
Interfering Nuclear Protein Laminb1 Induces DNA Damage and Reduces Vemurafenib Resistance in Melanoma Cells In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug Treatments
2.3. Lentivirus Transduction
2.4. Real-Time PCR
2.5. Western Blotting
2.6. Immunohistochemistry
2.7. Immunofluorescence
2.8. Bulk RNA Sequencing and Data Analysis
2.9. Statistical Analyses
3. Results
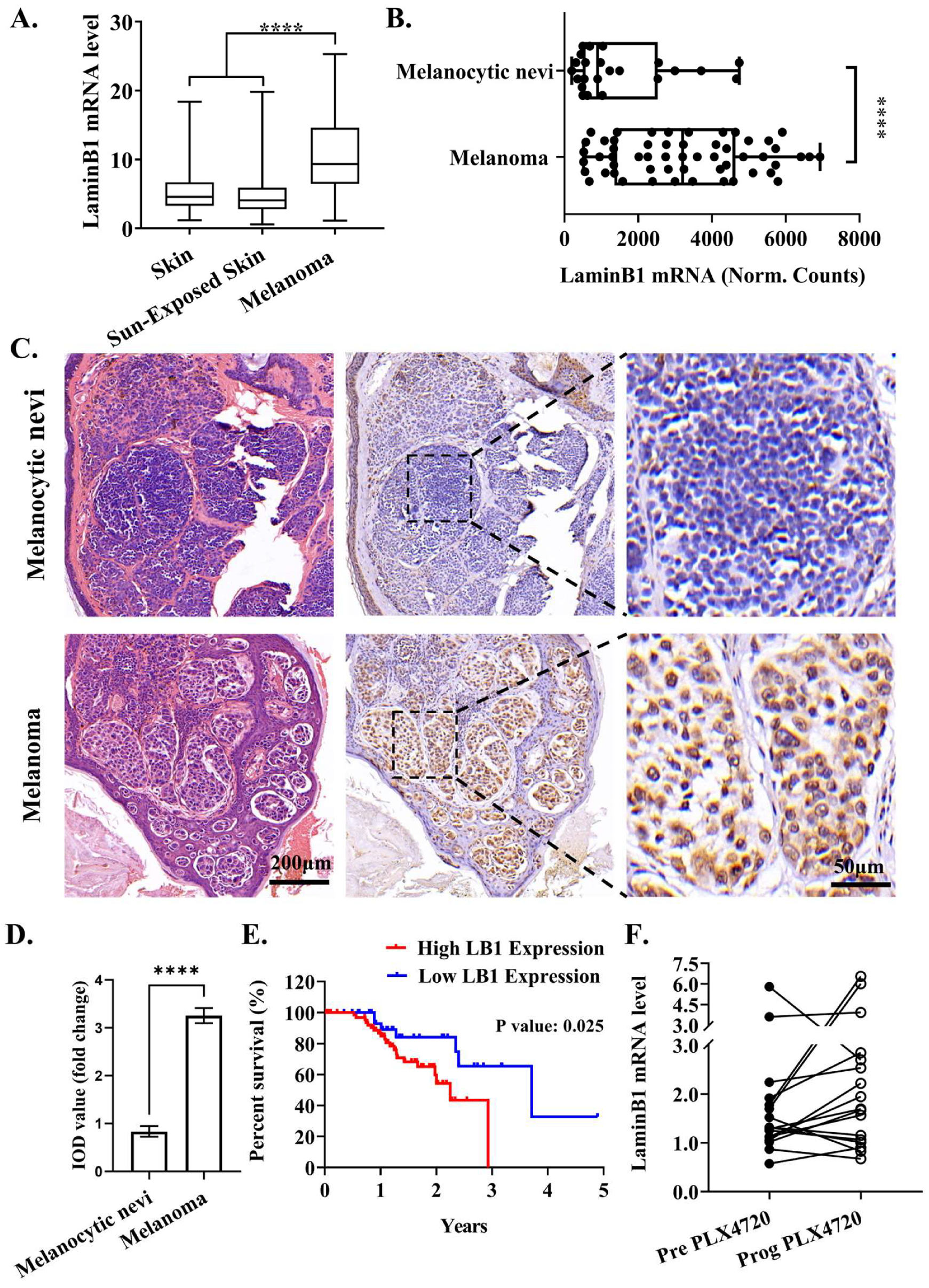
3.1. Elevated LaminB1 Expression Has Tight Clinical Relevance with Melanoma Progression
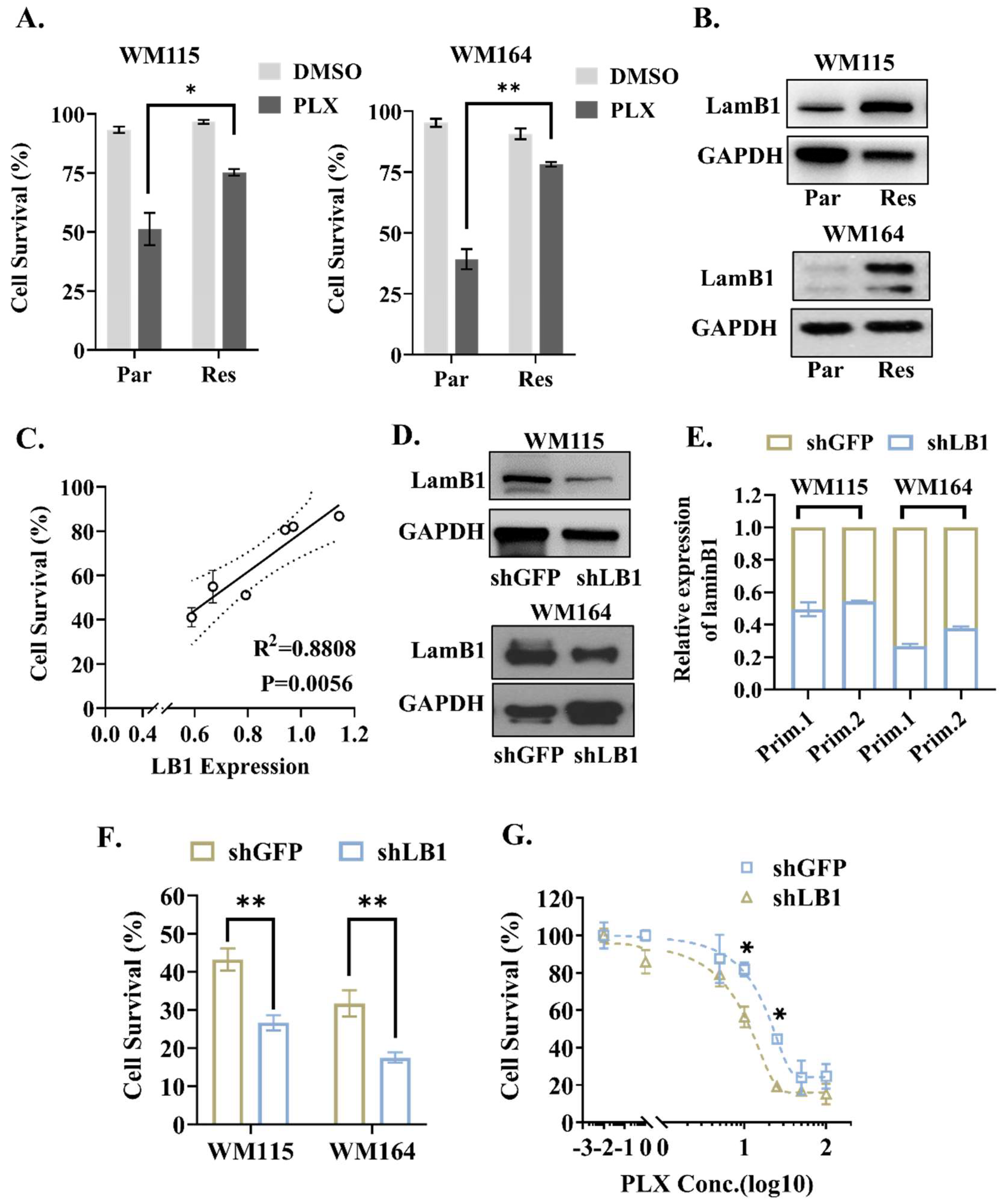
3.2. LaminB1 Positively Correlates with the Response of Melanoma Cells to PLX4720
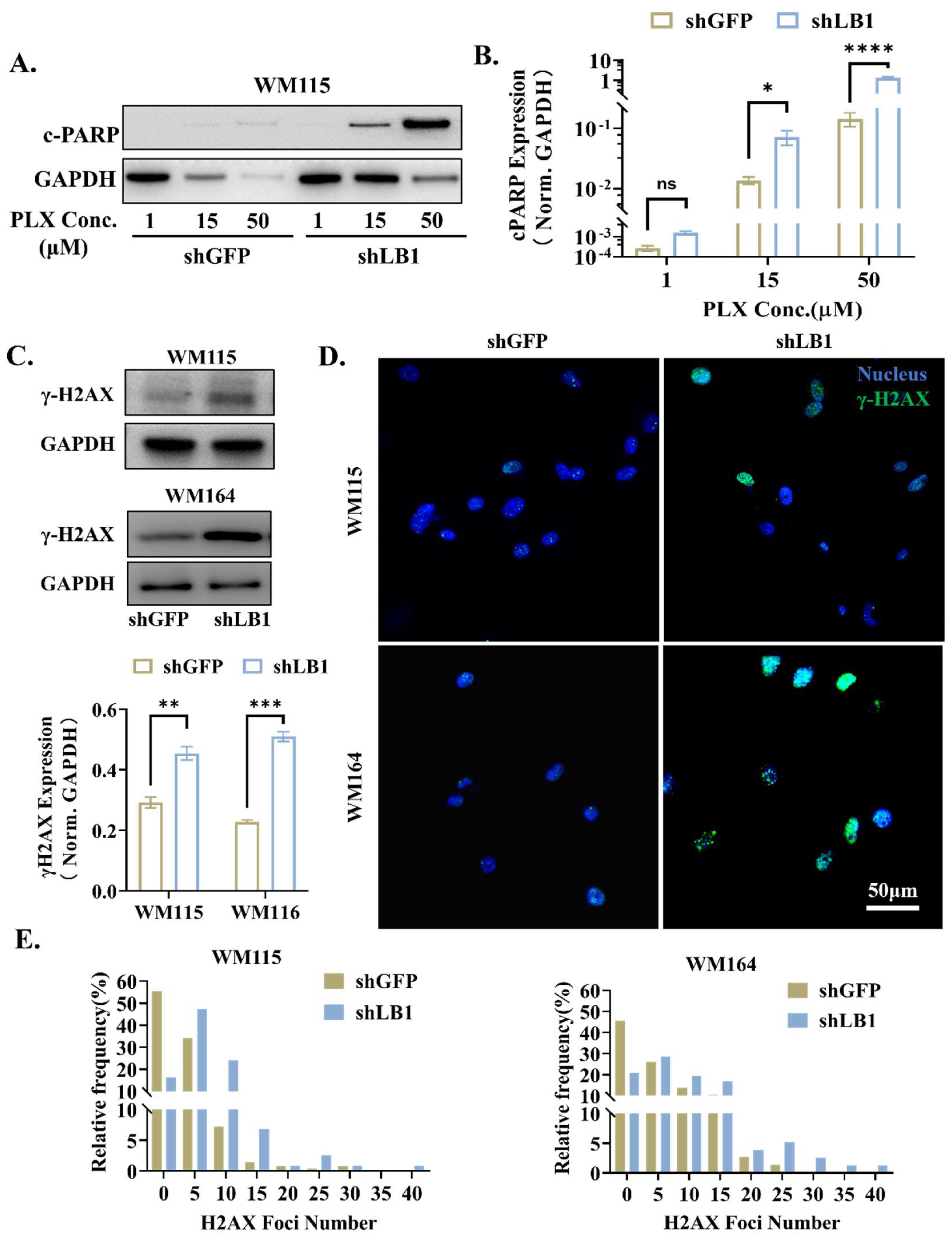
3.3. Loss of LaminB1 Promotes PLX4720-Induced Apoptosis in Melanoma Cells via Accumulation of DNA Damage
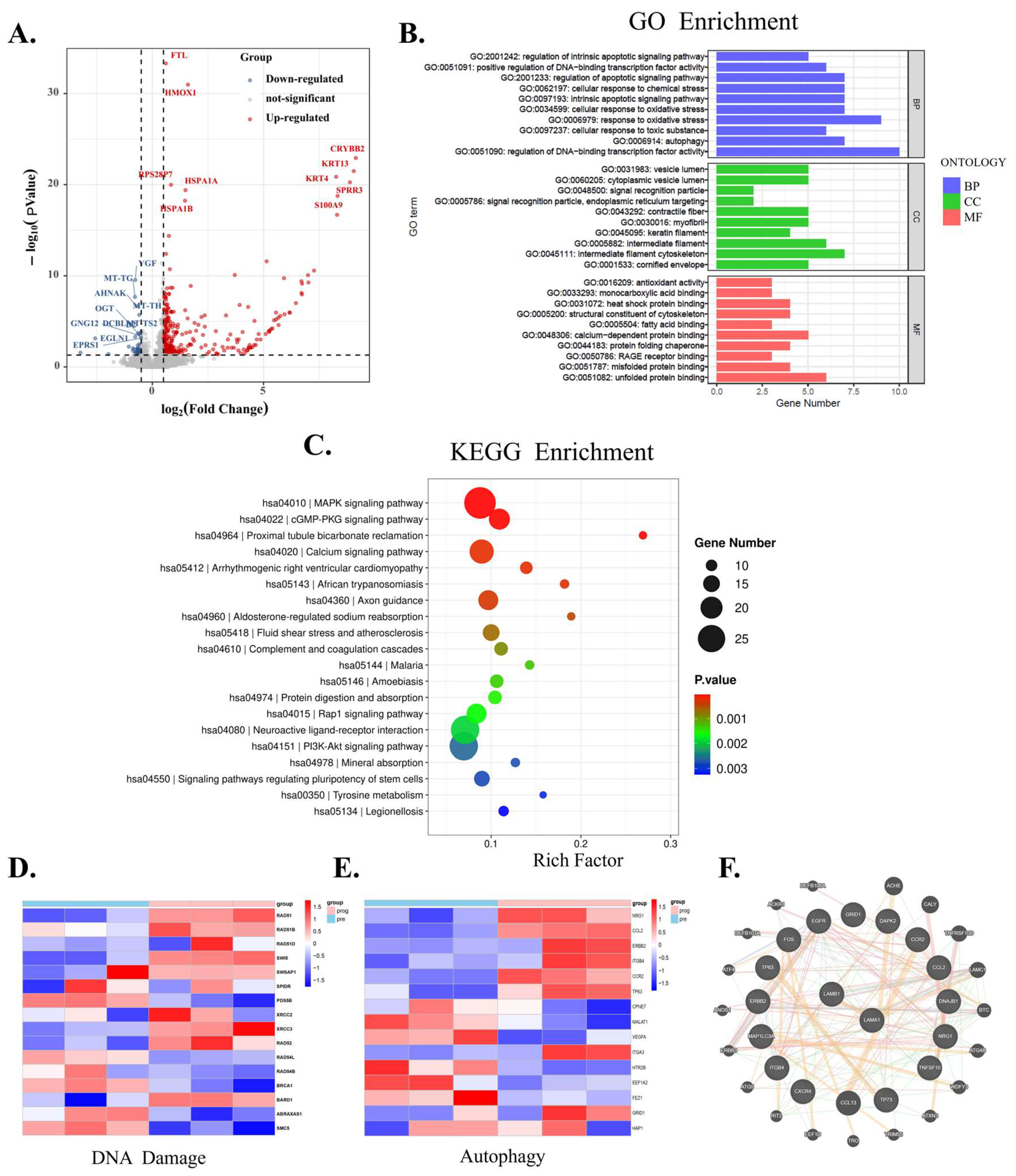
3.4. Regulatory Role of LaminB1 in Autophagy Is Unveiled by Bioinformatics Analysis in Melanoma Cells
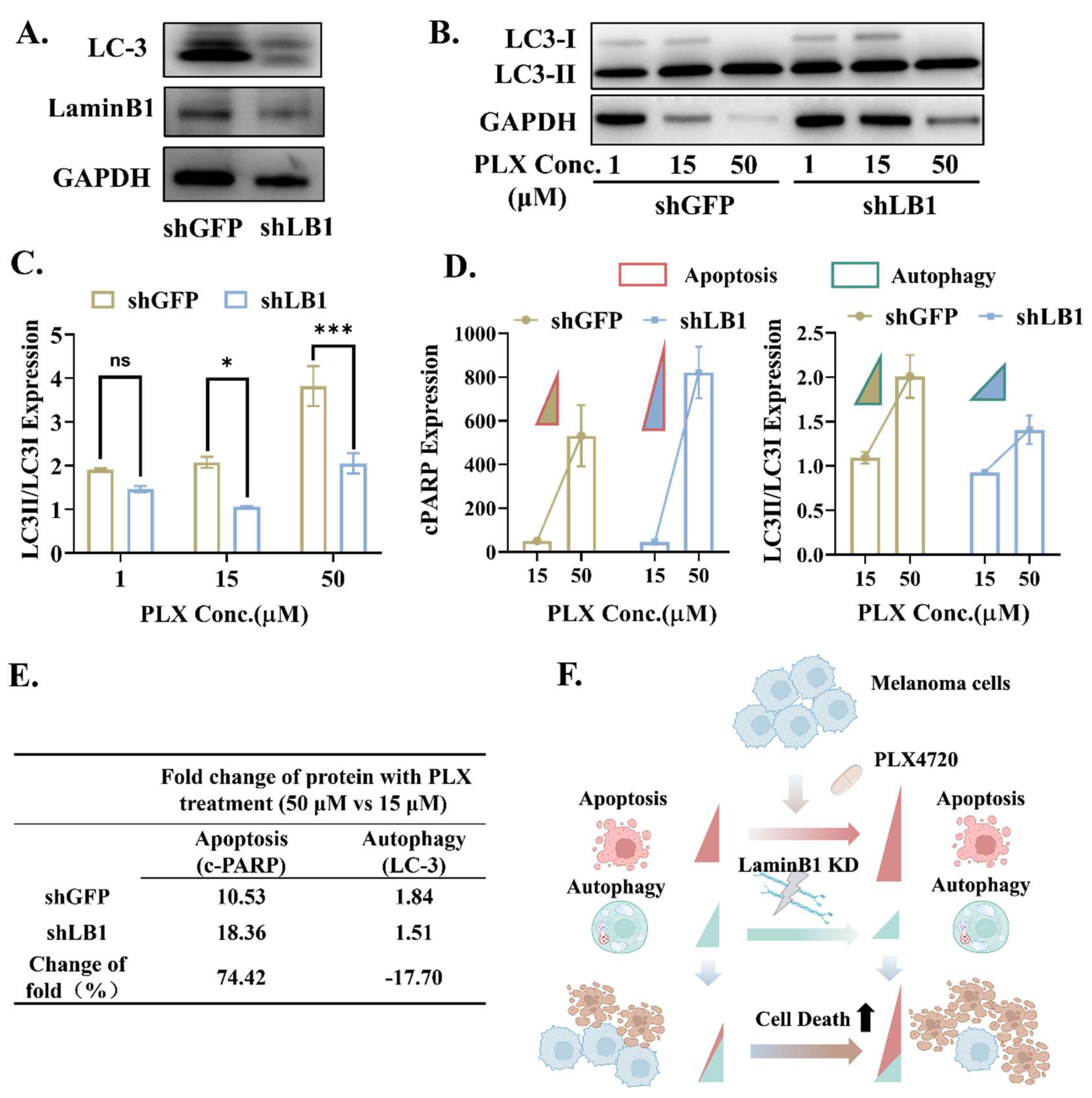
3.5. LaminB1 Is Essential to the Crosstalk Between Apoptosis and Autophagy in Responding to PLX4720
