
OVsignGenes: A Gene Expression-Based Neural Network Model Estimated Molecular Subtype of High-Grade Serous Ovarian Carcinoma
 ,
,  , and
, and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cohorts
2.2. Methods
3. Results
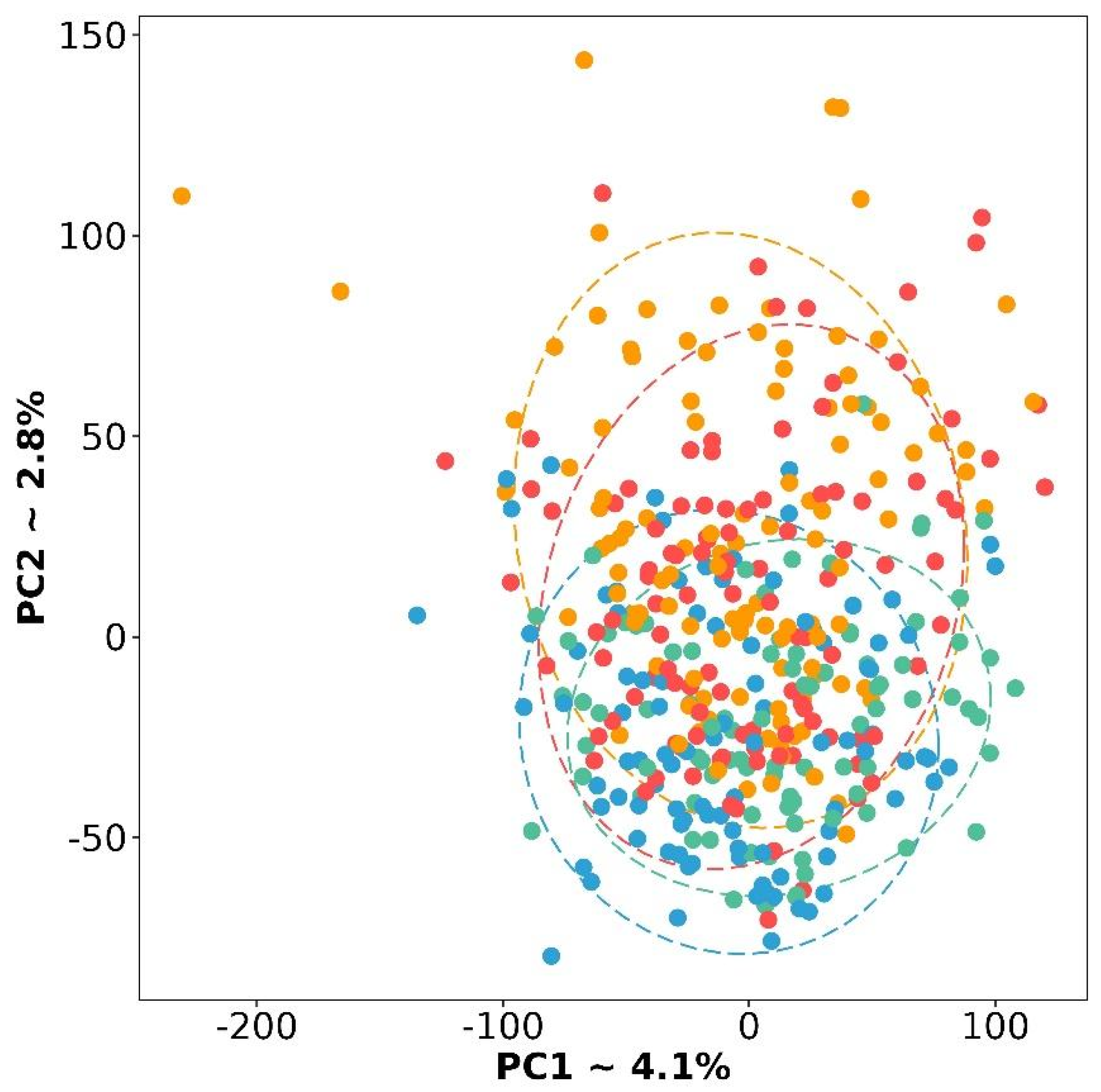
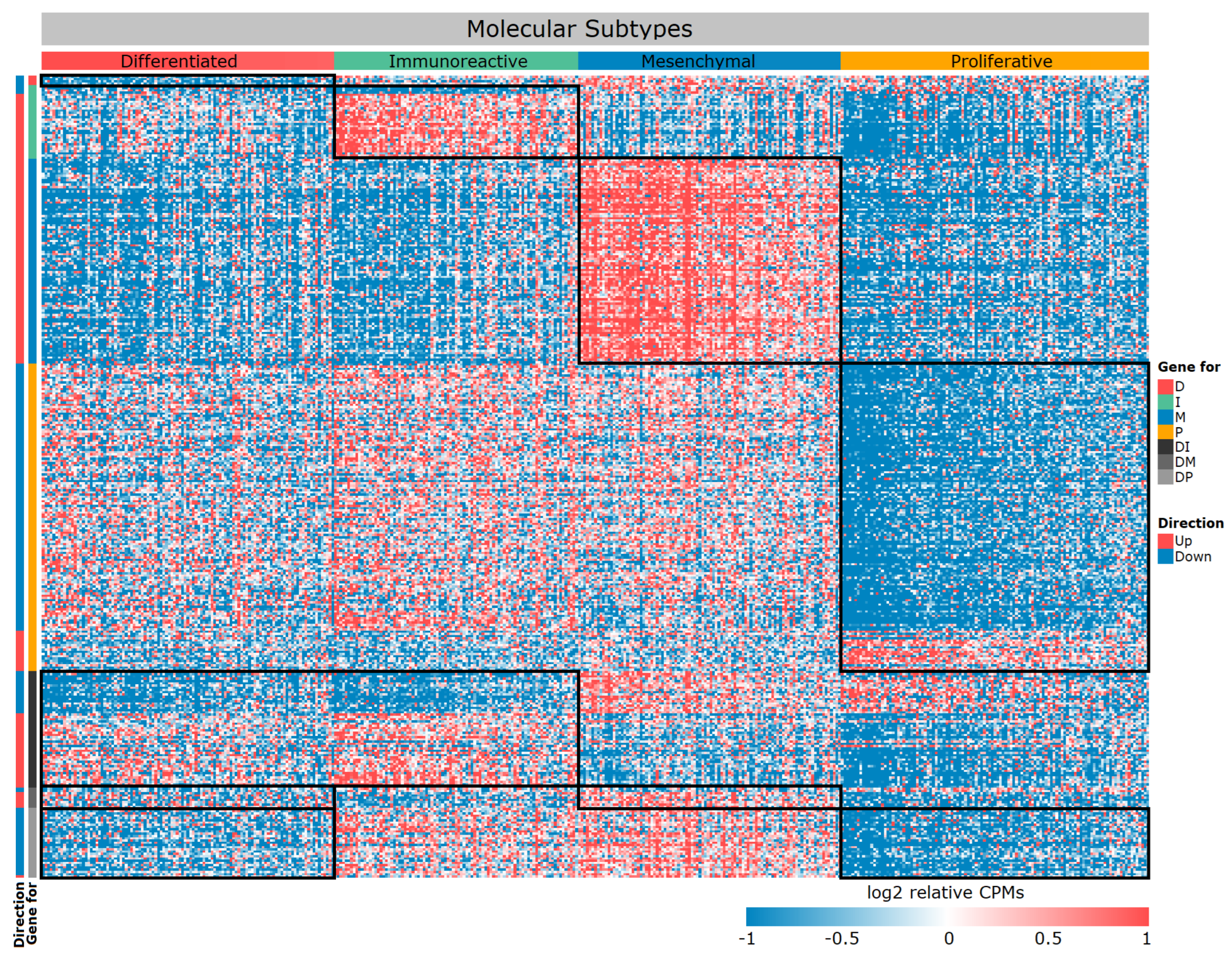
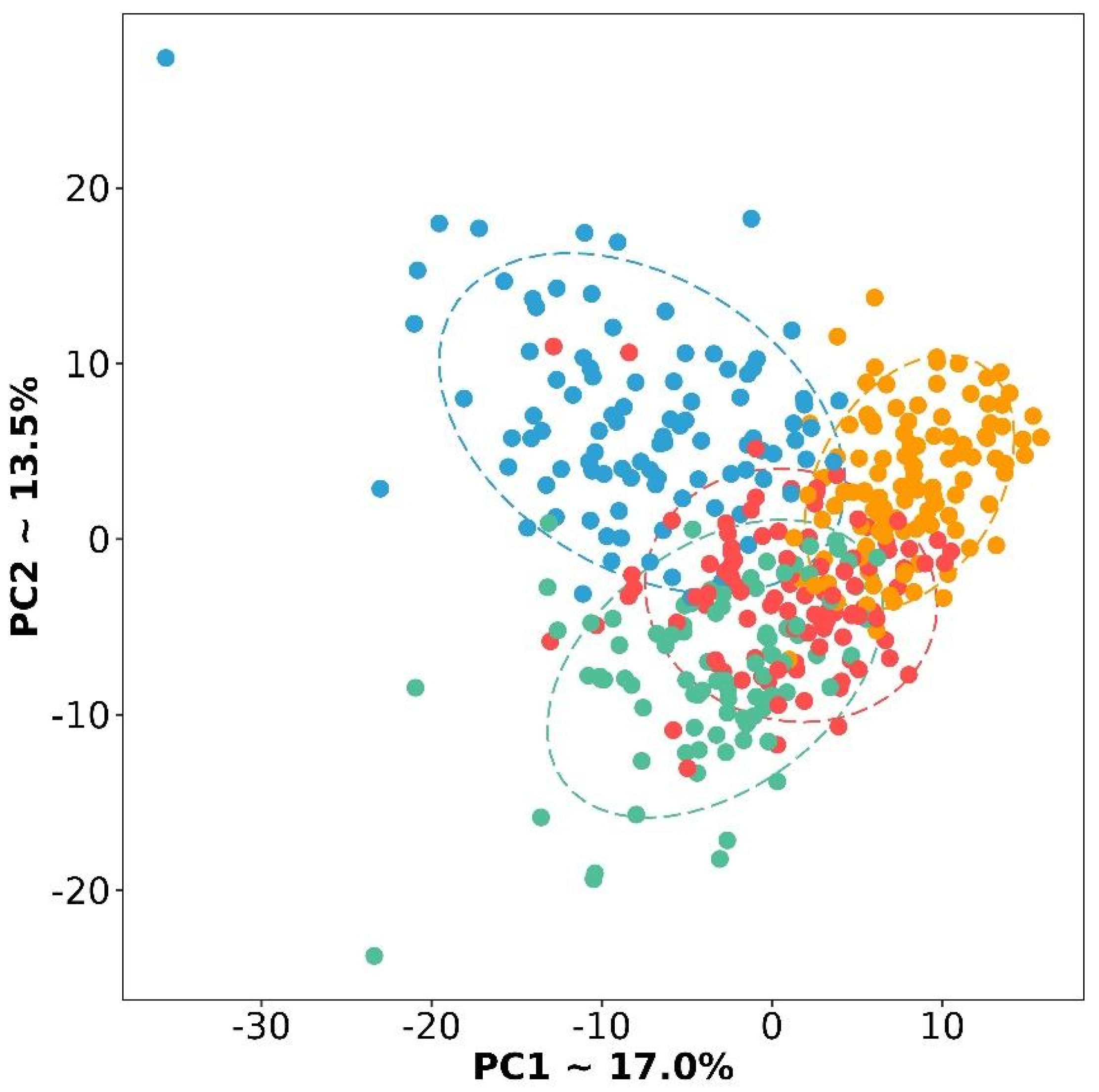
3.1. Gene Expression Signature of the Four Molecular Subtypes
3.2. Functional Characteristics of Differentially Expressed Genes
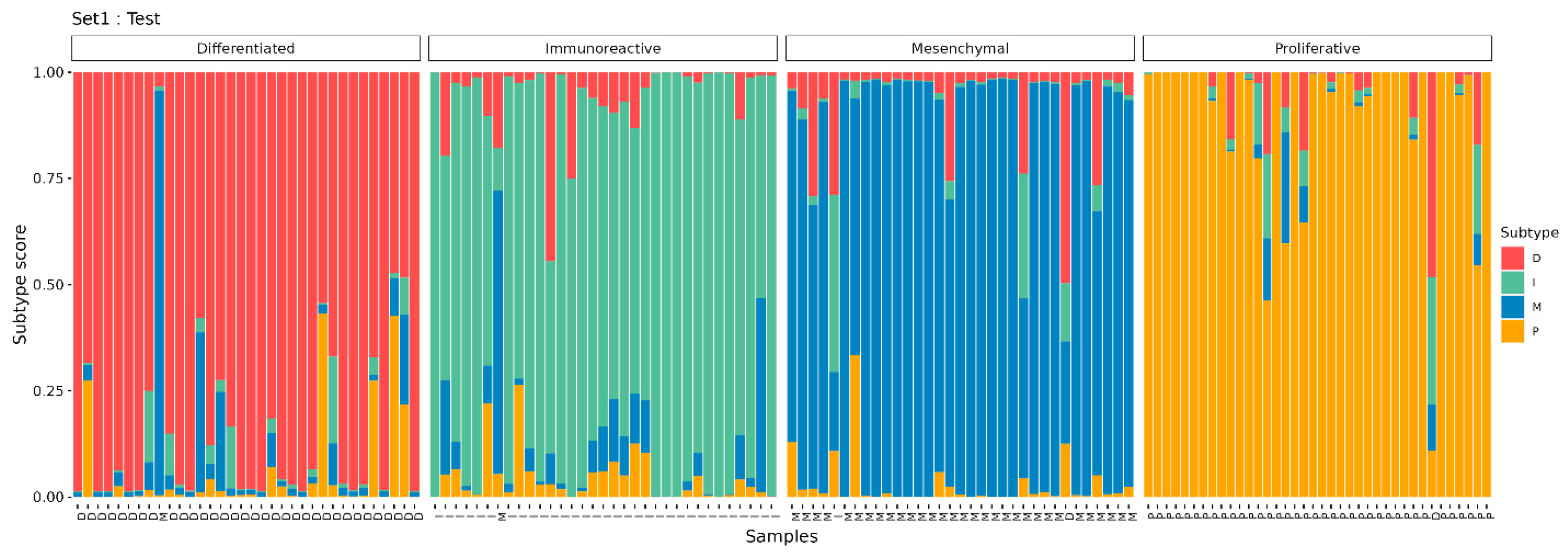
3.3. HGSC Molecular Subtype Neural Network Model
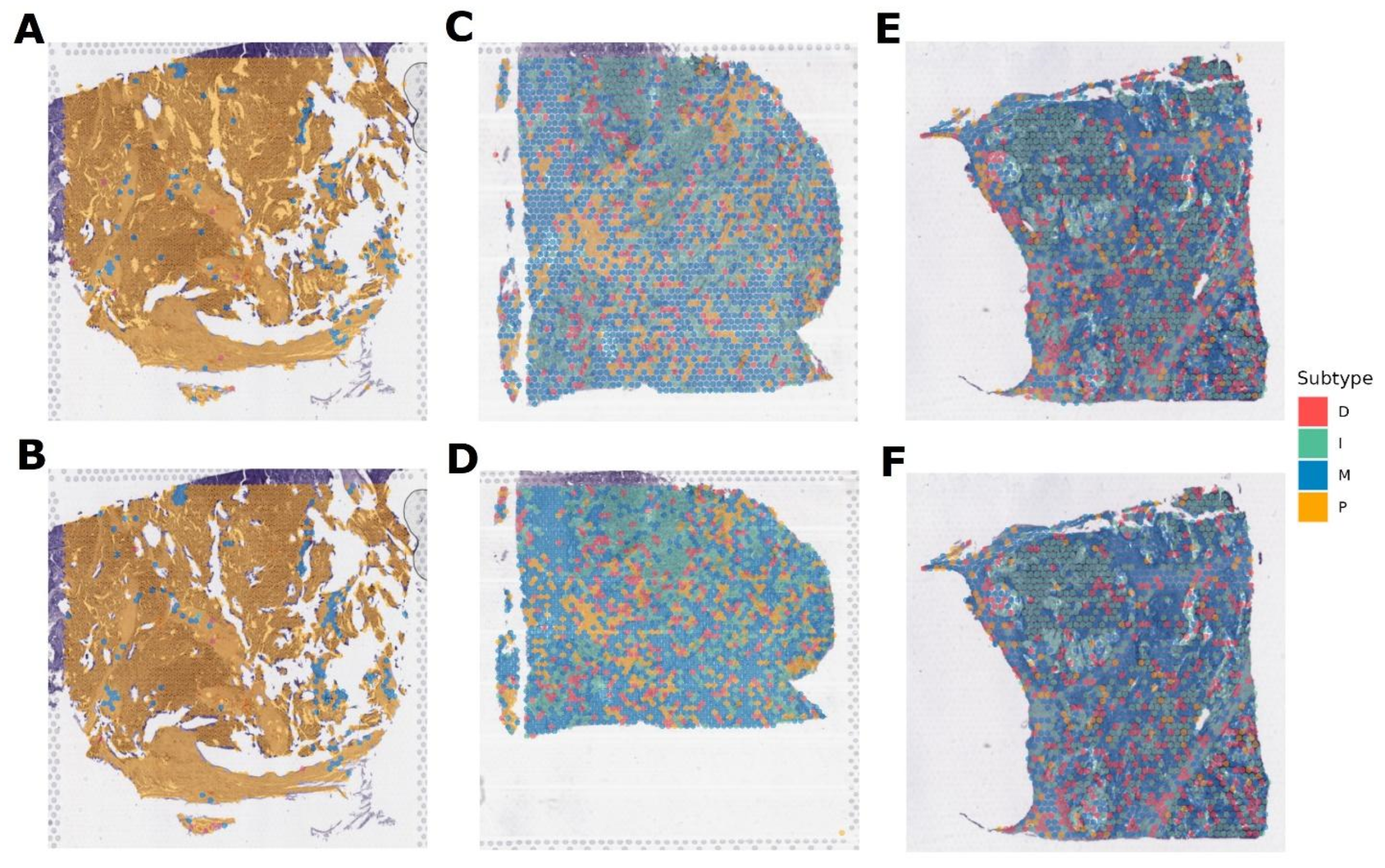
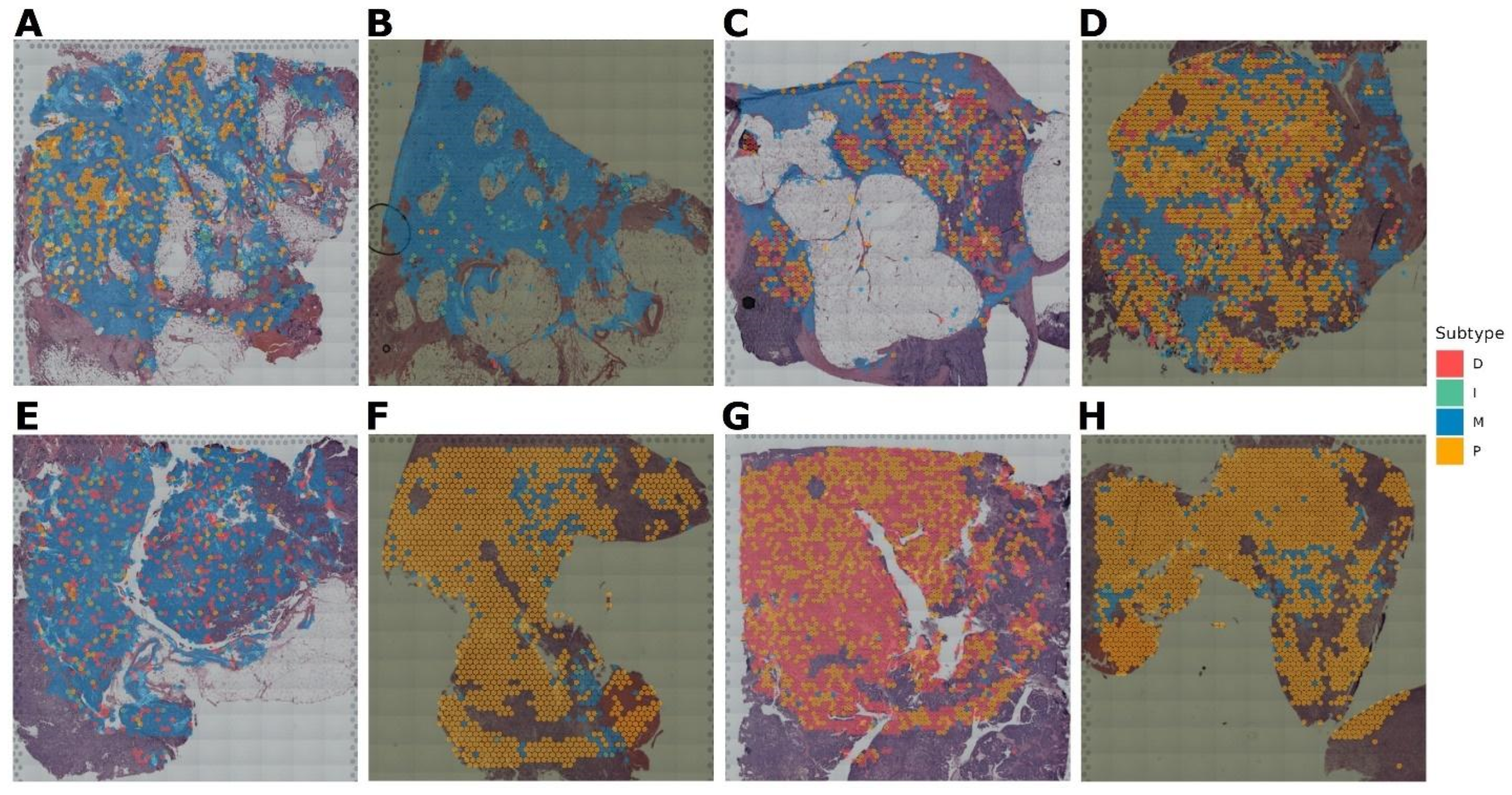
