
Altered Microbiome Promotes Pro-Inflammatory Pathways in Oesophago-Gastric Tumourigenesis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Risk Factors for Oesophago-Gastric Cancer
1.2. Initial Insights into the Oesophago-Gastric Microbiome
2. Materials and Methods
3. Results and Discussion
3.1. Profiling the Oesophago-Gastric Microbiome
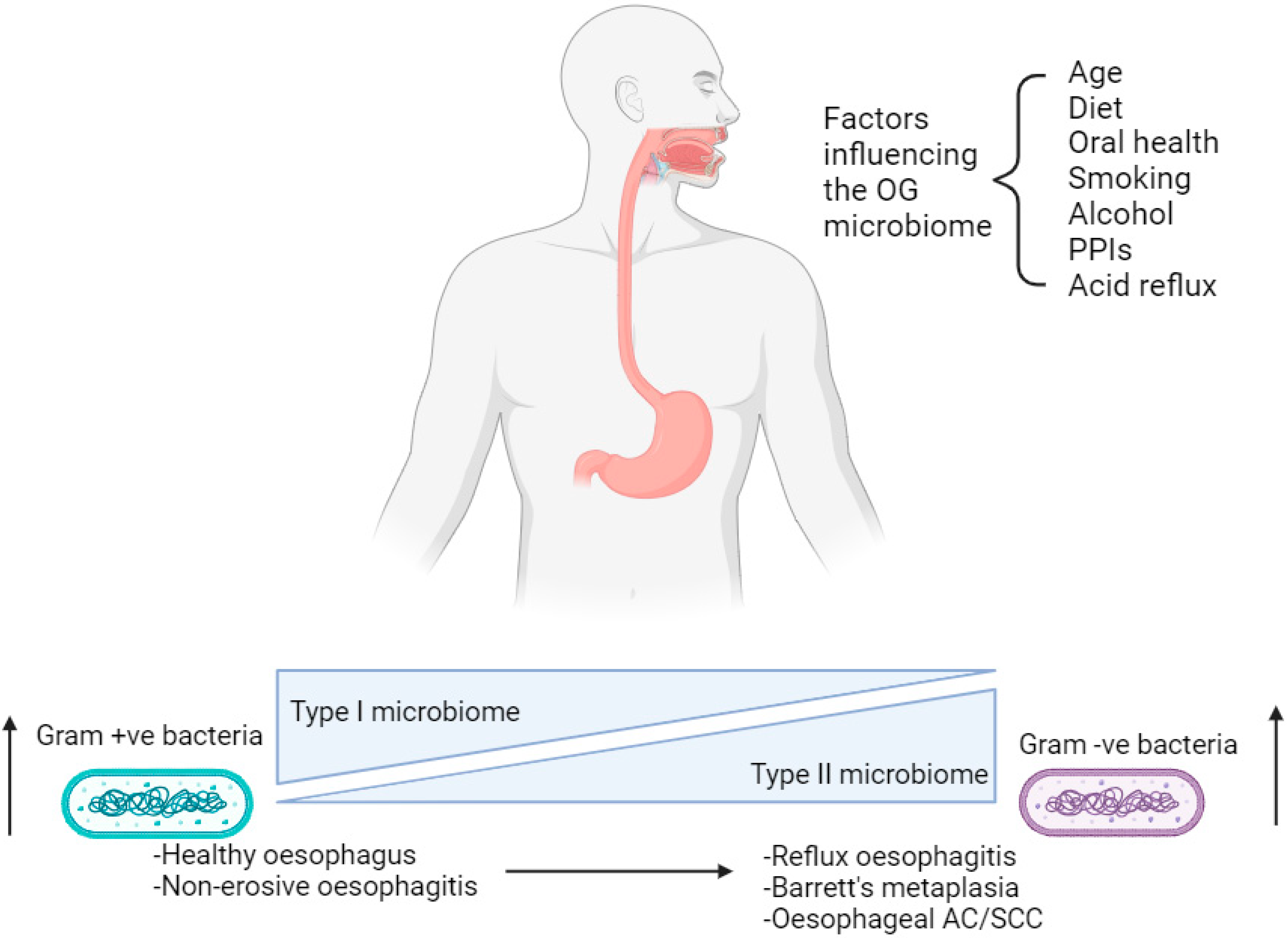
3.2. Factors Influencing the Oesophago-Gastric Microbiome
3.3. Pro-Inflammatory Pathways in Oesophago-Gastric Tumourigenesis
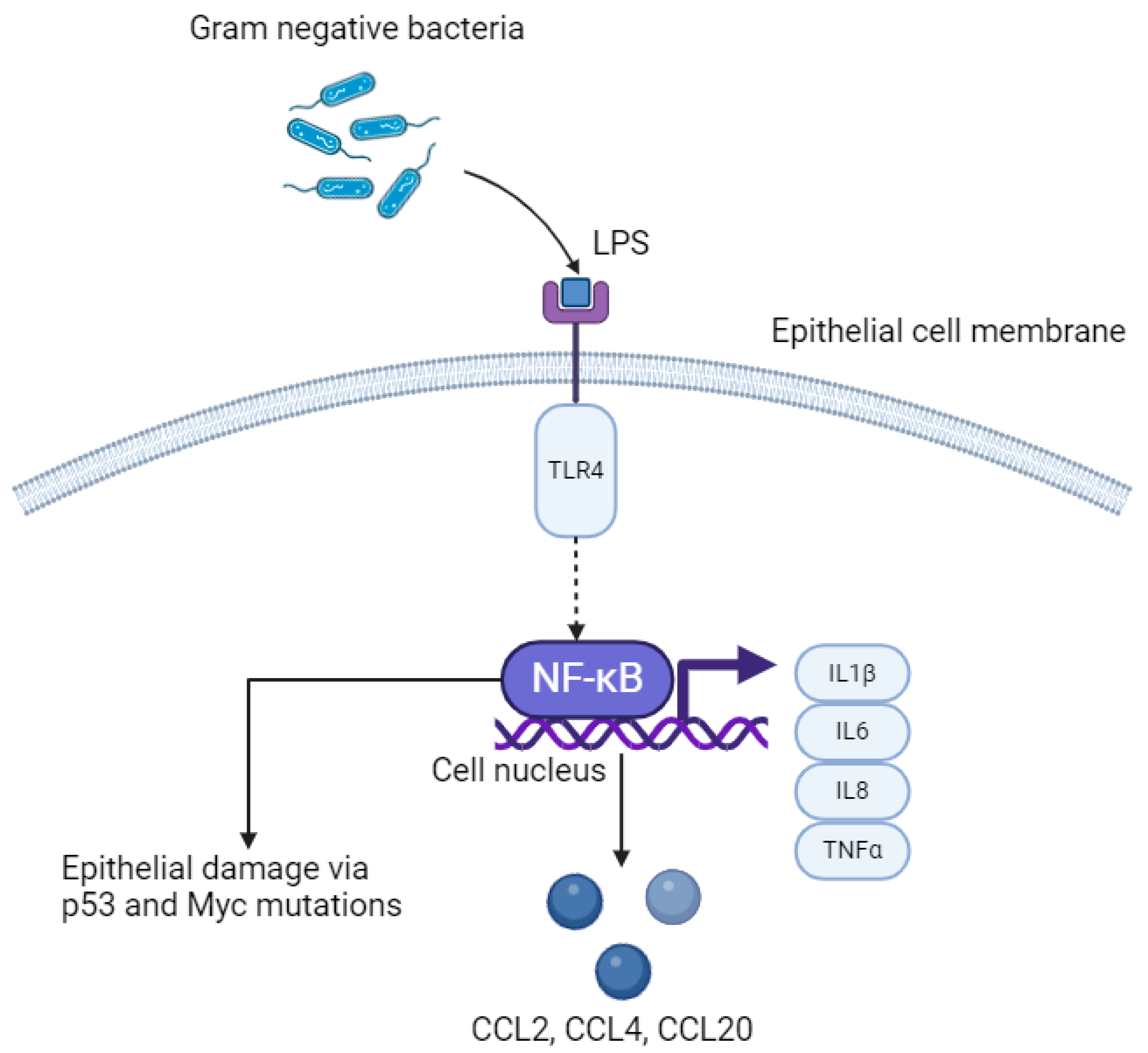
3.4. LPS–TLR4–NFκB Pathway
3.5. Inflammasome-Mediated Oesophago-Gastric Tumourigenesis
3.6. IL6–STAT3 Pathway
3.7. The Role of Immune Modulation in Oesophago-Gastric Cancer
3.8. Pro-Tumourigenic Pathways Induced by Diet
3.9. Barrett’s Metaplasia and Progression to Oesophageal and Gastro-Oesophageal Junction Adenocarcinoma
3.10. Systemic Effect of Dysbiosis in Oesophago-Gastric Tumourigenesis
3.11. Role of Short-Chain Fatty Acids in Oesophago-Gastric Tumourigenesis
3.12. Therapeutic Application of Insights into Microbiome-Mediated Oesophago-Gastric Tumourigenesis
4. Limitations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morgan, E.; Soerjomataram, I.; Rumgay, H.; Coleman, H.G.; Thrift, A.P.; Vignat, J.; Laversanne, M.; Ferlay, J.; Arnold, M. The global landscape of esophageal squamous cell carcinoma and esophageal adenocarcinoma incidence and mortality in 2020 and projections to 2040: New estimates from GLOBOCAN 2020. Gastroenterology 2022, 163, 649–658.e2. [Google Scholar] [CrossRef]
- Abnet, C.C.; Arnold, M.; Wei, W.-Q. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology 2018, 154, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Gokulan, R.C.; Garcia-Buitrago, M.T.; Zaika, A.I. From genetics to signaling pathways: Molecular pathogenesis of esophageal adenocarcinoma. Biochim. Biophys. Acta (BBA) Rev. Cancer 2019, 1872, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Fan, J.H.; Qiao, Y.L. Epidemiology, etiology, and prevention of esophageal squamous cell carcinoma in China. Cancer Biol. Med. 2017, 14, 33–41. [Google Scholar] [PubMed]
- Niu, C.; Liu, Y.; Wang, J.; Liu, Y.; Zhang, S.; Zhang, Y.; Zhang, L.; Zhao, D.; Liu, F.; Chao, L.; et al. Risk factors for esophageal squamous cell carcinoma and its histological precursor lesions in China: A multicenter cross-sectional study. BMC Cancer 2021, 21, 1034. [Google Scholar] [CrossRef]
- Hvid-Jensen, F.; Pedersen, L.; Drewes, A.M.; Sørensen, H.T.; Funch-Jensen, P. Incidence of Adenocarcinoma among Patients with Barrett’s Esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef]
- Jenkins, G.J.S.; Doak, S.H.; Parry, J.M.; D’Souza, F.R.; Griffiths, A.P.; Baxter, J.N. Genetic pathways involved in the progression of Barrett’s metaplasia to adenocarcinoma. Br. J. Surg. 2002, 89, 824–837. [Google Scholar] [CrossRef]
- Koppert, L.B.; Wijnhoven, B.P.L.; van Dekken, H.; Tilanus, H.W.; Dinjens, W.N.M. The molecular biology of esophageal adenocarcinoma. J. Surg. Oncol. 2005, 92, 1169–1190. [Google Scholar] [CrossRef]
- Lagergren, J.; Lagergren, P. Recent developments in esophageal adenocarcinoma. CA Cancer J. Clin. 2013, 63, 232–248. [Google Scholar] [CrossRef]
- Song, S.; Guha, S.; Liu, K.; Buttar, N.S.; Bresalier, R.S. COX-2 induction by unconjugated bile acids involves reactive oxygen species-mediated signalling pathways in Barrett’s oesophagus and oesophageal adenocarcinoma. Gut 2007, 56, 1512–1521. [Google Scholar] [CrossRef]
