
Enhancing the Efficacy of Breast Cancer Immunotherapy Using a Smac-Armed Oncolytic Virus



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus and Cells
2.2. Animal Studies
2.3. Flow Cytometry
2.4. Quantitative Real-Time PCR
2.5. Western Blot
2.6. Hematoxylin and Eosin (H&E) Staining
2.7. Immunohistochemistry (IHC) Staining
2.8. TUNEL Assay
2.9. Statistical Analysis
3. Results
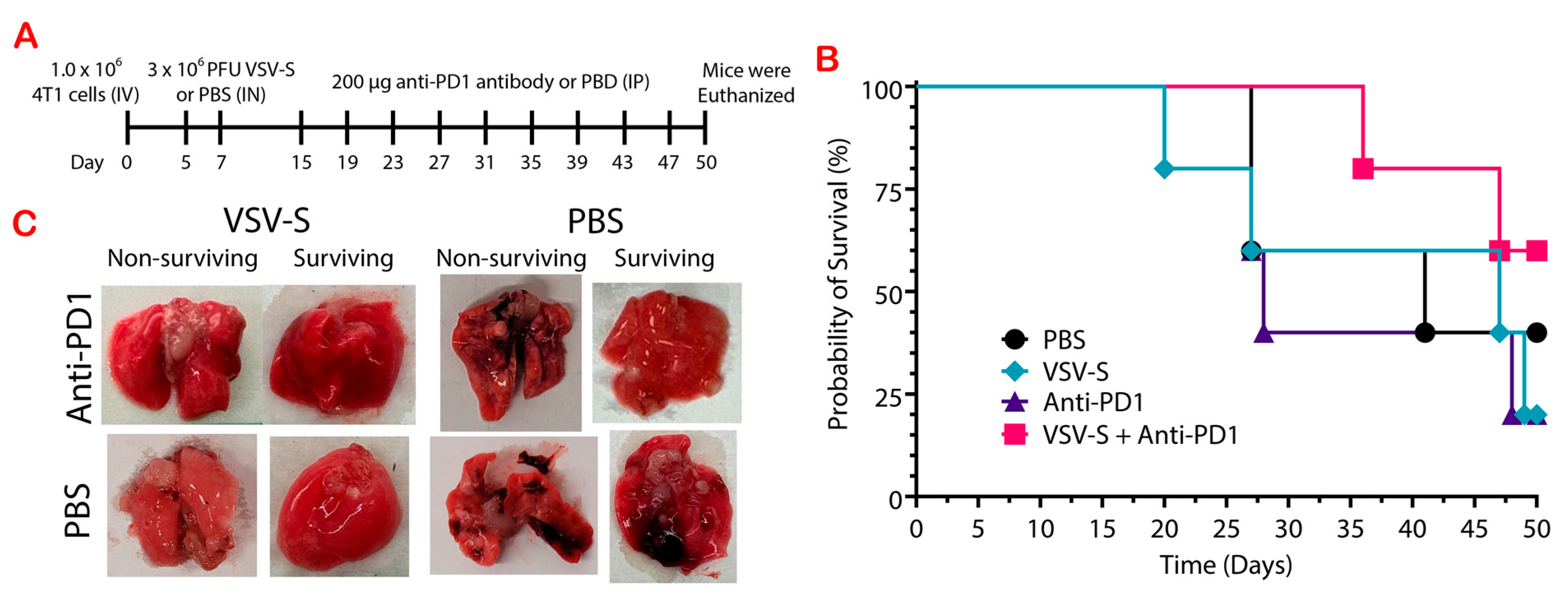
3.1. VSV-S Suppresses Breast Cancer Lung Metastasis as a Neoadjuvant Therapy
3.2. Adaptation Increases the Selective Infectivity of VSV-S in 4T1 Cells
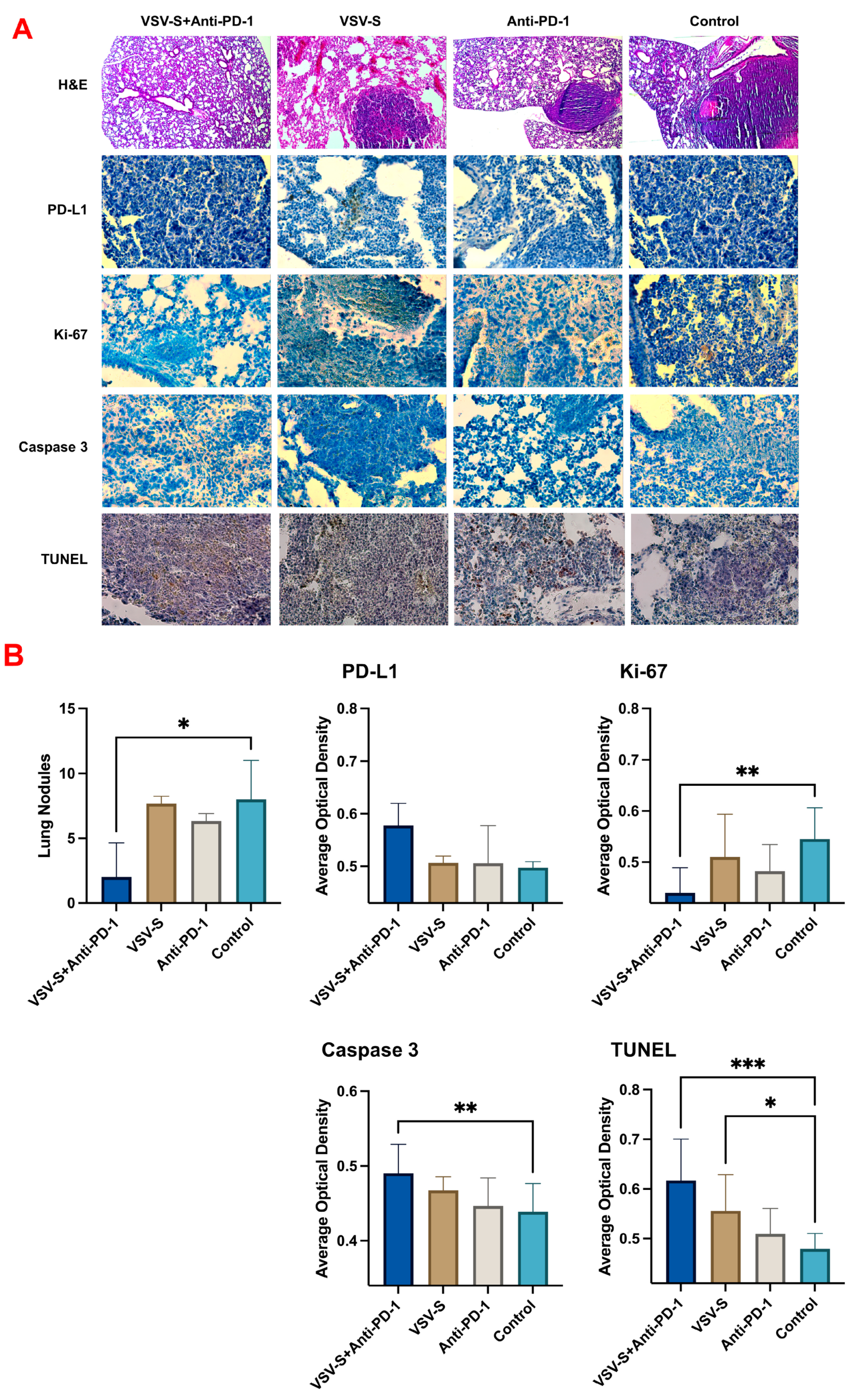
3.3. VSV-S Enhances the Efficacy of Immune Checkpoint Blockade
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hahn, E.E.; Ritzwoller, D.P.; Munoz-Plaza, C.E.; Gander, J.; Kushi, L.H.; McMullen, C.; Oshiro, C.; Roblin, D.W.; Wernli, K.J.; Staab, J. Incidence and Survival for Patients Diagnosed With Breast, Colorectal, and Lung Cancer in an Integrated System. Perm. J. 2023, 27, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Anayyat, U.; Ahad, F.; Muluh, T.A.; Zaidi, S.A.A.; Usmani, F.; Yang, H.; Li, M.; Hassan, H.A.; Wang, X. Immunotherapy: Constructive Approach for Breast Cancer Treatment. Breast Cancer (Dove Med. Press) 2023, 15, 925–951. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, F.; Liu, Z.; Fan, Z. Immunotherapy for Triple-Negative Breast Cancer: Combination Strategies to Improve Outcome. Cancers 2023, 15, 321. [Google Scholar] [CrossRef]
- Tarekegn, K.; Keskinkilic, M.; Kristoff, T.J.; Evans, S.T.; Kalinsky, K. The role of immune checkpoint inhibition in triple negative breast cancer. Expert. Rev. Anticancer. Ther. 2023, 23, 1095–1106. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Chen, X.; Feng, L.; Huang, Y.; Wu, Y.; Xie, N. Mechanisms and Strategies to Overcome PD-1/PD-L1 Blockade Resistance in Triple-Negative Breast Cancer. Cancers 2022, 15, 104. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; Romero-Garcia, S.; Lemini, C.; Prado-Garcia, H. Determining Factors in the Therapeutic Success of Checkpoint Immunotherapies against PD-L1 in Breast Cancer: A Focus on Epithelial-Mesenchymal Transition Activation. J. Immunol. Res. 2021, 2021, 6668573. [Google Scholar] [CrossRef] [PubMed]
