
Advancing Precision Oncology in Hereditary Paraganglioma-Pheochromocytoma Syndromes: Integrated Interpretation and Data Sharing of the Germline and Tumor Genomes

 , , ,
, , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Development of INT2GRATE|HPPGL Platform
2.1.1. INT2GRATE|HPPGL Variant Evidence Framework
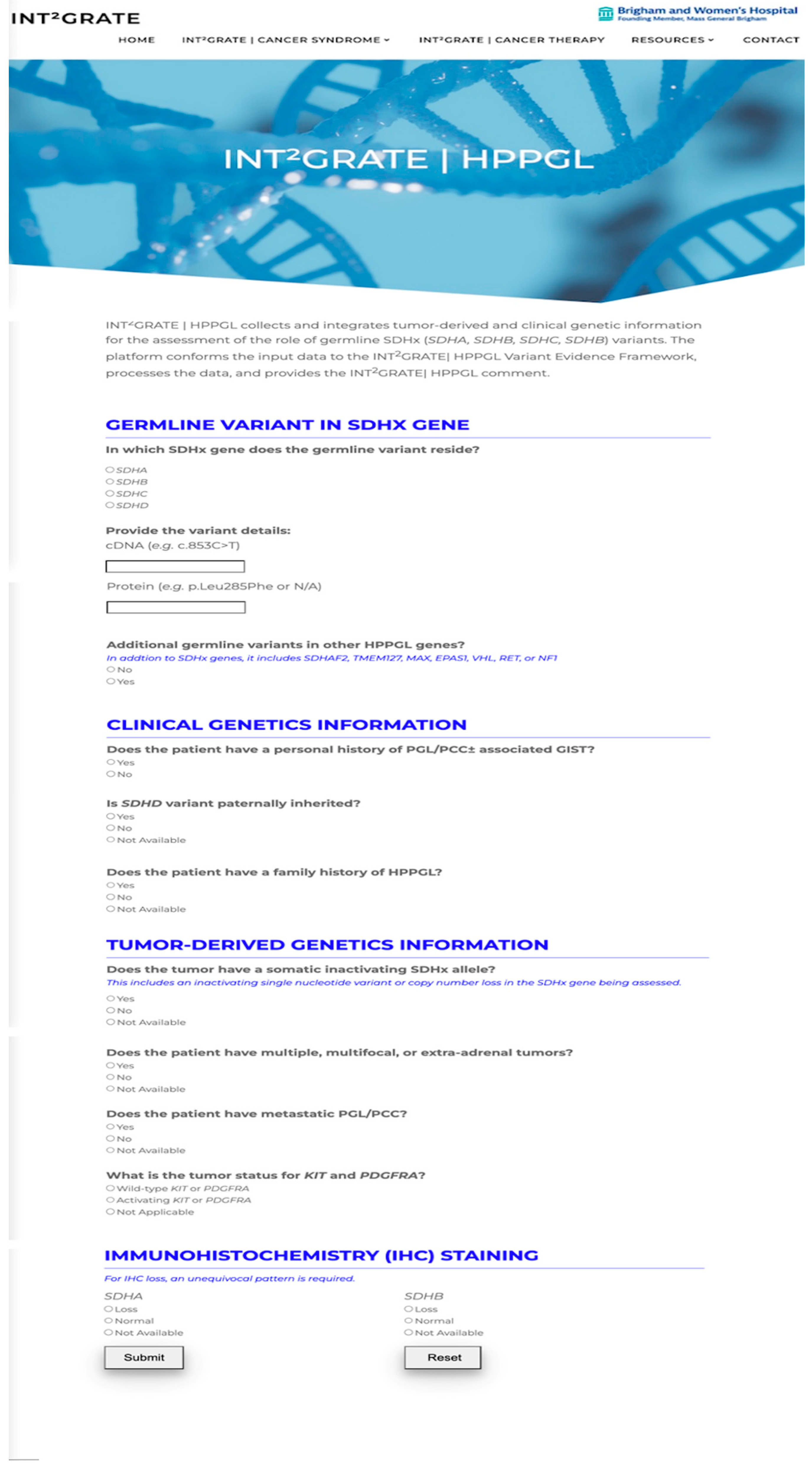
2.1.2. INT2GRATE Digital Web Portal for Single Variants
User Interface (UI)
INT2GRATE Backend
2.1.3. INT2GRATE|HPPGL Automated Batch Variant Processing
2.1.4. INT2GRATE|HPPGL Variant Data Sharing via API
INT2GRATE UI Variant Submission
Large-Scale INT2GRATE Variant Submission
2.2. Germline Genetic Laboratory Testing
2.3. Tumor Genetic Laboratory Testing
2.3.1. Single-Nucleotide Variant (SNV)/Indel Analysis
2.3.2. OncoPanel Copy Number Analysis
2.3.3. OncoPanel Structural Analysis
2.4. Laboratory Testing—Immunohistochemistry (IHC)
2.5. Patient Cohorts and Data Query
3. Results
3.1. Development of INT2GRATE|HPPGL Variant Evidence Framework and Rationale
3.1.1. Germline Variant in SDHx Genes and Rationale
3.1.2. Clinical Genetics Criteria and Rationale
Personal History
Family History
Parent of Origin
3.1.3. Tumor-Derived Genetic Information and Rationale
Somatic Inactivating Allele
KIT or PDGFRA Mutational Status
Multiple, Multifocal, or Extra-Adrenal Tumors and PGL/PCC Metastasis
3.1.4. Tumor Immunohistochemistry Pattern and Rationale
3.2. Assignment of INT2GRATE|HPPGL Categories
3.3. Application of INT2GRATE|HPPGL
3.4. Patient Clinical Presentations
3.5. INT2GRATE|HPPGL Variant Analysis
- Cohort 1
- Cohort 2
- Cohort 3
3.6. INT2GRATE|HPPGL Variant Submission to ClinVar
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adam, M.P.; Feldman, J.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Amemiya, A. GeneReviews; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Al Subhi, A.R.; Boyle, V.; Elston, M.S. Systematic Review: Incidence of Pheochromocytoma and Paraganglioma Over 70 Years. J. Endocr. Soc. 2022, 6, bvac105. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef]
- Moreno, C.; Santos, R.M.; Burns, R.; Zhang, W.C. Succinate Dehydrogenase and Ribonucleic Acid Networks in Cancer and Other Diseases. Cancers 2020, 12, 3237. [Google Scholar] [CrossRef]
- Manning, D.K.; Shivdasani, P.; Koeller, D.R.; Schwartz, A.; Rana, H.Q.; Garber, J.E.; Lindeman, N.I.; Ghazani, A.A. Assessment of genomic alterations in non-syndromic von Hippel-Lindau: Insight from integrating somatic and germline next generation sequencing genomic data. Data Brief 2021, 39, 107653. [Google Scholar] [CrossRef] [PubMed]
- Koeller, D.R.; Manning, D.K.; Schwartz, A.; Chittenden, A.; Hayes, C.P.; Abraamyan, F.; Rana, H.Q.; Lindeman, N.I.; Garber, J.E.; Ghazani, A.A. An optimized protocol for evaluating pathogenicity of. MethodsX 2022, 9, 101761. [Google Scholar] [CrossRef]
- Rana, H.Q.; Koeller, D.R.; Schwartz, A.; Manning, D.K.; Schneider, K.A.; Krajewski, K.M.; Choueiri, T.K.; Lindeman, N.I.; Garber, J.E.; Ghazani, A.A. Pathogenicity of VHL variants in families with non-syndromic von Hippel-Lindau phenotypes: An integrated evaluation of germline and somatic genomic results. Eur. J. Med. Genet. 2021, 64, 104359. [Google Scholar] [CrossRef]
- Schwartz, A.; Manning, D.K.; Koeller, D.R.; Chittenden, A.; Isidro, R.A.; Hayes, C.P.; Abraamyan, F.; Manam, M.D.; Dwan, M.; Barletta, J.A.; et al. An integrated somatic and germline approach to aid interpretation of germline variants of uncertain significance in cancer susceptibility genes. Front. Oncol. 2022, 12, 942741. [Google Scholar] [CrossRef] [PubMed]
- Abo, R.P.; Ducar, M.; Garcia, E.P.; Thorner, A.R.; Rojas-Rudilla, V.; Lin, L.; Sholl, L.M.; Hahn, W.C.; Meyerson, M.; Lindeman, N.I.; et al. BreaKmer: Detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 2015, 43, e19. [Google Scholar] [CrossRef]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F.; Society, E. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin. Diagn. Pathol. 2006, 23, 91–102. [Google Scholar] [CrossRef]
- Brcic, I.; Kashofer, K.; Skone, D.; Liegl-Atzwanger, B. KIT mutation in a naïve succinate dehydrogenase-deficient gastric GIST. Genes Chromosomes Cancer 2019, 58, 798–803. [Google Scholar] [CrossRef]