- Bhat, A.A.; Lu, H.; Soutto, M.; Capobianco, A.; Rai, P.; Zaika, A.; El-Rifai, W. Exposure of Barrett’s and esophageal adenocarcinoma cells to bile acids activates EGFR–STAT3 signaling axis via induction of APE1. Oncogene 2018, 37, 6011–6024. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T.; Peek, R.M. Gastrointestinal Malignancy and the Microbiome. Gastroenterology 2014, 146, 1534–1546.e3. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, A.; Wang, Y.; Zhang, Y. Intratumoral microbiota: Roles in cancer initiation, development and therapeutic efficacy. Signal Transduct. Target. Ther. 2023, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Hrncir, T. Gut Microbiota Dysbiosis: Triggers, Consequences, Diagnostic and Therapeutic Options. Microorganisms 2022, 10, 578. [Google Scholar] [CrossRef] [PubMed]
- Snider, E.J.; Freedberg, D.E.; Abrams, J.A. Potential Role of the Microbiome in Barrett’s Esophagus and Esophageal Adenocarcinoma. Dig. Dis. Sci. 2016, 61, 2217–2225. [Google Scholar] [CrossRef]
- D’Souza, S.M.; Houston, K.; Keenan, L.; Yoo, B.S.; Parekh, P.J.; Johnson, D.A. Role of microbial dysbiosis in the pathogenesis of esophageal mucosal disease: A paradigm shift from acid to bacteria? World J. Gastroenterol. 2021, 27, 2054–2072. [Google Scholar] [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile Acid and Inflammation Activate Gastric Cardia Stem Cells in a Mouse Model of Barrett-Like Metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef]
- Sharma, T.; Gupta, A.; Chauhan, R.; Bhat, A.A.; Nisar, S.; Hashem, S.; Akhtar, S.; Ahmad, A.; Haris, M.; Singh, M.; et al. Cross-talk between the microbiome and chronic inflammation in esophageal cancer: Potential driver of oncogenesis. Cancer Metastasis Rev. 2022, 41, 281–299. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, S.; Luan, S.; Xiao, X.; Yang, Y.; Mao, C.; Chen, L.; Zeng, X.; Zhang, Y.; Yuan, Y. Gut Microbiota for Esophageal Cancer: Role in Carcinogenesis and Clinical Implications. Front. Oncol. 2021, 11, 717242. [Google Scholar] [CrossRef]
- Mannell, A.; Plant, M.; Frolich, J. The microflora of the oesophagus. Ann. R. Coll. Surg. Engl. 1983, 65, 152–154. [Google Scholar]
- Hasan, A.; Hasan, L.K.; Schnabl, B.; Greytak, M.; Yadlapati, R. Microbiome of the Aerodigestive Tract in Health and Esophageal Disease. Dig. Dis. Sci. 2021, 66, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Bini, E.J.; Yang, L.; Zhou, M.; Francois, F.; Blaser, M.J. Bacterial biota in the human distal esophagus. Proc. Natl. Acad. Sci. USA 2004, 101, 4250–4255. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, J.; Chaudhary, N.; Pei, Z.; Yang, L. Microbiome, innate Immunity, and esophageal adenocarcinoma. Clin. Lab. Med. 2014, 34, 721–732. [Google Scholar] [CrossRef]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and Intestinal Metaplasia of the Distal Esophagus Are Associated with Alterations in the Microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef]
- Yu, G.; Gail, M.H.; Shi, J.; Klepac-Ceraj, V.; Paster, B.J.; Dye, B.A.; Wang, G.-Q.; Wei, W.-Q.; Fan, J.-H.; Qiao, Y.-L.; et al. Association between upper digestive tract microbiota and cancer-predisposing states in the esophagus and stomach. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 735–741. [Google Scholar] [CrossRef]