- Mohan, N.; Hosain, S.; Zhao, J.; Shen, Y.; Luo, X.; Jiang, J.; Endo, Y.; Wu, W.J. Atezolizumab potentiates Tcell-mediated cytotoxicity and coordinates with FAK to suppress cell invasion and motility in PD-L1(+) triple negative breast cancer cells. Oncoimmunology 2019, 8, e1624128. [Google Scholar] [CrossRef]
- Cheung, A.; Chenoweth, A.M.; Quist, J.; Sow, H.S.; Malaktou, C.; Ferro, R.; Hoffmann, R.M.; Osborn, G.; Sachouli, E.; French, E.; et al. CDK Inhibition Primes for Anti-PD-L1 Treatment in Triple-Negative Breast Cancer Models. Cancers 2022, 14, 3361. [Google Scholar] [CrossRef]
- Hu, X.; Li, G.; Li, S.; Wang, Q.; Wang, Y.; Zhang, P.; Yang, T.; Yang, B.; Yu, L.; Liu, Z. TTK inhibition activates STING signal and promotes anti-PD1 immunotherapy in breast cancer. Biochem. Biophys. Res. Commun. 2024, 694, 149388. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gan, C.; Yu, S.; Yao, S.; Li, W.; Cheng, H. Analysis of Immune Resistance Mechanisms in TNBC: Dual Effects Inside and Outside the Tumor. Clin. Breast Cancer 2024, 24, e91–e102. [Google Scholar] [CrossRef] [PubMed]
- Karn, T.; Jiang, T.; Hatzis, C.; Sanger, N.; El-Balat, A.; Rody, A.; Holtrich, U.; Becker, S.; Bianchini, G.; Pusztai, L. Association Between Genomic Metrics and Immune Infiltration in Triple-Negative Breast Cancer. JAMA Oncol. 2017, 3, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Peg, V.; Prat, A.; Moline, T.; Villacampa, G.; Pare, L.; Galvan, P.; Dientsmann, R.; Schmid, P.; Curigliano, G.; et al. Immune analysis of lymph nodes in relation to the presence or absence of tumor infiltrating lymphocytes in triple-negative breast cancer. Eur. J. Cancer 2021, 148, 134–145. [Google Scholar] [CrossRef]