3.4. Verification of the OVsignGenes Model on External Datasets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seidman, J.D.; Horkayne-Szakaly, I.; Haiba, M.; Boice, C.R.; Kurman, R.J.; Ronnett, B.M. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int. J. Gynecol. Pathol. 2004, 23, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Peres, L.C.; Cushing-Haugen, K.L.; Köbel, M.; Harris, H.R.; Berchuck, A.; Rossing, M.A.; Schildkraut, J.M.; Doherty, J.A. Invasive Epithelial Ovarian Cancer Survival by Histotype and Disease Stage. JNCI J. Natl. Cancer Inst. 2019, 111, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.-H.; Bast, R.C., Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Köbel, M.; Kalloger, S.E.; Carrick, J.; Huntsman, D.; Asad, H.; Oliva, E.; Ewanowich, C.A.; Soslow, R.A.; Gilks, C.B. A limited panel of immunomarkers can reliably distinguish between clear cell and high-grade serous carcinoma of the ovary. Am. J. Surg. Pathol. 2009, 33, 14–21. [Google Scholar] [CrossRef]
- Rafii, A.; Halabi, N.M.; Malek, J.A. High-prevalence and broad spectrum of Cell Adhesion and Extracellular Matrix gene pathway mutations in epithelial ovarian cancer. J. Clin. Bioinform. 2012, 2, 15. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef]
- Helland, Å.; Anglesio, M.S.; George, J.; Cowin, P.A.; Johnstone, C.N.; House, C.M.; Sheppard, K.E.; Etemadmoghadam, D.; Melnyk, N.; Rustgi, A.K.; et al. Deregulation of MYCN, LIN28B and LET7 in a Molecular Subtype of Aggressive High-Grade Serous Ovarian Cancers. PLoS ONE 2011, 6, e18064. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.-W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. JNCI J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef]
- Talhouk, A.; George, J.; Wang, C.; Budden, T.; Tan, T.Z.; Chiu, D.S.; Kommoss, S.; Leong, H.S.; Chen, S.; Intermaggio, M.P.; et al. Development and Validation of the Gene Expression Predictor of High-grade Serous Ovarian Carcinoma Molecular SubTYPE (PrOTYPE). Clin. Cancer Res. 2020, 26, 5411–5423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jing, Y.; Zhang, M.; Zhang, Z.; Ma, P.; Peng, H.; Shi, K.; Gao, W.-Q.; Zhuang, G. Stroma-associated master regulators of molecular subtypes predict patient prognosis in ovarian cancer. Sci. Rep. 2015, 5, 16066. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Liu, Z.; Deng, N.; Tan, T.Z.; Huang, R.Y.-J.; Taylor-Harding, B.; Cheon, D.-J.; Lawrenson, K.; Wiedemeyer, W.R.; Walts, A.E.; et al. A COL11A1-correlated pan-cancer gene signature of activated fibroblasts for the prioritization of therapeutic targets. Cancer Lett. 2016, 382, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Vargas, H.A.; Miccò, M.; Hong, S.I.; Goldman, D.A.; Dao, F.; Weigelt, B.; Soslow, R.A.; Hricak, H.; Levine, D.A.; Sala, E. Association between morphologic ct imaging traits and prognostically relevant gene signatures in women with