- Li, D.; He, R.; Hou, G.; Ming, W.; Fan, T.; Chen, L.; Zhang, L.; Jiang, W.; Wang, W.; Lu, Z.; et al. Characterization of the Esophageal Microbiota and Prediction of the Metabolic Pathways Involved in Esophageal Cancer. Front. Cell. Infect. Microbiol. 2020, 10, 268. [Google Scholar] [CrossRef]
- Park, C.H.; Lee, J.G.; Lee, A.-R.; Eun, C.S.; Han, D.S. Network construction of gastric microbiome and organization of microbial modules associated with gastric carcinogenesis. Sci. Rep. 2019, 9, 12444. [Google Scholar] [CrossRef]
- Gregersen, H.; Pedersen, J.; Drewes, A.M. Deterioration of Muscle Function in the Human Esophagus with Age. Dig. Dis. Sci. 2008, 53, 3065–3070. [Google Scholar] [CrossRef]
- Deshpande, N.P.; Riordan, S.M.; Castaño-Rodriguez, N.; Wilkins, M.R.; Kaakoush, N.O. Signatures within the esophageal microbiome are associated with hist genetics, age, and disease. Microbiome 2018, 6, 227. [Google Scholar] [CrossRef]
- Shin, J.M.; Kim, N. Pharmacokinetics and Pharmacodynamics of the Proton Pump Inhibitors. J. Neurogastroenterol. Motil. 2013, 19, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Amir, I.; Konikoff, F.M.; Oppenheim, M.; Gophna, U.; Half, E.E. Gastric microbiota is altered in oesophagitis and Barrett’s oesophagus and further modified by proton pump inhibitors. Environ. Microbiol. 2014, 16, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Rajilić–Stojanović, M.; Biagi, E.; Heilig, H.G.; Kajander, K.; Kekkonen, R.A.; Tims, S.; de Vos, W.M. Global and Deep Molecular Analysis of Microbiota Signatures in Fecal Samples from Patients with Irritable Bowel Syndrome. Gastroenterology 2011, 141, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Lucenteforte, E.; Garavello, W.; Bosetti, C.; Talamini, R.; Zambon, P.; Franceschi, S.; Negri, E.; La Vecchia, C. Diet diversity and the risk of squamous cell esophageal cancer. Int. J. Cancer 2008, 123, 2397–2400. [Google Scholar] [CrossRef]
- Bravi, F.; Edefonti, V.; Randi, G.; Garavello, W.; La Vecchia, C.; Ferraroni, M.; Talamini, R.; Franceschi, S.; Decarli, A. Dietary patterns and the risk of esophageal cancer. Ann. Oncol. 2012, 23, 765–770. [Google Scholar] [CrossRef]
- Choi, Y.; Song, S.; Song, Y.; Lee, J.E. Consumption of red and processed meat and esophageal cancer risk: Meta-analysis. World J. Gastroenterol. 2013, 19, 1020–1029. [Google Scholar] [CrossRef]
- Nobel, Y.R.; Snider, E.J.; Compres, G.; Freedberg, D.E.; Khiabanian, H.; Lightdale, C.J.; Toussaint, N.C.; Abrams, J.A. Increasing Dietary Fiber Intake Is Associated with a Distinct Esophageal Microbiome. Clin. Transl. Gastroenterol. 2018, 9, e199. [Google Scholar] [CrossRef]
- Fan, Y.; Yuan, J.-M.; Wang, R.; Gao, Y.-T.; Yu, M.C. Alcohol, Tobacco, and Diet in Relation to Esophageal Cancer: The Shanghai Cohort Study. Nutr. Cancer 2008, 60, 354–363. [Google Scholar] [CrossRef]
- Radojicic, J.; Zaravinos, A.; Spandidos, D.A. HPV, KRAS mutations, alcohol consumption and tobacco smoking effects on esophageal squamous-cell carcinoma carcinogenesis. Int. J. Biol. Markers 2012, 27, 1–12. [Google Scholar] [CrossRef]
