

CAR-T Cells in the Treatment of Nervous System Tumors



Abstract
Simple Summary
Abstract
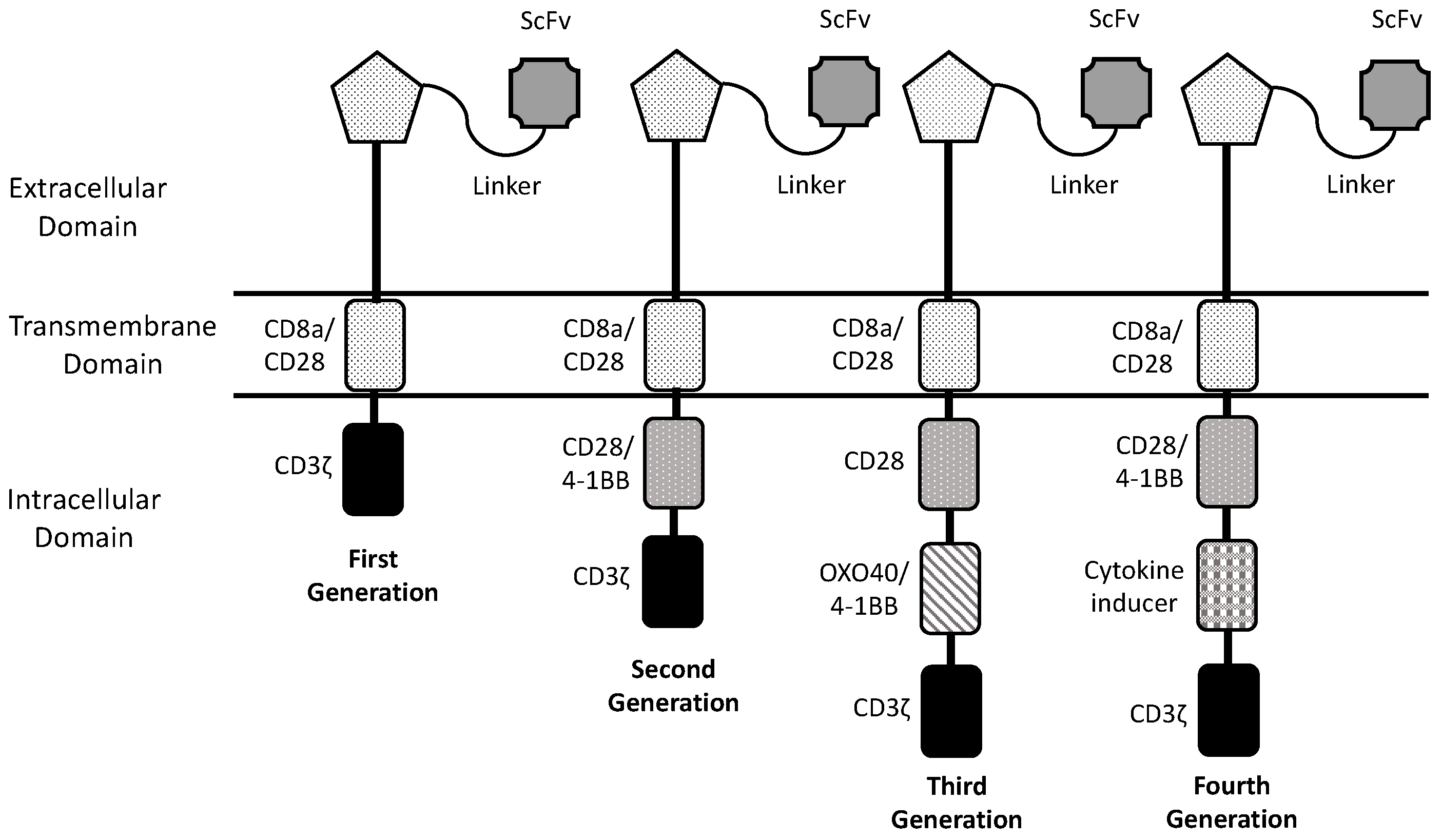
1. Introduction
2. Experimental and Clinical Studies of CAR-T Cell Therapy in Brain Tumors
2.1. HER-2
2.2. EGFRvIII
2.3. Interleukin-13 Receptor alpha2 (IL-13Rα2)
2.4. Disialogangloside (GD2)
2.5. B7-H3
3. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Roselli, E.; Faramand, R.; Davila, M.L. Insight into next-generation CAR therapeutics: Designing CAR T cells to improve clinical outcomes. J. Clin. Investig. 2021, 131, e142030. [Google Scholar] [CrossRef] [PubMed]
- Ceja, M.A.; Khericha, M.; Harris, C.M.; Puig-Saus, C.; Chen, Y.Y. CAR-T cell manufacturing: Major process parameters and next-generation strategies. J. Exp. Med. 2024, 221, e20230903. [Google Scholar] [CrossRef]
- Labanieh, L.; Mackall, C.I. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Savoldo, B.; Grover, N.; Dotti, G. CAR T cells for hematological malignancies. J. Clin. Investig. 2024, 134, e177160. [Google Scholar] [CrossRef]
- Khan, A.N.; Asija, S.; Pendhari, J.; Purwar, R. CAR-T cell therapy in hematological malignancies: Where are we now and are we heading for? Esur J. Haematol. 2024, 112, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Albelda, S.M. CAR T cell therapy for patients with solid tumors: Key lessons to learn and unlearn. Nat. Rev. Clin. Oncol. 2024, 21, 47–66. [Google Scholar] [CrossRef]
- Amoròs-Perez, B.; Rivas-Pardo, B.; del Moral, M.G.; Subiza, J.L.; Martinez-Naves, E. State of the art in CAR-T cell therapy for solid tumors: Is there a sweeter future? Cells 2024, 13, 725. [Google Scholar] [CrossRef]
- Schmidts, A.; Srivastava, A.A.; Ramaziyan, R.; Bailey, S.R.; Bouffard, A.A.; Cahill, D.P.; Carter, B.P.; Curey, W.T.; Dunn, G.P.; Frigault, M.J.; et al. Tandem chimeric antigen receptor (CAR) T cells targeting EGFRvIII and IL-13Rα2 are effective against heterogeneours glioblastoma. Neuro Oncol. Adv. 2022, 5, vdac185. [Google Scholar] [CrossRef]
- Cattaruzza, F.; Nareer, A.; To, M.; Hammond, M.; Koskin, C.; Liu, L.Y.; Yeung, V.P.; Remenfeldt, D.A.; Henkensiefken, A.; Fox, M.; et al. Precision-activated T-cell engagers targeting HER2 or EGFR and CD3 mitigate on target, off-tumor toxicity for immunotherapy in solid tumors. Nat. Cancer 2023, 4, 485–501. [Google Scholar] [CrossRef]
- Hosseinalizadeh, H.; Roudkenar, M.H.; Rouchandeh, A.M.; Kuwahone, Y.; Tomita, K.; Sato, T. Natural killer cell immunotherapy in glioblastoma. Discov. Oncol. 2022, 13, 113. [Google Scholar] [CrossRef]
- Xiong, Q.; Zhu, J.; Zhang, Y.; Deng, H. CAR-NK therapy for glioblastoma: What to de next? Front. Oncol. 2023, 13, 1192128. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Liu, J. Emerging roles of CAR-NK therapies in tumor immunotherapy. Cell Death Discov. 2024, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Uslu, U.; Castelli, S.; June, C.H. CAR T cell combination therapies to treat cancer. Cancer Cell 2024, 42, 1–7. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Y.; Liu, Z.; Liu, W.; Wu, H.; Whang, X.; Chen, Z. GD2 CAR-T cells in combination with Nivolumab exhibit enhanced antitumor efficacy. Transl. Oncol. 2023, 32, 101663. [Google Scholar] [CrossRef] [PubMed]