- Wang, Y.M.; Gu, M.L.; Ji, F. Succinate dehydrogenase-deficient gastrointestinal stromal tumors. World J. Gastroenterol. 2015, 21, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Wanna, G.B.; Ahmad, M.; Lussey-Lepoutre, C.; Perrier, N.D.; Nölting, S.; Amar, L.; Timmers, H.J.L.M.; Schwam, Z.G.; Estrera, A.L.; et al. Clinical consensus guideline on the management of phaeochromocytoma and paraganglioma in patients harbouring germline SDHD pathogenic variants. Lancet Diabetes Endocrinol. 2023, 11, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Del Rivero, J.; Else, T.; Howe, J.R.; Asa, S.L.; Cohen, D.L.; Dahia, P.L.M.; Fraker, D.L.; Goodman, K.A.; Hope, T.A.; et al. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma. Pancreas 2021, 50, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.A.; Ascher, D.B.; Pires, D.E.V.; Barnes, D.R.; Vialard, L.; Casey, R.T.; Bradshaw, N.; Adlard, J.; Aylwin, S.; Brennan, P.; et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes. J. Med. Genet. 2018, 55, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J.; Benn, D.E.; Chou, A.; Clarkson, A.; Muljono, A.; Meyer-Rochow, G.Y.; Richardson, A.L.; Sidhu, S.B.; Robinson, B.G.; Clifton-Bligh, R.J. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2010, 41, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Udager, A.M.; Magers, M.J.; Goerke, D.M.; Vinco, M.L.; Siddiqui, J.; Cao, X.; Lucas, D.R.; Myers, J.L.; Chinnaiyan, A.M.; McHugh, J.B.; et al. The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2018, 71, 47–54. [Google Scholar] [CrossRef]
- Papathomas, T.G.; Oudijk, L.; Persu, A.; Gill, A.J.; van Nederveen, F.; Tischler, A.S.; Tissier, F.; Volante, M.; Matias-Guiu, X.; Smid, M.; et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod. Pathol. 2015, 28, 807–821. [Google Scholar] [CrossRef]
- Amar, L.; Pacak, K.; Steichen, O.; Akker, S.A.; Aylwin, S.J.B.; Baudin, E.; Buffet, A.; Burnichon, N.; Clifton-Bligh, R.J.; Dahia, P.L.M.; et al. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat. Rev. Endocrinol. 2021, 17, 435–444. [Google Scholar] [CrossRef]
- Casey, R.T.; Ten Hoopen, R.; Ochoa, E.; Challis, B.G.; Whitworth, J.; Smith, P.S.; Martin, J.E.; Clark, G.R.; Rodger, F.; Maranian, M.; et al. SDHC epi-mutation testing in gastrointestinal stromal tumours and related tumours in clinical practice. Sci. Rep. 2019, 9, 10244. [Google Scholar] [CrossRef]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Moog, S.; Lussey-Lepoutre, C.; Favier, J. Epigenetic and metabolic reprogramming of SDH-deficient paragangliomas. Endocr. Relat. Cancer 2020, 27, R451–R463. [Google Scholar] [CrossRef] [PubMed]
- Bernardo-Castiñeira, C.; Valdés, N.; Sierra, M.I.; Sáenz-de-Santa-María, I.; Bayón, G.F.; Perez, R.F.; Fernández, A.F.; Fraga, M.F.; Astudillo, A.; Menéndez, R.; et al. SDHC Promoter Methylation, a Novel Pathogenic Mechanism in Parasympathetic Paragangliomas. J. Clin. Endocrinol. Metab. 2018, 103, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Idikio, H.A. Immunohistochemistry in diagnostic surgical pathology: Contributions of protein life-cycle, use of evidence-based methods and data normalization on interpretation of immunohistochemical stains. Int. J. Clin. Exp. Pathol. 2009, 3, 169–176. [Google Scholar] [PubMed]
- Ding, C.C.; Chan, S.; Mak, J.; Umetsu, S.E.; Simko, J.P.; Ruiz-Cordero, R.; Saunders, T.; Chan, E. An exploration in pitfalls in interpreting SDHB immunohistochemistry. Histopathology 2022, 81, 264–269. [Google Scholar] [CrossRef]
- Santi, R.; Rapizzi, E.; Canu, L.; Ercolino, T.; Baroni, G.; Fucci, R.; Costa, G.; Mannelli, M.; Nesi, G. Potential Pitfalls of SDH Immunohistochemical Detection in Paragangliomas and Phaeochromocytomas Harbouring Germline. Anticancer. Res. 2017, 37, 805–812. [Google Scholar] [CrossRef]


{kind=link}
| Germline Variant Information | Clinical Genetics Information | Tumor-Derived Information | Tumor Immunohistochemistry Staining | INT2GRATE Category | INT2GRATE Comment | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| INT2GRATE ID Codes | SDHx Gene | Additional Germline Variants in HPPGL- Associated Genes ❖ | Personal History of PGL/PCC +/− GIST Associated Tumors | Paternal Inheritance of Germline Variant | Family History of PGL/PCC /GIST | Multiple, Multifocal or Extra Adrenal Tumors | PGL/PCC Metastasis | Somatic Inactivating SDHx Allele | KIT and PDGFRA Mutation Status | SDHA | SDHB | |||