- Gao, Y.T.; McLaughlin, J.K.; Blot, W.J.; Ji, B.T.; Benichou, J.; Dai, Q.; Fraumeni, J.F., Jr. Risk factors for esophageal cancer in Shanghai, China. I. Role of cigarette smoking and alcohol drinking. Int. J. Cancer 1994, 58, 192–196. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral Microbiome Composition Reflects Prospective Risk for Esophageal Cancers. Cancer Res. 2017, 77, 6777–6787. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The Microbiome and Cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Maslenkina, K.; Mikhaleva, L.; Naumenko, M.; Vandysheva, R.; Gushchin, M.; Atiakshin, D.; Buchwalow, I.; Tiemann, M. Signaling Pathways in the Pathogenesis of Barrett’s Esophagus and Esophageal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 9304. [Google Scholar] [CrossRef] [PubMed]
- Neish, A.S. Mucosal Immunity and the Microbiome. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. S1), S28–S32. [Google Scholar] [CrossRef]
- McDermott, A.J.; Huffnagle, G.B. The microbiome and regulation of mucosal immunity. Immunology 2014, 142, 24–31. [Google Scholar] [CrossRef]
- Fu, K.; Cheung, A.H.K.; Wong, C.C.; Liu, W.; Zhou, Y.; Wang, F.; Huang, P.; Yuan, K.; Coker, O.O.; Pan, Y.; et al. Streptococcus anginosus promotes gastric inflammation, atrophy, and tumorigenesis in mice. Cell 2024, 187, 882–896.E17. [Google Scholar] [CrossRef]
- Abdel-Latif, M.M.; Duggan, S.; Reynolds, J.V.; Kelleher, D. Inflammation and esophageal carcinogenesis. Curr. Opin. Pharmacol. 2009, 9, 396–404. [Google Scholar] [CrossRef]
- An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Nieß, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. The Role of Gut-Derived Lipopolysaccharides and the Intestinal Barrier in Fatty Liver Diseases. J. Gastrointest. Surg. 2022, 26, 671–683. [Google Scholar] [CrossRef]
- Nadatani, Y.; Huo, X.; Zhang, X.; Yu, C.; Cheng, E.; Zhang, Q.; Dunbar, K.B.; Theiss, A.; Pham, T.H.; Wang, D.H.; et al. NOD-Like Receptor Protein 3 Inflammasome Priming and Activation in Barrett’s Epithelial Cells. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 439–453. [Google Scholar] [CrossRef]
- Yang, L.; Francois, F.; Pei, Z. Molecular Pathways: Pathogenesis and Clinical Implications of Microbiome Alteration in Esophagitis and Barrett Esophagus. Clin. Cancer Res. 2012, 18, 2138–2144. [Google Scholar] [CrossRef]
- Yamamura, K.; Baba, Y.; Nakagawa, S.; Mima, K.; Miyake, K.; Nakamura, K.; Sawayama, H.; Kinoshita, K.; Ishimoto, T.; Iwatsuki, M.; et al. Human Microbiome Fusobacterium nucleatum in Esophageal Cancer Tissue Is Associated with Prognosis. Clin. Cancer Res. 2016, 22, 5574–5581. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.W.; Karakasheva, T.A.; Hicks, P.D.; Bass, A.J.; Rustgi, A.K. The tumour microenvironment in esophageal cancer. Oncogene 2016, 35, 5337–5349. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, J.H.; Selander, K.S. Toll-Like Receptors in Esophageal Cancer. Front. Immunol. 2014, 5, 200. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, R.E.; Siersema, P.D.; Kate, F.J.T.; Fluiter, K.; Souza, R.F.; Vleggaar, F.P.; Bus, P.; van Baal, J.W.P.M. Toll-like receptor 4 activation in Barrett’s esophagus results in a strong increase in COX-2 expression. J. Gastroenterol. 2014, 49, 1121–1134. [Google Scholar] [CrossRef]
