweetenham, J.; Johnston, P.B.; Vose, J.M.; Lacasce, A.; Schaefer-Cutillo, J.; De Vos, S.; Sinha, R.; Leonard, J.P.; et al. Inhibition of Syk with Fostamatinib Disodium Has Significant Clinical Activity in Non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia. Blood 2010, 115, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular Subtypes of Small Cell Lung Cancer: A Synthesis of Human and Mouse Model Data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Carney, D.N.; Nau, M.M.; Minna, J.D. Characterization of Variant Subclasses of Cell Lines Derived from Small Cell Lung Cancer Having Distinctive Biochemical, Morphological, and Growth Properties. Cancer Res. 1985, 45, 2924–2930. [Google Scholar]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of Transcription Factor Programs and Immune Pathway Activation Define Four Major Subtypes of SCLC with Distinct Therapeutic Vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Dwivedi, B.; Chen, Z.; Zhang, C.; Barwick, B.; Ernani, V.; Zhang, G.; Gilbert-Ross, M.; Carlisle, J.; Khuri, F.R.; et al. YAP1 Expression in SCLC Defines a Distinct Subtype With T-Cell-Inflamed Phenotype. J. Thorac. Oncol. 2021, 16, 464–476. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-Cell Analyses Reveal Increased Intratumoral Heterogeneity after the Onset of Therapy Resistance in Small-Cell Lung Cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