| SDHA TABLE | A-I | SDHA | No | Yes | Yes, No, N/A | Yes, N/A | Yes, No, N/A | Yes, No, N/A | Yes | KIT and PDGFRA WT or N/A | Loss | Loss | INT2GRATE POSITIVE | The combination of clinical genetics evidence, tumor features, presence of somatic inactivating allele in the SDHA locus, and loss of SDHA and SDHB by immunohistochemistry support the likely relevance of this SDHA variant to Hereditary Paraganglioma-Pheochromocytoma. Correlation with clinical findings and other studies is advised. |
| A-II | SDHA | No | Yes | Yes, No, N/A | Yes, N/A | Yes, No, N/A | Yes, No, N/A | Yes | KIT and PDGFRA WT or N/A | Normal | Loss | INT2GRATE NEUTRAL | Immunohistochemistry for SDH proteins shows loss of SDHB and retained SDHA expression. This pattern is supportive of a defect in one of the members of the SDH complex, but does not specifically support the presence of biallelic inactivation of SDHA. Therefore, the significance of this germline variant is uncertain. Review of the immunohistochemical findings is advised and repeat immunohistochemistry could be considered for the assessment of this variant. | |
| A-III | SDHA | No | Yes | Yes, No, N/A | Yes, N/A | Yes, No, N/A | Yes, No, N/A | Yes | KIT and PDGFRA WT or N/A | Loss | Normal | INT2GRATE NEUTRAL | Immunohistochemistry for SDH proteins shows an unusual staining pattern (isolated loss of SDHA). Therefore, the significance of this germline variant is uncertain. Review of the immunohistochemical findings is advised and repeat immunohistochemistry could be considered for the assessment of this variant. | |
| A-IV | SDHA | No | Yes | Yes, No, N/A | Yes, N/A | Yes, No, N/A | Yes, No, N/A | Yes | KIT and PDGFRA WT or N/A | Normal | Normal | INT2GRATE NEUTRAL | INT2GRATE requires loss of SDHA and SDHB expression by immunohistochemistry for the assessment of this variant. Without additional evidence, the significance of this germline variant in relation to Hereditary Paraganglioma-Pheochromocytoma is uncertain. | |
| A-V | SDHA | No | No | Yes, No, N/A | No | No | No | No | KIT and PDGFRA WT or N/A | Normal | Normal | INT2GRATE NEGATIVE | The combination of the negative personal and family history of PGL/PCC and associated tumors, absence of somatic inactivating alteration in SDHA, and retained SDHA and SDHB expression by immunohistochemistry does not support the involvement of this germline variant in Hereditary Paraganglioma-Pheochromocytoma syndromes. Correlation with clinical findings and other studies is advised. | |
| SDHB TABLE | B-I | SDHB | No | Yes | Yes, No, N/A | Yes, N/A | Yes, N/A | Yes, N/A | Yes | KIT and PDGFRA WT | Normal | Loss | INT2GRATE POSITIVE | The combination of clinical genetics evidence, tumor features, presence of somatic inactivating allele in the SDHB locus, and loss of SDHB by immunohistochemistry support the likely relevance of this SDHB variant to Hereditary Paraganglioma-Pheochromocytoma. Correlation with clinical findings and other studies is advised. |
| B-II | SDHB | No | Yes | Yes, No, N/A | Yes, N/A | Yes, No, N/A, Any | Yes, No, N/A (Any) | Yes | KIT and PDGFRA WT | Loss | Loss | INT2GRATE NEUTRAL | Immunohistochemistry for SDH proteins shows loss of both SDHA and SDHB, suggesting biallelic inactivation of SDHA. Therefore, the significance of this germline variant in relation to Hereditary Paraganglioma-Pheochromocytoma is uncertain. Review of the immunohistochemical findings is advised and repeat immunohistochemistry could be considered for the assessment of this variant. | |
| B-III | SDHB | No | Yes | Yes, No, N/A | Yes, N/A | Yes, No, N/A, Any | Yes, No, N/A (Any) | Yes | KIT and PDGFRA WT | Normal | Normal | INT2GRATE NEUTRAL | INT2GRATE requires loss of SDHB expression by immunohistochemistry for the assessment of this variant. Without additional evidence, the significance of this germline variant in relation to Hereditary Paraganglioma-Pheochromocytoma is uncertain. | |