- Puri, R.N.; Fan, Y.P.; Rattan, S. Role of pp60(c-src) and p(44/42) MAPK in ANG II_induced contraction of rat tonic gastrointestinal smooth muscles. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G390–G399. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Molla, M.D.; Akalu, Y.; Geto, Z.; Dagnew, B.; Ayelign, B.; Shibabaw, T. Role of Caspase-1 in the Pathogenesis of Inflammatory-Associated Chronic Noncommunicable Diseases. J. Inflamm. Res. 2020, 13, 749–764. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Verbeke, H.; Geboes, K.; Van Damme, J.; Struyf, S. The role of CXC chemokines in the transition of chronic inflammation to esophageal and gastric cancer. Biochim. Biophys. Acta 2012, 1825, 117–129. [Google Scholar]
- Blank, S.; Nienhüser, H.; Dreikhausen, L.; Sisic, L.; Heger, U.; Ott, K.; Schmidt, T. Inflammatory cytokines are associated with response and prognosis in patients with esophageal cancer. Oncotarget 2017, 8, 47518–47532. [Google Scholar] [CrossRef]
- Shrivastava, M.S.; Hussain, Z.; Giricz, O.; Shenoy, N.; Polineni, R.; Maitra, A.; Verma, A. Targeting chemokine pathways in esophageal adenocarcinoma. Cell Cycle 2014, 13, 3320–3327. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.-J.; Ma, C.; Hu, K.; Zhao, M.-M.; Zhang, N.; Sun, Z.-G. Molecular mechanism, regulation, and therapeutic targeting of the STAT3 signaling pathway in esophageal cancer (Review). Int. J. Oncol. 2022, 61, 105. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.; Hughes, M.; Chang, W. The role of microbiota in esophageal squamous cell carcinoma: A review of the literature. Thorac. Cancer 2023, 14, 2821–2829. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Sakamoto, E.; Kutsuna, H.; Kimura-Eto, A.; Hato, F.; Kitagawa, S. Proteolytic conversion of STAT3alpha to STAT3gamma in human neutrophils: Role of granule-derived serine proteases. J. Biol. Chem. 2004, 279, 31076–31080. [Google Scholar] [CrossRef]
- Bromberg, J.H.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Bernard, A.; Chevrier, S.; Beltjens, F.; Dosset, M.; Viltard, E.; Lagrange, A.; Derangère, V.; Oudot, A.; Ghiringhelli, F.; Collin, B.; et al. Cleaved Caspase-3 Transcriptionally Regulates Angiogenesis-Promoting Chemotherapy Resistance. Cancer Res. 2019, 79, 5958–5970. [Google Scholar] [CrossRef]
- Su, W.; Guo, C.; Wang, L.; Wang, Z.; Yang, X.; Niu, F.; Tzou, D.; Yang, X.; Huang, X.; Wu, J.; et al. LncRNA MIR22HG abrogation inhibits proliferation and induces apoptosis in esophageal adenocarcinoma cells via activation of the STAT3/c-Myc/FAK signalling. Aging 2019, 11, 4587–4596. [Google Scholar] [CrossRef]
- Chen, M.; Ye, A.; Wei, J.; Wang, R.; Poon, K. Deoxycholic acid upregulates the reprogramming factors KFL4 and OCT4 through the IL-6/STAT3 pathway in esophageal adenocarcinoma cells. Technol. Cancer Res. Treat. 2020, 19, 1533033820945302. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, M.; Wang, Z.; Gao, W.; Sun, Z.-G. Activated STAT3 Could Reduce Survival in Patients with Esophageal Squamous Cell Carcinoma by Up-regulating VEGF and Cyclin D1 Expression. J. Cancer 2020, 11, 1859–1868. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Li, R.; Ma, L.; Liu, L.; Lai, X.; Yang, D.; Wei, J.; Ma, D.; Li, Z. Porphyromonas gingivalis promotes the motility of esophageal squamous cell carcinoma by activating NF-κB signaling pathway. Microbes Infect. 2019, 21, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-F.; Lu, M.-S.; Hsieh, C.-C.; Chen, W.-C. Porphyromonas gingivalis promotes tumor progression in esophageal squamous cell carcinoma. Cell. Oncol. 2021, 44, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pan, Z. Influence of microbiota on immunity and immunotherapy for gastric and esophageal cancers. Gastroenterol. Rep. 2020, 8, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Xie, J.; Bedolla, R.; Rivas, P.; Chakravarthy, D.; Freeman, J.W.; Reddick, R.; Kopetz, S.; Peterson, A.; Wang, H.; et al. Combined targeting of STAT3/NF-κB/COX-2/EP4 for effective management of pancreatic cancer. Clin. Cancer Res. 2014, 20, 1259–1273. [Google Scholar] [CrossRef]