- Timperi, E.; Gueguen, P.; Molgora, M.; Magagna, I.; Kieffer, Y.; Lopez-Lastra, S.; Sirven, P.; Baudrin, L.G.; Baulande, S.; Nicolas, A.; et al. Lipid-Associated Macrophages Are Induced by Cancer-Associated Fibroblasts and Mediate Immune Suppression in Breast Cancer. Cancer Res. 2022, 82, 3291–3306. [Google Scholar] [CrossRef]
- Kajihara, N.; Kobayashi, T.; Otsuka, R.; Nio-Kobayashi, J.; Oshino, T.; Takahashi, M.; Imanishi, S.; Hashimoto, A.; Wada, H.; Seino, K.I. Tumor-derived interleukin-34 creates an immunosuppressive and chemoresistant tumor microenvironment by modulating myeloid-derived suppressor cells in triple-negative breast cancer. Cancer Immunol. Immunother. 2023, 72, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Nief, C.A.; Swartz, A.M.; Chelales, E.; Sheu, L.Y.; Crouch, B.T.; Ramanujam, N.; Nair, S.K. Ethanol Ablation Therapy Drives Immune-Mediated Antitumor Effects in Murine Breast Cancer Models. Cancers 2022, 14, 4669. [Google Scholar] [CrossRef]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef]
- Qian, X.; Zhang, Q.; Shao, N.; Shan, Z.; Cheang, T.; Zhang, Z.; Su, Q.; Wang, S.; Lin, Y. Respiratory hyperoxia reverses immunosuppression by regulating myeloid-derived suppressor cells and PD-L1 expression in a triple-negative breast cancer mouse model. Am. J. Cancer Res. 2019, 9, 529–545. [Google Scholar]
- Li, W.; Turaga, R.C.; Li, X.; Sharma, M.; Enadi, Z.; Dunham Tompkins, S.N.; Hardy, K.C.; Mishra, F.; Tsao, J.; Liu, Z.R.; et al. Overexpression of Smac by an Armed Vesicular Stomatitis Virus Overcomes Tumor Resistance. Mol. Ther. Oncolytics 2019, 14, 188–195. [Google Scholar] [CrossRef]
- Tang, S.; Shi, L.; Luker, B.T.; Mickler, C.; Suresh, B.; Lesinski, G.B.; Fan, D.; Liu, Y.; Luo, M. Modulation of the tumor microenvironment by armed vesicular stomatitis virus in a syngeneic pancreatic cancer model. Virol. J. 2022, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Hodge, J.; Saaoud, F.; Wang, J.; Iwanowycz, S.; Wang, Y.; Hui, Y.; Evans, T.D.; Razani, B.; Fan, D. Transcriptional factor EB regulates macrophage polarization in the tumor microenvironment. Oncoimmunology 2017, 6, e1312042. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Q.; Guan, Y.; Sun, Y.; Wang, X.; Lively, K.; Wang, Y.; Luo, M.; Kim, J.A.; Murphy, E.A.; et al. Breast cancer cell-derived microRNA-155 suppresses tumor progression via enhancing immune cell recruitment and anti-tumor function. J. Clin. Invest. 2022, 132, e157248. [Google Scholar] [CrossRef]
- Al-Sabawy, H.B.; Rahawy, A.M.; Al-Mahmood, S.S. Standard techniques for formalin-fixed paraffin-embedded tissue: A pathologist’s perspective. Iraqi J. Vet. Sci. 2021, 35, 127–135. [Google Scholar] [CrossRef]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 2001, 39, 20.2.1–20.2.16. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Huang, J.S.; Yang, C.M.; Wang, J.S.; Liou, H.H.; Hsieh, I.C.; Li, G.C.; Huang, S.J.; Shu, C.W.; Fu, T.Y.; Lin, Y.C.; et al. Caspase-3 expression in tumorigenesis and prognosis of buccal mucosa squamous cell carcinoma. Oncotarget 2017, 8, 84237–84247. [Google Scholar] [CrossRef]
- Pu, X.; Storr, S.J.; Zhang, Y.; Rakha, E.A.; Green, A.R.; Ellis, I.O.; Martin, S.G. Caspase-3 and caspase-8 expression in breast cancer: Caspase-3 is associated with survival. Apoptosis Int. J. Program. Cell Death 2017, 22, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Qu, F.; Wang, G.; Wen, P.; Liu, X.; Zeng, X. Knowledge mapping of immunotherapy for breast cancer: A bibliometric analysis from 2013 to 2022. Hum. Vaccines Immunother. 2024, 20, 2335728. [Google Scholar] [CrossRef]
- Harris, M.A.; Savas, P.; Virassamy, B.; O’Malley, M.M.R.; Kay, J.; Mueller, S.N.; Mackay, L.K.; Salgado, R.; Loi, S. Towards targeting the breast cancer immune microenvironment. Nat. Rev. Cancer 2024, 24, 554–577. [Google Scholar] [CrossRef]