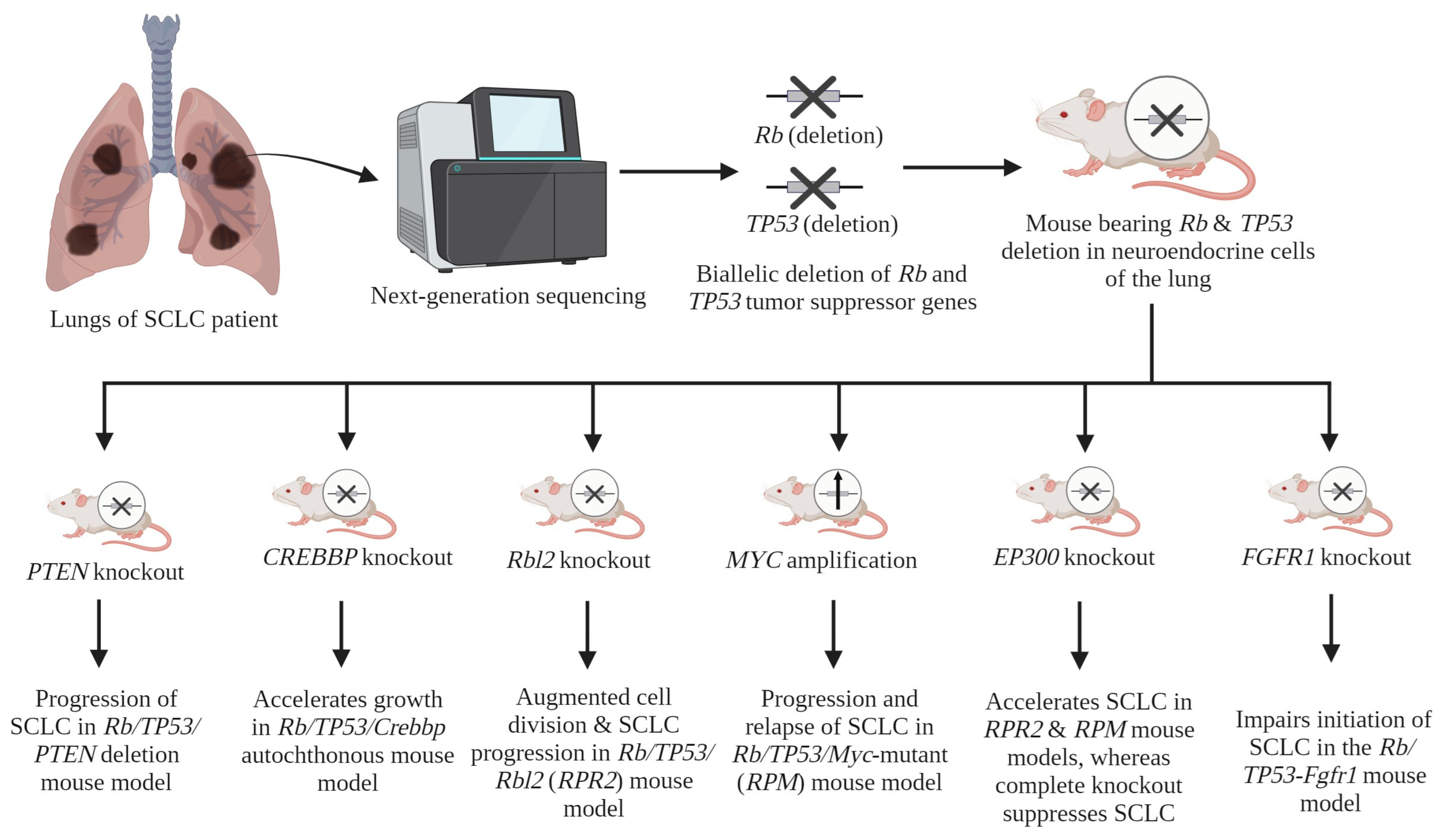
| Serial No. | Gene(s) | Contribution in SCLC Progression | Reference(s) |
|---|---|---|---|
| 1 | Rb and TP53 | Inactivation of Rb and TP53 genes in a conditional mouse model induces SCLC. | [24] |
| 2 | PTEN | Inactivation of one allele of PTEN in Rb/TP53-deleted mouse model leads to the progression of SCLC. | [47] |
| 3 | CREBBP | The deletion of CREBBP accelerates growth in the Rb/TP53/Crebbp autochthonous mouse model. | [34] |
| 4 | Rbl2 | The deletion of this gene results in augmented cell division and a notable increase in the progression of SCLC in vivo, in the Rb/TP53/Rbl2 (RPR2) mouse model. | [37] |
| 5 | MYC | MYC amplification leads to the promotion of aggressive, highly metastatic, and refractory SCLC tumors that are initially responsive to chemotherapy. This effect has been observed in vivo in the Rb/TP53/Myc-mutant (RPM) mouse model. | [42] |
| 6 | EP300 | A mutated EP300 gene within the RPR2 and RPM models accelerates SCLC growth, while the complete KO of EP300 suppresses SCLC growth. A mechanistic study showed the tumor-suppressive role of the HAT domain of the EP300 protein, whereas other domains (i.e., KIX, BAD, and TAZ) showed tumor-promoting activity. | [36] |