- Nabeki, B.; Ishigami, S.; Uchikado, Y.; Sasaki, K.; Kita, Y.; Okumura, H.; Arigami, T.; Kijima, Y.; Kurahara, H.; Maemura, K.; et al. Interleukin-32 expression and Treg infiltration in esophageal squamous cell carcinoma. Anticancer Res. 2015, 35, 2941–2947. [Google Scholar]
- Zou, W.; Restifo, N.P. T(H)17 cells in tumour immunity and immunotherapy. Nat. Rev. Immunol. 2010, 10, 248–256. [Google Scholar] [CrossRef]
- Shitara, K.; Rha, S.Y.; Wyrwicz, L.S.; Oshima, T.; Karaseva, N.; Osipov, M.; Yasui, H.; Yabusaki, H.; Afanasyev, S.; Park, Y.-K.; et al. Neoadjuvant and adjuvant pembrolizumab plus chemotherapy in locally advanced gastric or gastro-oesophageal cancer (KEYNOTE-585): An interim analysis of the multicentre, double-blind, randomised phase 3 study. Lancet Oncol. 2024, 25, 212–224. [Google Scholar] [CrossRef]
- Bouras, E.; Tsilidis, K.K.; Triggi, M.; Siargkas, A.; Chourdakis, M.; Haidich, A.-B. Diet and Risk of Gastric Cancer: An Umbrella Review. Nutrients 2022, 14, 1764. [Google Scholar] [CrossRef]
- Tricker, A.R.; Preussmann, R. Carcinogenic N-nitrosamines in the diet: Occurrence, formation, mechanisms and carcinogenic potential. Mutat. Res. Genet. Toxicol. 1991, 259, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Jaroenlapnopparat, A.; Bhatia, K.; Coban, S. Inflammation and Gastric Cancer. Diseases 2022, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Seyyedsalehi, M.S.; Mohebbi, E.; Tourang, F.; Sasanfar, B.; Boffetta, P.; Zendehdel, K. Association of Dietary Nitrate, Nitrite, and N-Nitroso Compounds Intake and Gastrointestinal Cancers: A Systematic Review and Meta-Analysis. Toxics 2023, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Moriya, A.; Grant, J.; Mowat, C.; Williams, C.; Carswell, A.; Preston, T.; Anderson, S.; Iijima, K.; McColl, K.E.L. In vitro Studies Indicate that Acid Catalysed Generation of N-Nitrosocompounds from Dietary Nitrate Will be Maximal at the Gastro-oesophageal Junction and Cardia. Scand. J. Gastroenterol. 2002, 37, 253–261. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E.; Cole, J.A.; Benjamin, N. Nitrate, bacteria and human health. Nat. Rev. Microbiol. 2004, 2, 593–602. [Google Scholar] [CrossRef]
- Iijima, K.; Henry, E.; Moriya, A.; Wirz, A.; Kelman, A.; McColl, K. Dietary nitrate generates potentially mutagenic concentrations of nitric oxide at the gastroesophageal junction. Gastroenterology 2002, 122, 1248–1257. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, X.; Zhang, C.; Zhu, C.; Tao, G.; Zhao, L.; Tang, S.; Shu, Z.; Cai, J.; Dai, S.; et al. Red and Processed Meat Intake Is Associated with Higher Gastric Cancer Risk: A Meta-Analysis of Epidemiological Observational Studies. PLoS ONE 2013, 8, e70955. [Google Scholar] [CrossRef]
- Tobey, N.; Hosseini, S.; Argote, C.; Dobrucali, A.; Awayda, M.S.; Orlando, R. Dilated Intercellular Spaces and Shunt Permeability in Nonerosive Acid-Damaged Esophageal Epithelium. Am. J. Gastroenterol. 2004, 99, 13–22. [Google Scholar] [CrossRef]