| B-IV | SDHB | No | No | Yes, No, N/A | No | No | No | No | KIT and PDGFRA WT or NA | Normal | Normal | INT2GRATE NEGATIVE | The combination of the negative personal and family history of PGL/PCC and associated tumors, absence of somatic inactivating alteration in SDHB, and absence of SDHB deficiency status by immunohistochemistry does not support the involvement of this germline variant in Hereditary Paraganglioma-Pheochromocytoma syndromes. Correlation with clinical findings and other studies is advised. | |
| B-V | SDHB | No | No | Yes, No, N/A | No | No | No | No | KIT and PDGFRA WT or NA | N/A | N/A | INT2GRATE NEGATIVE | The combination of the negative personal and family history of PGL/PCC and associated tumors, and absence of somatic inactivating alteration in SDHB does not support the involvement of this germline variant in Hereditary Paraganglioma-Pheochromocytoma syndromes. Correlation with clinical findings, age-related penetrance and other studies is advised. | |
| SDHC TABLE | C-I | SDHC | No | Yes | Yes, No, N/A | Yes, N/A | Yes, N/A | Yes, No, N/A (Any) | Yes | KIT and PDGFRA WT | Normal | Loss | INT2GRATE POSITIVE | The combination of clinical genetics evidence, tumor features, presence of somatic inactivating allele in the SDHC locus, and loss of SDHB by immunohistochemistry support the likely relevance of this SDHC variant to Hereditary Paraganglioma-Pheochromocytoma. Correlation with clinical findings and other studies is advised. |
| C-II | SDHC | No | Yes | Yes, No, N/A | Yes, N/A | Yes, N/A | N/A | Yes | KIT and PDGFRA WT | Loss | Loss | INT2GRATE NEUTRAL | Immunohistochemistry for SDH proteins shows loss of both SDHA and SDHB, suggesting biallelic inactivation of SDHA. Therefore, the significance of this germline variant in relation to Hereditary Paraganglioma-Pheochromocytoma is uncertain. Review of the immunohistochemical findings is advised and repeat immunohistochemistry could be considered for the assessment of this variant. | |
| C-III | SDHC | No | Yes | Yes, No, N/A | Yes, N/A | Yes, N/A | N/A | Yes | KIT and PDGFRA WT | Normal | Normal | INT2GRATE NEUTRAL | INT2GRATE requires loss of SDHB expression by immunohistochemistry for the assessment of this VUS. Without additional evidence, the significance of this germline variant in relation to Hereditary Paraganglioma-Pheochromocytoma is uncertain. | |
| C-IV | SDHC | No | No | Yes, No, N/A | No | No | No | No | KIT and PDGFRA WT or NA | Normal | Normal | INT2GRATE NEGATIVE | The combination of the negative personal and family history of PGL/PCC and associated tumors, absence of somatic inactivating alteration in SDHC, and absence of SDHB deficiency status by immunohistochemistry does not support the involvement of this germline variant in Hereditary Paraganglioma-Pheochromocytoma syndromes. Correlation with clinical findings and other studies is advised. | |
| SDHD TABLE | D-I | SDHD | No | Yes | Yes | Yes, N/A | Yes, N/A | N/A | Yes | KIT and PDGFRA WT | Normal | Loss | INT2GRATE POSITIVE | The combination of clinical genetics evidence, tumor features, parent-of-origin effect (paternal inheritance of this SDHD variant), presence of somatic inactivating allele in the SDHD locus, and loss of SDHB by immunohistochemistry support the likely relevance of this SDHD variant to Hereditary Paraganglioma-Pheochromocytoma. Correlation with clinical findings and other studies is advised. |
| D-II | SDHD | No | Yes | Yes | Yes, N/A | Yes, N/A | N/A | Yes | KIT and PDGFRA WT | Loss | Loss | INT2GRATE NEUTRAL | Immunohistochemistry for SDH proteins shows loss of both SDHA and SDHB, suggesting biallelic inactivation of SDHA. Therefore, the significance of this germline variant in relation to Hereditary Paraganglioma-Pheochromocytoma is uncertain. Review of the immunohistochemical findings is advised and repeat immunohistochemistry could be considered for the assessment of this variant. | |
| D-III | SDHD | No | Yes | Yes | Yes, N/A | Yes, N/A | N/A | Yes | KIT and PDGFRA WT | Normal | Normal | INT2GRATE NEUTRAL | INT2GRATE requires loss of SDHB expression by immunohistochemistry for the assessment of this VUS. Without additional evidence, the significance of this germline variant in relation to Hereditary Paraganglioma-Pheochromocytoma is uncertain. | |