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poulikakos, P.I.; Sullivan, R.J.; Yaeger, R. Molecular Pathways and Mechanisms of BRAF in Cancer Therapy. Clin. Cancer Res. 2022, 28, 4618–4628. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.; Lee, H.; Chen, Z.; Lee, M.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance toB-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Samarkina, A.; Youssef, M.K.; Ostano, P.; Ghosh, S.; Ma, M.; Tassone, B.; Proust, T.; Chiorino, G.; Levesque, M.P.; Goruppi, S.; et al. Androgen receptor is a determinant of melanoma targeted drug resistance. Nat. Commun. 2023, 14, 6498. [Google Scholar] [CrossRef] [PubMed]
- Marusak, C.; Thakur, V.; Li, Y.; Freitas, J.T.; Zmina, P.M.; Thakur, V.S.; Chang, M.; Gao, M.; Tan, J.F.; Xiao, M.; et al. Targeting Extracellular Matrix Remodeling Restores BRAF Inhibitor Sensitivity in BRAFi-resistant Melanoma. Clin. Cancer Res. 2020, 26, 6039–6050. [Google Scholar] [CrossRef]
- Das, S.; Shukla, N.; Singh, S.S.; Kushwaha, S.; Shrivastava, R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis 2021, 26, 512–533. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Morana, O.; Wood, W.; Gregory, C.D. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 1328. [Google Scholar] [CrossRef]