- Ajayi, T.A.; Cantrell, S.; Spann, A.; Garman, K.S. Barrett’s esophagus and esophageal cancer: Links to microbes and the microbiome. PLoS Pathog. 2018, 14, e1007384. [Google Scholar] [CrossRef]
- Souza, R.F.; Spechler, S.J. Mechanisms and pathophysiology of Barrett oesophagus. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 605–620. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, J.M.; Abdel-Latif, M.M.; Ravi, N.; McNamara, D.; Byrne, P.J.; McDonald, G.S.A.; Keeling, P.W.N.; Kelleher, D.; Reynolds, J.V. Proinflammatory Cytokine and Nuclear Factor Kappa-B Expression along the Inflammation-Metaplasia-Dysplasia-Adenocarcinoma Sequence in the Esophagus. Am. J. Gastroenterol. 2005, 100, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-kappaB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866.e24. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef]
- Scott, A.J.; Alexander, J.L.; Merrifield, C.A.; Cunningham, D.; Jobin, C.; Brown, R.; Alverdy, J.; O’Keefe, S.J.; Gaskins, H.R.; Teare, J.; et al. International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut 2019, 68, 1624–1632. [Google Scholar] [CrossRef]
- Cestari, R.; Villanacci, V.; Bassotti, G.; Rossi, E.; Casa, D.D.; Missale, G.; Minelli, L.; Cengia, P.; Gambarotti, M.; Pirali, F.; et al. The pathology of gastric cardia: A prospective, endoscopic, and morphologic study. Am. J. Surg. Pathol. 2007, 31, 706–710. [Google Scholar] [CrossRef]
- Özetkin, M.; Yilmaz, B.; Ağagündüz, D.; Capasso, R. Overview of Helicobacter pylori Infection: Clinical Features, Treatment and Nutritional Aspects. Diseases 2021, 9, 66. [Google Scholar] [CrossRef]
- Lax, A.J. Opinion: Bacterial Toxins and Cancer—A Case to Answer? Nat. Rev. Microbiol. 2005, 3, 343–349. [Google Scholar] [CrossRef]
- Pickett, C.L.; Pesci, E.C.; Cottle, D.L.; Russell, G.; Erdem, A.N.; Zeytin, H. Prevalence of Cytolethal Distending Toxin Production in Campylobacter jejuni and Relatedness of Campylobacter sp. cdtB Gene. Infect. Immun. 1996, 64, 2070–2078. [Google Scholar] [CrossRef]
- Backert, S.; Clyne, M.; Tegtmeyer, N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun. Signal. 2011, 9, 28. [Google Scholar] [CrossRef]
- Javaheri, A.; Kruse, T.; Moonens, K.; Mejías-Luque, R.; Debraekeleer, A.; Asche, C.I.; Tegtmeyer, N.; Kalali, B.; Bach, N.C.; Sieber, S.A.; et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2016, 2, 16189. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Kanemitsu, A.; Knight, C.T.; Hatakeyama, M. Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis. Cell. Mol. Immunol. 2020, 17, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Blanke, S.R. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 2005, 3, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Bakhti, S.Z.; Latifi-Navid, S. Interplay and cooperation of Helicobacter pylori and gut microbiota in gastric carcinogenesis. BMC Microbiol. 2021, 21, 258. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.; Peek, R.; Tham, K.; Cover, T.; Blaser, M. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology 1997, 112, 92–99. [Google Scholar] [CrossRef]
- Mann, E.R.; Lam, Y.K.; Uhlig, H.H. Short-chain fatty acids: Linking diet, the microbiome and immunity. Nat. Rev. Immunol. 2024, 24, 577–595. [Google Scholar] [CrossRef]