| 7 | FGFR1 | In vivo, within the RP-Fgfr1 mouse model, FGFR1 demonstrates a context-dependent impact. The impairment of SCLC formation from CGRPPOS NE cells is observed. Conversely, it is noted that it promotes the growth of SCLC and low-grade NE bronchial lesions from tracheobronchial-basal cells. | [46] |
| 8 | PLCG2 | Using clinical samples and in vivo models, PLCG2 has been reported to be associated with higher SCLC metastatic potential, thereby emerging as a potential driver of SCLC progression. | [48] |
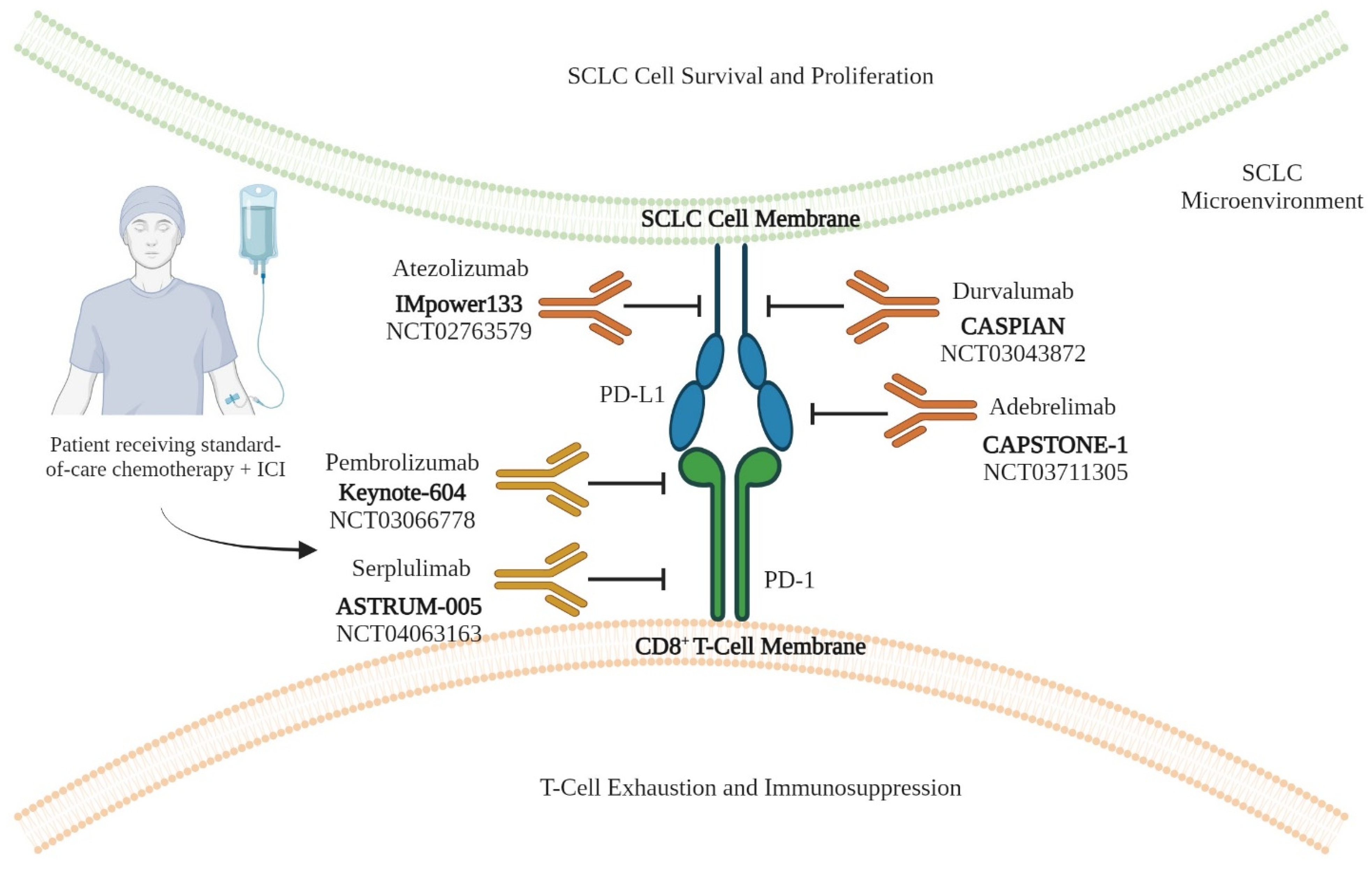
| S. No. | Clinical Trial Identifier (Name) | Treatment Group | Primary End Point(s) | Outcome | Reference(s) |
|---|---|---|---|---|---|
| 1 | NCT02763579 (IMpower133) | Group I (Treatment): Atezolizumab (PD-L1 inhibitor) + carboplatin-etoposide (C/E) chemotherapy Group II (Control): Placebo + C/E chemotherapy | PFS and OS | PFS: Group I: 5.2 months Group II: 4.3 months OS: Group I: 12.3 months Group II: 10.3 months | [10,11,12,13] |
| 2 | NCT03043872 (CASPIAN) | Group I (Treatment): Durvalumab (PD-L1 inhibitor) + carboplatin-etoposide (C/E) chemotherapy Group II (Control): C/E chemotherapy | PFS and OS | PFS: Group I: 5.1 months Group II: 5.4 months OS: Group I: 13.0 months Group II: 10.3 months | [50,51] |