- Domagala, A.; Stachura, J.; Gabrysiak, M.; Muchowicz, A.; Zagozdzon, R.; Golab, J.; Firczuk, M. Inhibition of autophagy sensitizes cancer cells to Photofrin-based photodynamic therapy. BMC Cancer 2018, 18, 210. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.; Lotze, M.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef]
- Germain, M.; Mathai, J.; Shore, G. BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J. Biol. Chem. 2002, 277, 18053–18060. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Cheng, X.; Kuang, J.; Chen, L.; Yuen, S.; Shi, M.; Liang, J.; Shen, B.; Jin, Z.; Yan, J.; et al. Apatinib-induced protective autophagy and apoptosis through the AKT-mTOR pathway in anaplastic thyroid cancer. Cell Death Dis. 2018, 9, 1030. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xia, G.; Li, N.; Su, R.; Chen, X.; Zhong, L. Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. J. Cancer 2018, 9, 3407–3416. [Google Scholar] [CrossRef]
- Chen, N.Y.; Yang, Y.; Weston, T.A.; Belling, J.N.; Heizer, P.; Tu, Y.; Kim, P.; Edillo, L.; Jonas, S.J.; Weiss, P.S.; et al. An absence of lamin B1 in migrating neurons causes nuclear membrane ruptures and cell death. Proc. Natl. Acad. Sci. USA 2019, 116, 25870–25879. [Google Scholar] [CrossRef]