- Martin, N.T.; Roy, D.G.; Workenhe, S.T.; van den Wollenberg, D.J.M.; Hoeben, R.C.; Mossman, K.L.; Bell, J.C.; Bourgeois-Daigneault, M.C. Pre-surgical neoadjuvant oncolytic virotherapy confers protection against rechallenge in a murine model of breast cancer. Sci. Rep. 2019, 9, 1865. [Google Scholar] [CrossRef] [PubMed]
- Niavarani, S.R.; Lawson, C.; Boudaud, M.; Simard, C.; Tai, L.H. Oncolytic vesicular stomatitis virus-based cellular vaccine improves triple-negative breast cancer outcome by enhancing natural killer and CD8(+) T-cell functionality. J. Immunother. Cancer 2020, 8, e000465. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.J.; Sangsuwannukul, T.; van Vloten, J.; Evgin, L.; Kendall, B.; Tonne, J.; Thompson, J.; Metko, M.; Moore, M.; Chiriboga Yerovi, M.P.; et al. Expression of tumor antigens within an oncolytic virus enhances the anti-tumor T cell response. Nat. Commun. 2024, 15, 5442. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VSV-S Stock | TCID50/mL (4T1 Cells) | PFU/mL (HeLa Cells) |
|---|---|---|
| adapted to 4T1 cells | 7.92 ± 1.01 × 107 | 3.13 ± 0.2 × 107 |
| grown in HeLa cells | 1.32 ± 0.32 × 105 | 8 ± 0.5 × 106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, S.; Lyles, K.V.; Wang, Y.; Fan, D.; Luo, M. Enhancing the Efficacy of Breast Cancer Immunotherapy Using a Smac-Armed Oncolytic Virus. Cancers 2024, 16, 3248. https://doi.org/10.3390/cancers16193248
Tang S, Lyles KV, Wang Y, Fan D, Luo M. Enhancing the Efficacy of Breast Cancer Immunotherapy Using a Smac-Armed Oncolytic Virus. Cancers. 2024; 16(19):3248. https://doi.org/10.3390/cancers16193248
Chicago/Turabian StyleTang, Sijia, Kristin V. Lyles, Yuzhen Wang, Daping Fan, and Ming Luo. 2024. "Enhancing the Efficacy of Breast Cancer Immunotherapy Using a Smac-Armed Oncolytic Virus" Cancers 16, no. 19: 3248. https://doi.org/10.3390/cancers16193248
APA StyleTang, S., Lyles, K. V., Wang, Y., Fan, D., & Luo, M. (2024). Enhancing the Efficacy of Breast Cancer Immunotherapy Using a Smac-Armed Oncolytic Virus. Cancers, 16(19), 3248. https://doi.org/10.3390/cancers16193248