- Carretta, M.D.; Quiroga, J.; López, R.; Hidalgo, M.A.; Burgos, R.A. Participation of Short-Chain Fatty Acids and Their Receptors in Gut Inflammation and Colon Cancer. Front. Physiol. 2021, 12, 662739. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Medhat, A.; Spector, I. Short-chain fatty acids in cancer pathogenesis. Cancer Metastasis Rev. 2023, 42, 677–698. [Google Scholar] [CrossRef]
- Kim, C.H.; Park, J.; Kim, M. Gut Microbiota-Derived Short-Chain Fatty Acids, T Cells, and Inflammation. Immune Netw. 2014, 14, 277–288. [Google Scholar] [CrossRef]
- Liang, W.; Yang, Y.; Wang, H.; Wang, H.; Yu, X.; Lu, Y.; Shen, S.; Teng, L. Gut microbiota shifts in patients with gastric cancer in perioperative period. Medicine 2019, 98, e16626–e16635. [Google Scholar] [CrossRef]
- Gordon, A.; Johnston, E.; Lau, D.K.; Starling, N. Targeting FGFR2 Positive Gastroesophageal Cancer: Current and Clinical Developments. OncoTargets Ther. 2022, 15, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti- and Pro-inflammatory Roles of TGF-β, IL-10, and IL-22 In Immunity and Autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, K.-N.; Vitetta, L. Effects of Intestinal Microbial–Elaborated Butyrate on Oncogenic Signaling Pathways. Nutrients 2019, 11, 1026–1051. [Google Scholar] [CrossRef] [PubMed]
- Pahle, J.; Menzel, L.; Niesler, N.; Kobelt, D.; Aumann, J.; Rivera, M.; Walther, W. Rapid eradication of colon carcinoma by Clostridium perfringens Enterotoxin suicidal gene therapy. BMC Cancer 2017, 17, 129. [Google Scholar] [CrossRef]
- Zhao, L.-Y.; Mei, J.-X.; Yu, G.; Lei, L.; Zhang, W.-H.; Liu, K.; Chen, X.-L.; Kołat, D.; Yang, K.; Hu, J.-K. Role of the gut microbiota in anticancer therapy: From molecular mechanisms to clinical applications. Signal Transduct. Target. Ther. 2023, 8, 201. [Google Scholar] [CrossRef]
- Park, E.M.; Chlvanambi, M.; Bhutiani, N.; Kroemer, G.; Zitvogel, L.; Wargo, J.A. Targeting the gut and tumor microbiota in cancer. Nat. Med. 2022, 28, 690–703. [Google Scholar] [CrossRef]
- Si, W.; Liang, H.; Bugno, J.; Xu, Q.; Ding, X.; Yang, K.; Fu, Y.; Weichselbaum, R.R.; Zhao, X.; Wang, L. Lactobacillus rhamnosus GG induces cGAS/STING- dependent type I interferon and improves response to immune checkpoint blockade. Gut 2022, 71, 521–533. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Panthee, B.; Gyawali, S.; Panthee, P.; Techato, K. Environmental and Human Microbiome for Health. Life 2022, 12, 456. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, N.M.; Patel, P.H.; Bhogal, R.H.; Harrington, K.J.; Singanayagam, A.; Kumar, S. Altered Microbiome Promotes Pro-Inflammatory Pathways in Oesophago-Gastric Tumourigenesis. Cancers 2024, 16, 3426. https://doi.org/10.3390/cancers16193426
Patel NM, Patel PH, Bhogal RH, Harrington KJ, Singanayagam A, Kumar S. Altered Microbiome Promotes Pro-Inflammatory Pathways in Oesophago-Gastric Tumourigenesis. Cancers. 2024; 16(19):3426. https://doi.org/10.3390/cancers16193426
Chicago/Turabian StylePatel, Nikhil Manish, Pranav Harshad Patel, Ricky Harminder Bhogal, Kevin Joseph Harrington, Aran Singanayagam, and Sacheen Kumar. 2024. "Altered Microbiome Promotes Pro-Inflammatory Pathways in Oesophago-Gastric Tumourigenesis" Cancers 16, no. 19: 3426. https://doi.org/10.3390/cancers16193426
APA StylePatel, N. M., Patel, P. H., Bhogal, R. H., Harrington, K. J., Singanayagam, A., & Kumar, S. (2024). Altered Microbiome Promotes Pro-Inflammatory Pathways in Oesophago-Gastric Tumourigenesis. Cancers, 16(19), 3426. https://doi.org/10.3390/cancers16193426