high-grade serous ovarian cancer: A hypothesis-generating study. Radiology 2015, 274, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.; Kumar, A.; Wallace, S.K.; Bakkum-Gamez, J.N.; Konecny, G.E.; Weaver, A.L.; McGree, M.E.; Goode, E.L.; Cliby, W.A.; Wang, C. Intraperitoneal disease dissemination patterns are associated with residual disease, extent of surgery, and molecular subtypes in advanced ovarian cancer. Gynecol. Oncol. 2017, 147, 503–508. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Tamayo, P.; Yang, J.-Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef]
- Leong, H.S.; Galletta, L.; Etemadmoghadam, D.; George, J.; Köbel, M.; Ramus, S.J.; Bowtell, D. The Australian Ovarian Cancer Study Efficient molecular subtype classification of high-grade serous ovarian cancer. J. Pathol. 2015, 236, 272–277. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing [Electronic resource]/December 2023. Mode of Access. Available online: https://www.r-project.org/ (accessed on 10 September 2024).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Chollet, F. Keras. 2015/December 2023. Mode of Access. Available online: https://keras.io (accessed on 10 September 2024).
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. Tensorflow: A System for Large-Scale Machine Learning. 2016/December 2023. Mode of Access. Available online: https://tensorflow.rstudio.com/ (accessed on 10 September 2024).