| 3 | NCT03066778 (KEYNOTE-604) | Group I (Treatment): Pembrolizumab (PD-1 inhibitor) + carboplatin-etoposide (C/E) chemotherapy Group II (Control): Placebo + C/E chemotherapy | PFS and OS | PFS: Group I: 4.5 months Group II: 4.3 months OS: Group I: 10.8 months Group II: 9.7 months | [52] |
| 4 | NCT03711305 (CAPSTONE-1) | Group I (Treatment): Adebrelimab (PD-L1 inhibitor) + carboplatin-etoposide (C/E) chemotherapy Group II (Control): Placebo + C/E chemotherapy | OS | OS: Group I: 15.3 months Group II: 12.8 months | [53] |
| 5 | NCT04063163 (ASTRUM-005) | Group I (Treatment): Serplulimab (PD-1 inhibitor) + carboplatin-etoposide (C/E) chemotherapy Group II (Control): Placebo + C/E chemotherapy | OS | OS: Group I: 15.4 months Group II: 10.9 months | [54] |
| PK Inhibitor | Main Target | Role in Immunotherapy in SCLC | Clinical Trial Identifier | Reference(s) |
|---|---|---|---|---|
| Trilaciclib (G1T28) | CDK4/6 | Has a pivotal role in governing the advancement of the cell cycle in cancer cells. The administration of chemotherapy to patients with SCLC leads to an increased peripheral lymphocyte count and improved activation of T cells when CDK4/6 is inhibited. Furthermore, it has been observed that it augments the levels of PD-L1 expression, hence sensitizing its blockage, in syngeneic mouse models conducted in vivo. | NCT03041311 | [57], [62,63] |