- Karoutas, A.; Akhtar, A. Functional mechanisms and abnormalities of the nuclear lamina. Nat. Cell Biol. 2021, 23, 116–126. [Google Scholar] [CrossRef]
- Butin-Israeli, V.; Adam, S.A.; Jain, N.; Otte, G.L.; Neems, D.; Wiesmüller, L.; Berger, S.L.; Goldman, R.D. Role of lamin b1 in chromatin instability. Mol. Cell Biol. 2015, 35, 884–898. [Google Scholar] [CrossRef]
- Liu, N.A.; Sun, J.; Kono, K.; Horikoshi, Y.; Ikura, T.; Tong, X.; Haraguchi, T.; Tashiro, S. Regulation of homologous recombinational repair by lamin B1 in radiation-induced DNA damage. FASEB J. 2015, 29, 2514–2525. [Google Scholar] [CrossRef]
- Thakur, V.; Tiburcio de Freitas, J.; Li, Y.; Zhang, K.; Savadelis, A.; Bedogni, B. MT1-MMP-dependent ECM processing regulates laminB1 stability and mediates replication fork restart. PLoS ONE 2021, 16, e0253062. [Google Scholar] [CrossRef]
- Molenberghs, F.; Verschuuren, M.; Vandeweyer, L.; Peeters, S.; Bogers, J.J.; Novo, C.P.; Vanden Berghe, W.; De Reu, H.; Cools, N.; Schelhaas, M.; et al. Lamin B1 curtails early human papillomavirus infection by safeguarding nuclear compartmentalization and autophagic capacity. Cell Mol. Life Sci. 2024, 81, 141. [Google Scholar] [CrossRef]