| D-IV | SDHD | No | No | No | No | No | No | No | KIT and PDGFRA WT or NA | Normal | Normal | INT2GRATE NEGATIVE | The combination of the negative personal and family history of PGL/PCC and associated tumors, absence of somatic inactivating alteration in SDHD, absence of parent-of-origin effect (paternal inheritance of this SDHD variant), and absence of SDHB deficiency status by immunohistochemistry does not support the involvement of this germline variant in Hereditary Paraganglioma-Pheochromocytoma syndromes. Correlation with clinical findings and other studies is advised. | |
| D-V | SDHD | No | No | No | No | No | No | No | KIT and PDGFRA WT or NA | NA | NA | INT2GRATE NEGATIVE | The combination of the negative personal and family history of PGL/PCC and associated tumors, absence of somatic inactivating alteration in SDHD, absence of parent-of-origin effect (paternal inheritance of this SDHD variant) does not support the involvement of this germline variant in Hereditary Paraganglioma-Pheochromocytoma syndromes. Correlation with clinical findings, age-related penetrance and other studies is advised. | |
| Category | Number | Percent (%) |
|---|---|---|
| Total Patient Number | 109 | 100 |
| Male | 37 | 0.34 |
| Female | 72 | 0.66 |
| Median age at cancer diagnosis | 47 (2–84) | |
| Personal Cancer History | ||
| HPPGL-related cancer | 30 | 0.28 |
| Non-HPPGL-related cancer | 77 | 0.71 |
| Unaffected | 2 | 0.02 |
| FamilyCancer History | ||
| Positive for HPPGL-related cancer | 4 | 0.04 |
| Negative for HPPGL-related cancer | 105 | 0.96 |
| Self-Reported Ancestry | ||
| European | 80 | 0.73 |
| African/African American | 5 | 0.05 |
| Latino/Admixed American | 3 | 0.03 |
| East Asian/South Asian | 6 | 0.06 |
| Ashkenazi Jewish | 5 | 0.05 |
| Other | 7 | 0.06 |
| Not reported | 3 | 0.03 |
| Category | Number | Percent |
|---|---|---|
| Total Patient Number | 494 | 100 |
| Male | 142 | 28.74 |
| Female | 352 | 71.26 |
| Personal Cancer History | ||
| Positive for HPPGL-related cancer | 36 | 7.29 |
| Negative for HPPGL-related cancer | 458 | 92.71 |
| Familyhistory of cancer | ||
| Positive for HPPGL-related cancer | 12 | 2.43 |
| Negative for HPPGL-related cancer | 482 | 97.57 |
| Subject | Germline Variant In HPPGL Genes | Clinical Genetics Information | Tumor-Derived Information | Tumor Immunohistochemistry Staining | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Germline SDHx Allele | Other Germline Alteration in HPPGL | Personal History of PGL/PCC ± Associated GIST | Parent-of-Origin Inheritance | Family History of PGL/PCC | Multiple, Multifocal or Extra Adrenal Tumors | PGL/PCC Metastasis | Somatic SDHx Allele | KIT and PDGFRA Mutation Status | SDHA and SDHB | |
| 1 | SDHC:c.397C>T (p.Arg133Ter) | Neg | No (GIST) | NT | Neg | Neg | Neg | Neg | WT | Intact SDHA |
| 2 | SDHA:c.615T>A (p.Tyr205Ter) | Neg | No (GIST) | NT | Neg | Neg | Neg | Neg | WT | Loss of SDHB and SDHA |
| 5 | SDHB:c.380T>G (p.Ile127Ser) | Neg | No (GIST) | NT | Yes, PGL in mother | Neg | Neg | Neg | WT | NT |
| 7 | SDHA:c.1753C>T (p.Arg585Trp) | Neg | Yes | NT | Neg | Neg | Yes, PGL metastasis | SDHA:c.1534C>T (p.Arg512Ter) SDHA:c.1091T>C (p.Val364Ala) | WT | Loss of SDHB, equivocal SDHA (areas of tumor that are negative) |
| 8 | SDHB:c.600G>T (p.Trp200Cys) | Neg | No (GIST) | NT | Neg | Neg | Neg | Neg | WT | Loss of SDHB |
| 11 | SDHC:c.224G>A (p.Gly75Asp) | Neg | No (GIST) | NT | Neg | Neg | Neg | Neg | WT | NT |
| 12 | SDHA:c.91C>T (p.Arg31Ter) | Neg | No (GIST) | NT | Neg | Neg | Neg | Neg | WT | NT |
| 13 | SDHC:c.43C>T (p.Arg15Ter) | Neg | Yes | NT | Neg | Neg | Neg | Neg | WT | Intact SDHA, loss of SDHB |
| 14 | SDHB:c.380T>G (p.Ile127Ser) | Neg | Yes | NT | Neg | Neg | Neg | SDHB One Copy Deletion | WT | NT |
| 15 | SDHC:c.397C>T (p.Arg133Ter) | Neg | No (GIST) | NT | Neg | Neg | Neg | Neg | WT | Loss of SDHB |
| 18 | SDHB:c.605_609dupACGGA (p.Asp204fs) | Neg | No | NT | Yes, PGL in son | Neg | Neg | SDHB One Copy Deletion | WT | NT |
| 20 | SDHC:c.223G>C (p.Gly75Arg) | Neg | Yes | NT | Neg | Neg | Neg | Neg | WT | Intact SDHB and SDHA |
| 22 | SDHC:c.397C>T (p.Arg133Ter) | Neg | Yes | NT | Neg | Neg | Neg | Neg | WT | Loss of SDHB |
| 23 | SDHC:c.397C>T (p.Arg133Ter) | Neg | No | NT | Yes, PGL in Father | Neg | Neg | Neg | WT | Intact SDHA and SDHB |
| 28 | SDHB:c.72+1G>T | Neg | Yes | NT | Neg | Neg | Neg | SDHB One Copy Deletion | WT | Loss of SDHB |