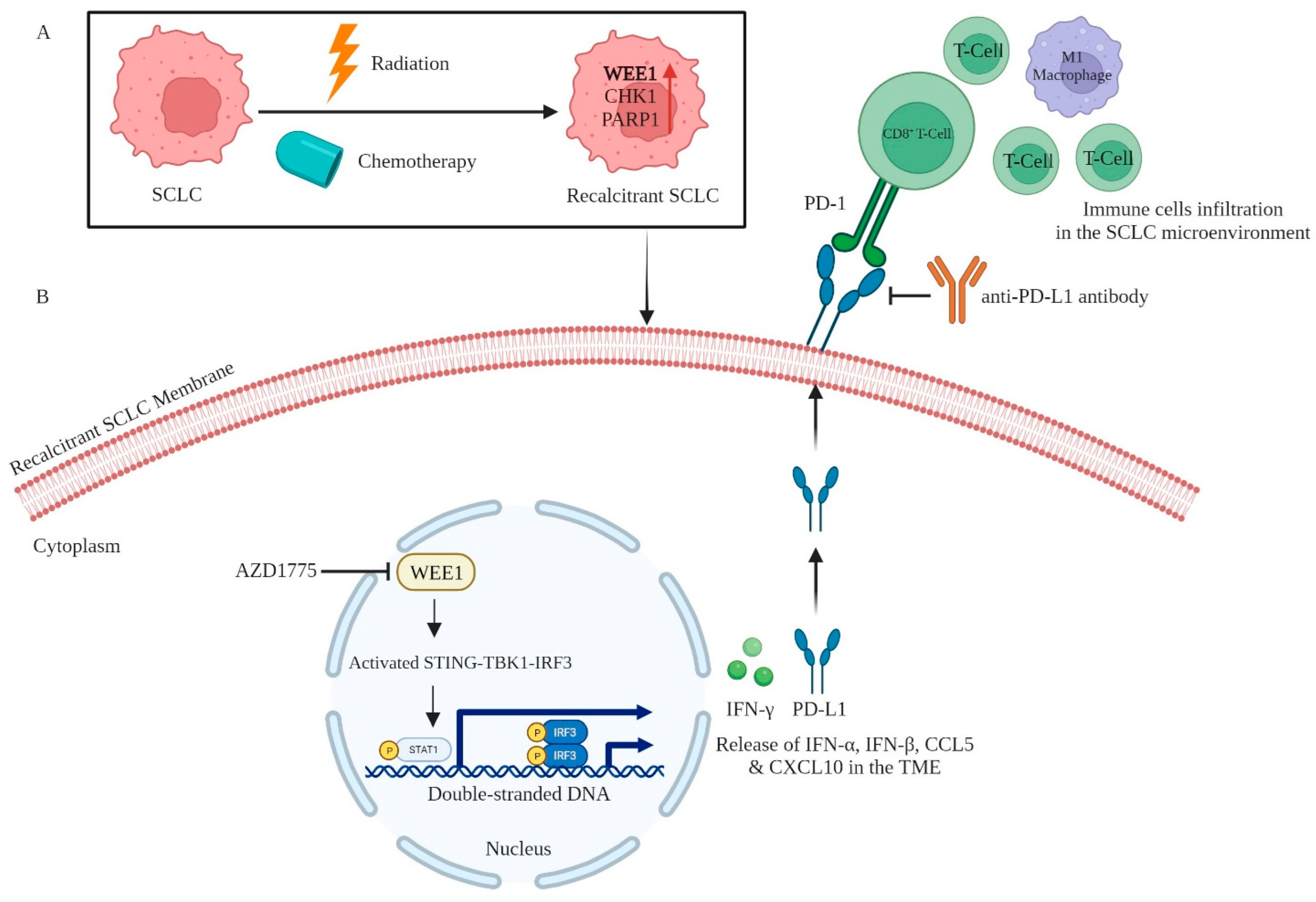
| AZD1775 (Adavosertib) | WEE1 | Regulates cell cycle checkpoint at G2/M transition in cancer. The inhibition of WEE1 is linked to the surged infiltration of CD8+ T cells, facilitating immune responses. Concurrently, this inhibition also elevates IFN-γ and PD-L1 (sensitizing its blockade), via STAT1 activation in in vivo SCLC mouse models. | NCT02937818 | [19] |
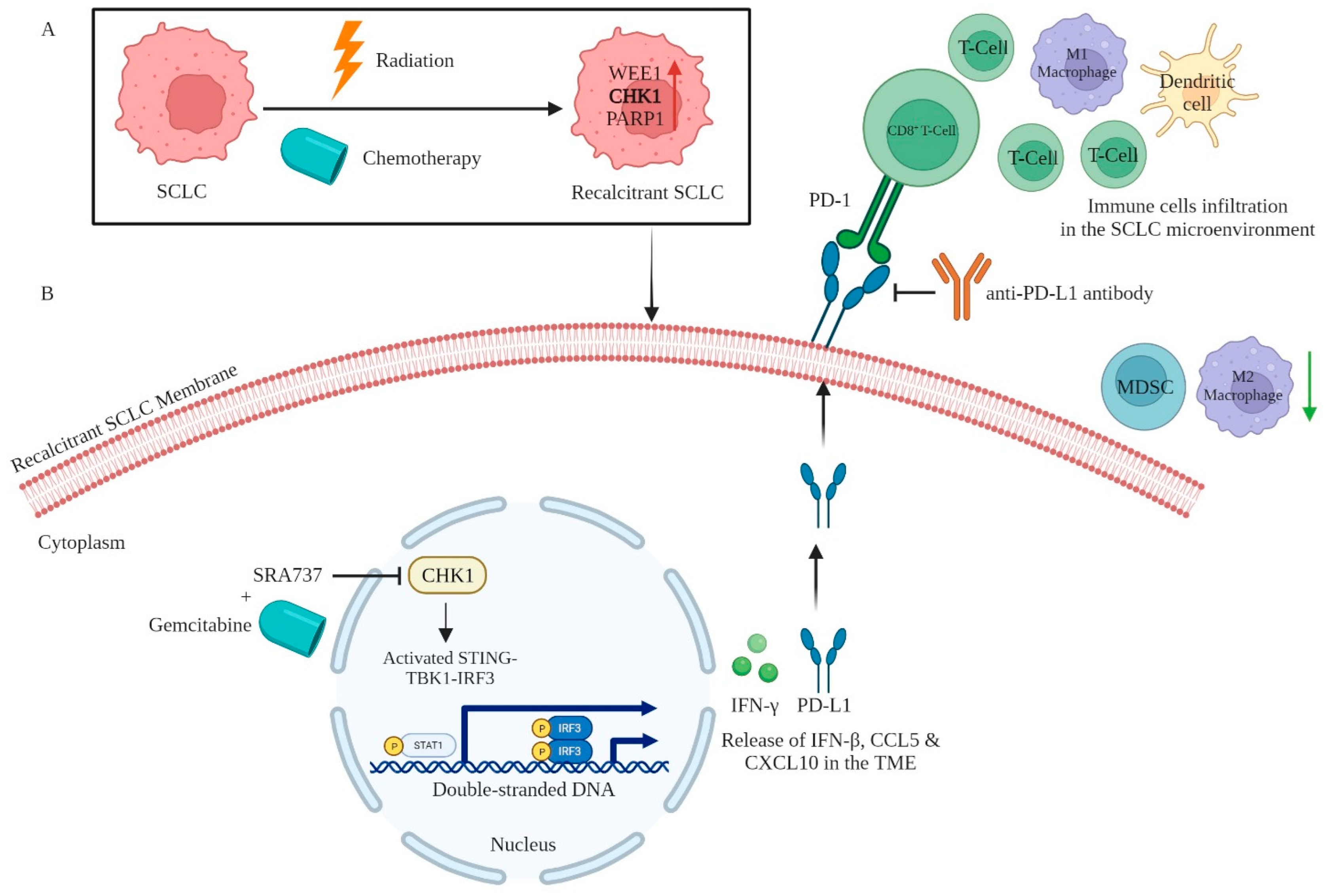
| SRA737 Prexasertib (LY2606368) Rabusertib (LY2603618) | CHK1 | Involved in the regulation of the cell cycle in cancer. The inhibition of CHK1 augments the effect of PD-L1 blockade in an RPP tumor-bearing immunocompetent SCLC mouse model by significantly augmenting the infiltration of CD8+ T cells, DCs, and M1-like macrophages (pro-inflammatory/anti-tumor), in the TME | [20] [80] | |
| Olaparib Pamiparib (BGB290) Fluzoparib (SHR-3162) | PARP (non-kinase) | A non-kinase involved in the DDR in cancer. The inhibition of PARP is also associated with the increased infiltration of CD8+ T cells in the TME and elevated expression of PD-L1 (sensitizing its blockade) in an RPP tumor-bearing immunocompetent SCLC mouse model. | NCT02484404 NCT02734004 NCT02660034 