- Ding, J.; Chen, Y.; Zhao, Y.J.; Chen, F.; Dong, L.; Zhang, H.L.; Hu, W.R.; Li, S.F.; Zhou, R.P.; Hu, W. Acid-sensitive ion channel 1a mediates osteoarthritis chondrocyte senescence by promoting Lamin B1 degradation. Biochem. Pharmacol. 2022, 202, 115107. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef] [PubMed]
- Niessner, H.; Sinnberg, T.; Kosnopfel, C.; Smalley, K.S.M.; Beck, D.; Praetorius, C.; Mai, M.; Beissert, S.; Kulms, D.; Schaller, M.; et al. BRAF Inhibitors Amplify the Proapoptotic Activity of MEK Inhibitors by Inducing ER Stress in NRAS-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 6203–6214. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef]
- Fimia, G.M.; Corazzari, M.; Antonioli, M.; Piacentini, M. Ambra1 at the crossroad between autophagy and cell death. Oncogene 2013, 32, 3311–3318. [Google Scholar] [CrossRef]
- Yang, B.; Liu, Q.; Bi, Y. Autophagy and apoptosis are regulated by stress on Bcl2 by AMBRA1 in the endoplasmic reticulum and mitochondria. Theor. Biol. Med. Model. 2019, 16, 18. [Google Scholar] [CrossRef]
- Dai, Y.; Grant, S. BCL2L11/Bim as a dual-agent regulating autophagy and apoptosis in drug resistance. Autophagy 2015, 11, 416–418. [Google Scholar] [CrossRef]