| 30 | SDHB:c.194T>A (p.Leu65His) | Yes, SDHB:c.200+3G>C | Yes | NT | Neg | Neg | Yes, PGL metastasis | Neg | WT | Loss of SDHB |
| 32 | SDHB:c.137G>A (p.Arg46Gln) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 33 | SDHB:c.137G>A (p.Arg46Gln) | Neg | Yes | NT | Neg | Neg | Neg | SDHB One Copy Deletion | WT | Loss of SDHB, intact SDHA |
| 35 | SDHC:c.397C>T (p.Arg133Ter) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | SDHB is intact |
| 37 | SDHB EX8_3’UTR Del | Neg | Yes | NT | Neg | Neg | Neg | SDHB One Copy Deletion | WT | SDHB is intact |
| 38 | SDHA:c.91C>T (p.Arg31Ter) | Neg | No (GIST) | NT | Neg | Neg | Neg | SDHA One Copy Deletion | WT | NT |
| 43 | SDHC:c.397C>T (p.Arg133Ter) | Neg | Yes | NT | Neg | Neg | Neg | Neg | WT | SDHB is intact |
| 46 | SDHC:c.397C>T (p.Arg133Ter) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 51 | SDHA EX1_9 Del | Neg | No (GIST) | NT | Neg | Neg | Neg | SDHA One Copy Deletion | WT | Loss of SDHB and SDHA |
| 53 | SDHC EX6 Del | Yes, RET:c.1771G>A | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 54 | SDHA:c.1794+1G>A | Yes, SDHA:c.1246A>G (p.Asn416Asp) | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 59 | SDHC:c.43C>T (p.Arg15Ter) | Neg | Yes | NT | Neg | Neg | Neg | Neg | WT | Loss of SDHB |
| 60 | SDHA:c.563G>A (p.Arg188Gln) | Neg | No | NT | Neg | Neg | Neg | SDHA Copy Number Gain (<6 copies) | WT | NT |
| 61 | SDHA:c.688delG (p.Glu230fs) | Neg | Yes (+GIST) | NT | Neg | Neg | Neg | SDHA:c.1432+1G>C | WT | Loss of SDHB and SDHA |
| 63 | SDHB:c.487T>C (p.Ser163Pro) | Yes, SDHA:c.553C>T (p.Gln185Ter) SDHA:c.622-7C>T | Yes (+GIST) | NT | Neg | Neg | Neg | SDHB One Copy Deletion SDHA:c.1043_1055del (p.Thr348LysfsTer37) | WT | Loss of SDHB |
| 72 | SDHA:c.91C>T (p.Arg31Ter) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 79 | SDHA:c.91C>T (p.Arg31Ter) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 80 | SDHB Ex1 Del | Neg | No | NT | Neg | Neg | Neg | Neg | WT | Loss of SDHB |
| 82 | SDHB:c.137G>A (p.Arg46Gln) | Neg | Yes (+GIST) | NT | Yes, PGL in Brother | Yes | Yes, PGL metastasis | SDHB One Copy Deletion | WT | Loss of SDHB |
| 92 | SDHC:c.397C>T (p.Arg133Ter) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 93 | SDHD:c.106C>T (p.Gln36Ter) | Neg | Yes | SDHD variant paternally inherited | Yes, PGL in Father | Neg | Neg | SDHD One Copy Deletion | WT | Loss of SDHB |
| 107 | SDHB:c.418G>T (p.Val140Phe) | Neg | Yes | NT | Neg | Neg | Yes, PGL metastasis | SDHB One Copy Deletion | WT | Loss of SDHB |
| Subject | Germline Variant in HPPGL Genes | Clinical Genetics Information | Tumor-Derived Information | Tumor Immunohistochemistry Staining | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Germline SDHx Allele | Other Germline Alteration in HPPGL | Personal History of PGL/PCC ± Associated GIST | Parent-of-Origin Inheritance | Family History of PGL/PCC/GIST | Multiple, Multifocal or Extra Adrenal Tumors | PGL/PCC Metastasis | Somatic SDHx Allele | KIT and PDGFRA Mutation Status | SDHA and SDHB | |
| 6 | SDHA:c.853C>T (p.Leu285Phe) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 9 | SDHA:c.1216G>A (p.Val406Met) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 17 | SDHB:c.170A>G (p.His57Arg) | Yes, SDHA:c.-4A, RET:c.405C>T (p.Gly135=) | Yes | NA | Neg | Neg | Neg | Neg | WT | Intact SDHB |
| 21 | SDHA:c.1894G>T (p.Val632Phe) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 24 | SDHA:c.1523C>T (p.Thr508Ile) | Neg | Yes | NA | Neg | Neg | Yes, PGL metastasis | Neg | WT | Intact SDHB |
| 31 | SDHA:c.596C>T (p.Ser199Leu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 34 | SDHA:c.287C>T (p.Thr96Ile) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 39 | SDHA:c.133G>A (p.Ala45Thr) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 41 | SDHA:c.872A>T (p.Glu291Val) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 44 | SDHA:c.955A>C (p.Ile319Leu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 45 | SDHA:c.1006G>T (p.Asp336Tyr) | Neg | Yes (+GIST) | NA | Neg | Neg | Neg | SDHA:c.511C>T (p.Arg171Cys) | WT | Intact SDHA, intact SDHB |