NCT02937818 NCT04782089 | [82] [83] [84] [80] [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiwari, A.; Kumari, B.; Nandagopal, S.; Mishra, A.; Shukla, K.K.; Kumar, A.; Dutt, N.; Ahirwar, D.K. Promises of Protein Kinase Inhibitors in Recalcitrant Small-Cell Lung Cancer: Recent Scenario and Future Possibilities. Cancers 2024, 16, 963. https://doi.org/10.3390/cancers16050963
Tiwari A, Kumari B, Nandagopal S, Mishra A, Shukla KK, Kumar A, Dutt N, Ahirwar DK. Promises of Protein Kinase Inhibitors in Recalcitrant Small-Cell Lung Cancer: Recent Scenario and Future Possibilities. Cancers. 2024; 16(5):963. https://doi.org/10.3390/cancers16050963
Chicago/Turabian StyleTiwari, Aniket, Beauty Kumari, Srividhya Nandagopal, Amit Mishra, Kamla Kant Shukla, Ashok Kumar, Naveen Dutt, and Dinesh Kumar Ahirwar. 2024. "Promises of Protein Kinase Inhibitors in Recalcitrant Small-Cell Lung Cancer: Recent Scenario and Future Possibilities" Cancers 16, no. 5: 963. https://doi.org/10.3390/cancers16050963
APA StyleTiwari, A., Kumari, B., Nandagopal, S., Mishra, A., Shukla, K. K., Kumar, A., Dutt, N., & Ahirwar, D. K. (2024). Promises of Protein Kinase Inhibitors in Recalcitrant Small-Cell Lung Cancer: Recent Scenario and Future Possibilities. Cancers, 16(5), 963. https://doi.org/10.3390/cancers16050963