- Al-Odat, O.S.; Guirguis, D.A.; Schmalbach, N.K.; Yao, G.; Budak-Alpdogan, T.; Jonnalagadda, S.C.; Pandey, M.K. Autophagy and Apoptosis: Current Challenges of Treatment and Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2022, 24, 644. [Google Scholar] [CrossRef]
- Lin, J.F.; Lin, Y.C.; Tsai, T.F.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des. Devel Ther. 2017, 11, 1517–1533. [Google Scholar] [CrossRef]
- Singh, A.K.; Sonawane, P.; Kumar, A.; Singh, H.; Naumovich, V.; Pathak, P.; Grishina, M.; Khalilullah, H.; Jaremko, M.; Emwas, A.H.; et al. Challenges and Opportunities in the Crusade of BRAF Inhibitors: From 2002 to 2022. ACS Omega 2023, 8, 27819–27844. [Google Scholar] [CrossRef]
- Menzer, C.; Menzies, A.M.; Carlino, M.S.; Reijers, I.; Groen, E.J.; Eigentler, T.; de Groot, J.W.B.; van der Veldt, A.A.M.; Johnson, D.B.; Meiss, F.; et al. Targeted Therapy in Advanced Melanoma With Rare BRAF Mutations. J. Clin. Oncol. 2019, 37, 3142–3151. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Ashrafizadeh, M.; Mongiardini, V.; Grimaldi, B.; Crea, F.; Rietdorf, K.; Győrffy, B.; Klionsky, D.J.; Ren, J.; Zhang, W.; et al. Autophagy and cancer drug resistance in dialogue: Pre-clinical and clinical evidence. Cancer Lett. 2023, 570, 216307. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Zahedi, S.; Fitzwalter, B.E.; Morin, A.; Grob, S.; Desmarais, M.; Nellan, A.; Green, A.L.; Vibhakar, R.; Hankinson, T.C.; Foreman, N.K.; et al. Effect of early-stage autophagy inhibition in BRAFV600E autophagy-dependent brain tumor cells. Cell Death Dis. 2019, 10, 679. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Feng, Y.; Chen, D. Interfering Nuclear Protein Laminb1 Induces DNA Damage and Reduces Vemurafenib Resistance in Melanoma Cells In Vitro. Cancers 2024, 16, 4060. https://doi.org/10.3390/cancers16234060
Li Y, Feng Y, Chen D. Interfering Nuclear Protein Laminb1 Induces DNA Damage and Reduces Vemurafenib Resistance in Melanoma Cells In Vitro. Cancers. 2024; 16(23):4060. https://doi.org/10.3390/cancers16234060
Chicago/Turabian StyleLi, Yuan, Yuqing Feng, and Dan Chen. 2024. "Interfering Nuclear Protein Laminb1 Induces DNA Damage and Reduces Vemurafenib Resistance in Melanoma Cells In Vitro" Cancers 16, no. 23: 4060. https://doi.org/10.3390/cancers16234060
APA StyleLi, Y., Feng, Y., & Chen, D. (2024). Interfering Nuclear Protein Laminb1 Induces DNA Damage and Reduces Vemurafenib Resistance in Melanoma Cells In Vitro. Cancers, 16(23), 4060. https://doi.org/10.3390/cancers16234060