| 47 | SDHA:c.1908+6T>C | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 48 | SDHA:c.464A>G (p.Asn155Ser) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 49 | SDHA:c.1472A>C (p.Glu491Ala) | Neg | No | NA | Neg | Neg | Neg | Neg | KIT:c.1091A>G; PDGFRA:WT | NT |
| 50 | SDHA:c.530G>C (p.Ser177Thr) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 55 | SDHA:c.737G>A (p.Arg246His) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 56 | SDHA:c.830C>T (p.Thr277Met) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 57 | SDHA:c.464A>G (p.Asn155Ser) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 62 | SDHA:c.1064+5G>A | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 65 | SDHA:c.1064+5G>A | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 66 | SDHA:c.1325C>T (p.Ala442Val) | Neg | Yes | NA | Neg | Neg | Neg | Neg | WT | Loss of SDHB |
| 67 | SDHA:c.955A>C (p.Ile319Leu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 68 | SDHA:c.1115C>G (p.Pro372Arg) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 70 | SDHA:c.1340A>G (p.His447Arg) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 74 | SDHA:c.1648A>C (p.Lys550Gln) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 75 | SDHA:c.1763C>T (p.Ser588Leu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 81 | SDHA:c.872A>T (p.Glu291Val) | Yes, SDHC:c.54T>G (p.Phe18Leu) | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 84 | SDHA:c.1835G>A (p.Gly612Glu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 85 | SDHA:c.499A>C (p.Lys167Gln) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 86 | SDHA:c.613T>C (p.Tyr205His) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 88 | SDHA:c.737G>A (p.Arg246His) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 89 | SDHA:c.287C>T (p.Thr96Ile) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 90 | SDHA:c.391G>A (p.Asp131Asn) | Neg | No | NA | Neg | Neg | Neg | SDHA Copy Number Gain(6n) | WT | NT |
| 91 | SDHA:c.1429C>T (p.Pro477Ser) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 95 | SDHA:c.1115C>G (p.Pro372Arg) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 96 | SDHA:c.830C>T (p.Thr277Met) | Yes, SDHB:c.523G>A (p.Glu175Lys) | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 99 | SDHA:c.935G>T (p.Arg312Leu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 100 | SDHA:c.830C>T (p.Thr277Met) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 101 | SDHA:c.1908+6T>C | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 105 | SDHA:c.724G>A (p.Gly242Arg) | Neg | No | NA | Neg | Neg | Neg | Neg | KIT:WT; PDGFRA:c.369C>G | NT |
| Subject | Germline Variant in HPPGL Genes | Clinical Genetics Information | Tumor-Derived Information | Tumor Immunohistochemistry Staining | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Germline SDHx Allele | Other Germline Alteration in HPPGL | Personal History of PGL/PCC ± Associated GIST | Parent-of-Origin Inheritance | Family History of PGL/PCC/GIST | Multiple, Multifocal or Extra Adrenal Tumors | PGL/PCC Metastasis | Somatic SDHx Allele | KIT and PDGFRA Mutation Status | SDHA and SDHB | |
| 3 | SDHB:159_184delIns25 (p.Gly53fs) | Neg | Yes (+GIST) | NA | Neg | Neg | Neg | Neg | WT | NT |
| 4 | SDHB:c.72+3G>A | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 10 | SDHB:c.158G>A (p.Gly53Glu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 19 | SDHB:c.487T>C (p.Ser163Pro) | Neg | No | NA | Neg | Neg | Neg | SDHB One Copy Deletion | WT | Intact SDHB (IHC on RCC) |
| 25 | SDHB:c.478_480del (p.Lys160del) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 26 | SDHB:c.553G>A (p.Glu185Lys) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 27 | SDHB Duplication | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 36 | SDHB:c.687G>C (p.Glu229Asp) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 58 | SDHB:c.79C>G (p.Arg27Gly) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 69 | SDHB:c.329C>T (p.Thr110Ile) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 71 | SDHB:c.317A>G (p.Asn106Ser) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 78 | SDHB:c.67C>G (p.Leu23Val) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 83 | SDHB:c.478_480del (p.Lys160del) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 97 | SDHB:c.529C>A (p.Arg177Ser) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 109 | SDHB:c.178A>G (p.Thr60Ala) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| Subject | Germline Variant in HPPGL Genes | Clinical Genetics Information | Tumor-Derived Information | Tumor Immunohistochemistry Staining | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Germline SDHx Allele | Other Germline Alteration in HPPGL | Personal History of PGL/PCC ± Associated GIST | Parent-of-Origin Inheritance | Family History of PGL/PCC/GIST | Multiple, Multifocal or Extra Adrenal Tumors | PGL/PCC Metastasis | Somatic SDHx Allele | KIT and PDGFRA Mutation Status | SDHA and SDHB | |