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization/December 2023. Mode of Access. Available online: https://arxiv.org/abs/1412.6980 (accessed on 10 September 2024).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. 2022. _caret: Classification and Regression Training_. R Package Version 6.0-93. Available online: https://github.com/topepo/caret (accessed on 10 September 2024).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Winterhoff, B.J.; Maile, M.; Mitra, A.K.; Sebe, A.; Bazzaro, M.; Geller, M.A.; Abrahante, J.E.; Klein, M.; Hellweg, R.; Mullany, S.A.; et al. Single cell sequencing reveals heterogeneity within ovarian cancer epithelium and cancer associated stromal cells. Gynecol. Oncol. 2017, 144, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.M.; Kannan, L.; Geistlinger, L.; Kofia, V.; Safikhani, Z.; Gendoo, D.M.; Parmigiani, G.; Birrer, M.J.; Haibe-Kains, B.; Waldron, L. Consensus on Molecular Subtypes of High-Grade Serous Ovarian Carcinoma. Clin. Cancer Res. 2018, 24, 5037–5047. [Google Scholar] [CrossRef] [PubMed]
- Geistlinger, L.; Oh, S.; Ramos, M.; Schiffer, L.; LaRue, R.S.; Henzler, C.M.; Munro, S.A.; Daughters, C.; Nelson, A.C.; Winterhoff, B.J.; et al. Multiomic Analysis of Subtype Evolution and Heterogeneity in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2020, 80, 4335–4345. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | Description | Cases, n | PID |
|---|---|---|---|
| 1 | TCGA-OV, Bulk RNA-seq | 413 | 21720365 |
| 2 | CPTAC, Bulk RNA-seq | 62 | 25873244 |
| 3 | PTRC-HGSOC, scRNA-seq | 5 | 37541199 |
| 4 | Spatial ovarian cancer 6, 10x Genomics Visium spatial transcriptomics | 6 | 36882687 |
| 5 | Spatial ovarian cancer 8, 10x Genomics Visium spatial transcriptomics | 8 | 36788074 |
| 6 | Ovarian cancer, scRNA-seq | 41 | 36517593 |
| Genes, n | D | I | M | P | |||
|---|---|---|---|---|---|---|---|
| D vs. Other | DI vs. MP | DM vs. IP | DP vs. IM | ||||
| Up | 0 | 33 | 7 | 1 | 29 | 91 | 18 |
| Down | 4 | 19 | 2 | 30 | 4 | 0 | 119 |
| Total | 96 | 33 | 91 | 137 | |||
| Subtype | Sensitivity | Specificity | Precision | Accuracy | Kappa | AUC |
|---|---|---|---|---|---|---|
| D | 0.98 | 0.97 | 0.99 | 0.98 | 0.94 | 0.976 |
| I | 0.99 | 0.97 | 0.99 | 0.99 | 0.96 | 0.980 |
| M | 0.98 | 0.94 | 0.98 | 0.96 | 0.92 | 0.960 |
| P | 0.99 | 0.97 | 0.99 | 0.99 | 0.98 | 0.987 |
| All | 0.96 | 0.99 | 0.96 | 0.98 | 0.95 | 0.969 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobelyatskaya, A.; Tregubova, A.; Palicelli, A.; Badlaeva, A.; Asaturova, A. OVsignGenes: A Gene Expression-Based Neural Network Model Estimated Molecular Subtype of High-Grade Serous Ovarian Carcinoma. Cancers 2024, 16, 3951. https://doi.org/10.3390/cancers16233951
Kobelyatskaya A, Tregubova A, Palicelli A, Badlaeva A, Asaturova A. OVsignGenes: A Gene Expression-Based Neural Network Model Estimated Molecular Subtype of High-Grade Serous Ovarian Carcinoma. Cancers. 2024; 16(23):3951. https://doi.org/10.3390/cancers16233951
Chicago/Turabian StyleKobelyatskaya, Anastasiya, Anna Tregubova, Andrea Palicelli, Alina Badlaeva, and Aleksandra Asaturova. 2024. "OVsignGenes: A Gene Expression-Based Neural Network Model Estimated Molecular Subtype of High-Grade Serous Ovarian Carcinoma" Cancers 16, no. 23: 3951. https://doi.org/10.3390/cancers16233951
APA StyleKobelyatskaya, A., Tregubova, A., Palicelli, A., Badlaeva, A., & Asaturova, A. (2024). OVsignGenes: A Gene Expression-Based Neural Network Model Estimated Molecular Subtype of High-Grade Serous Ovarian Carcinoma. Cancers, 16(23), 3951. https://doi.org/10.3390/cancers16233951