| 16 | SDHC:c.20+6T>G | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 52 | SDHC:c.476C>T (p.Thr159Ile) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 64 | SDHC:c.292T>G (p.Ser98Ala) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 77 | SDHC:c.292T>G (p.Ser98Ala) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 87 | SDHC:c.103A>G (p.Lys35Glu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 94 | SDHC:c.15G>T (p.Leu5Phe) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 102 | SDHC:c.40C>T (p.Leu14Phe) | Neg | No | NA | Neg | Neg | Neg | SDHC Gain | WT | NT |
| 103 | SDHC:c.20+20G>T | Neg | No | NA | Neg | Neg | Neg | SDHC Gain (estimated 6 copies) | WT | NT |
| 104 | SDHC:c.32G>T (p.Arg11Leu) | Neg | No | NA | Neg | Neg | Neg | Neg | WT | NT |
| 106 | SDHC:c.-30-?_*2318+?dup | Neg | No | NA | Neg | Neg | Neg | SDHC Gain (estimated 8 copies) | WT | NT |
| Subject | Germline Variant in HPPGL Genes | Clinical Genetics Information | Tumor-Derived Information | Tumor Immunohistochemistry Staining | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Germline SDHx Allele | Other Germline Alteration in HPPGL | Personal History of PGL/PCC ± Associated GIST | Parent of Origin Inheritance | Family History of PGL/PCC/GIST | Multiple, Multifocal or Extra Adrenal Tumors | PGL/PCC Metastasis | Somatic SDHx Allele | KIT and PDGFRA Mutation Status | SDHA and SDHB IHC | |
| 29 | SDHD:c.453A>C (p.Lys151Asn) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 40 | SDHD:c.400T>G (p.L134V) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 42 | SDHD:c.53C>T (p.Ala18Val) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 73 | SDHD:c.80G>A (p.Arg27Lys) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 76 | SDHD:c.101T>G (p.Phe34Cys) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 98 | SDHD:c.53C>T (p.A18V) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
| 108 | SDHD:c.428A>G (p.Asn143Ser) | Neg | No | NT | Neg | Neg | Neg | Neg | WT | NT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, H.Q.; Koeller, D.R.; Walker, M.; Unal, B.; Levine, A.S.; Chittenden, A.; Isidro, R.A.; Hayes, C.P.; Manam, M.D.; Buehler, R.M.; et al. Advancing Precision Oncology in Hereditary Paraganglioma-Pheochromocytoma Syndromes: Integrated Interpretation and Data Sharing of the Germline and Tumor Genomes. Cancers 2024, 16, 947. https://doi.org/10.3390/cancers16050947
Rana HQ, Koeller DR, Walker M, Unal B, Levine AS, Chittenden A, Isidro RA, Hayes CP, Manam MD, Buehler RM, et al. Advancing Precision Oncology in Hereditary Paraganglioma-Pheochromocytoma Syndromes: Integrated Interpretation and Data Sharing of the Germline and Tumor Genomes. Cancers. 2024; 16(5):947. https://doi.org/10.3390/cancers16050947
Chicago/Turabian StyleRana, Huma Q., Diane R. Koeller, McKenzie Walker, Busra Unal, Alison Schwartz Levine, Anu Chittenden, Raymond A. Isidro, Connor P. Hayes, Monica D. Manam, Ryan M. Buehler, and et al. 2024. "Advancing Precision Oncology in Hereditary Paraganglioma-Pheochromocytoma Syndromes: Integrated Interpretation and Data Sharing of the Germline and Tumor Genomes" Cancers 16, no. 5: 947. https://doi.org/10.3390/cancers16050947
APA StyleRana, H. Q., Koeller, D. R., Walker, M., Unal, B., Levine, A. S., Chittenden, A., Isidro, R. A., Hayes, C. P., Manam, M. D., Buehler, R. M., Manning, D. K., Barletta, J. A., Hornick, J. L., Garber, J. E., Ghazani, A. A., & INT2GRATE Oncology Consortium. (2024). Advancing Precision Oncology in Hereditary Paraganglioma-Pheochromocytoma Syndromes: Integrated Interpretation and Data Sharing of the Germline and Tumor Genomes. Cancers, 16(5), 947. https://doi.org/10.3390/cancers16050947