- Asija, S.; Chatterjee, A.; Goda, J.S.; Yadav, S.; Chekuri, S.; Purwar, R. Oncolytic immunotherapy for high-grade gliomas: A novel and an evolving therapeutic option. Front. Immunol. 2023, 14, 1118246. [Google Scholar] [CrossRef]
- Hamad, A.; Yusubalieva, G.M.; Baklaushev, V.P.; Chumakov, P.M.; Lipatova, A.V. Recent development in glioblastoma therapy: Oncolytic viruses and emerging features strategies. Viruses 2023, 15, 547. [Google Scholar] [CrossRef]
- Ma, R.; Lu, T.; Li, Z.; Teng, K.Y.; Mansour, A.G.; Yu, M.; Tian, L.; Xu, B.; Ma, S.; Zhang, J.; et al. An oncolytic virus expressingIL-15/IL-15Rα combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 2021, 81, 3635–3648. [Google Scholar] [CrossRef]
- Huang, J.; Zheng, M.; Zhang, Z.; Tang, X.; Chen, Y.; Peng, A.; Peng, X.; Tong, A.; Zhou, L. Interleukin-7-loaded oncolytic adenovirus improves CAR-T cell therapy for glioblastoma. Cancer Immunol. Immunother. 2021, 70, 2453–2465. [Google Scholar] [CrossRef]
- Del Baldo, G.; Del Bufalo, F.; Pinacchio, C.; Carai, A.; Quintarelli, C.; De Angelis, B.; Merli, P.; Cacchione, A.; Locatelli, F.; Mastronuzzi, A. The peculiar challenge of bringing CAR-T cells into the brain: Perspective in the clinical application to the treatment of pediatric central nervous system tumors. Front. Immunol. 2023, 14, 1142597. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Ronsley, R.; Choe, M.; Hewrison, C.; Breedt, M.; Barrios-Anderson, A.; Wein, A.; Brown, C.; Beebe, A.; Kong, A.; et al. Locoregional CAR T cells for children with NNS tumors: Clinical procedure and catether safety. Neoplasia 2023, 36, 100870. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hedge, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directe CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Trans. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Zheng, Z.; Xi, L.; et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Choi, B.D.; Gerstner, E.R.; Frigault, M.J.; Leick, M.B.; Mount, C.W.; Balaj, L.; Nikiforow, S.; Carter, B.S.; Curry, W.T.; Gallagher, K.; et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N. Engl. J. Med. 2024, 390, 1290–1298. [Google Scholar] [CrossRef]
- Bagley, S.J.; Binder, Z.A.; Lamrani, L.; Marinari, E.; Desai, A.S.; Nasrallah, M.L.; Maloney, E.; Brem, S.; Lustig, R.A.; Kurtz, G.; et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: A phase 1 trial. Nat. Cancer 2024, 5, 517–531. [Google Scholar] [CrossRef]
- Bagley, S.J.; Logun, M.; Fraietta, J.A.; Wang, X.; Desai, A.S.; Bagley, L.J.; Nabavizadeh, A.; Jarocha, D.; Martins, R.; Maloney, F.; et al. Intratechal bivalent CART cell targeting EGFR and IL13Rα2 in recurrent glioblastoma: Phase 1 trial interim results. Nat. Med. 2024, 30, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Hibbard, J.C.; Alidazeh, D.; Blanchard, M.S.; Natri, H.M.; Wang, D.; Ostberg, J.R.; Aguilar, B.; Wagner, J.R.; Paul, J.A.; et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: A phase 1 trial. Nat. Med. 2024, 30, 1001–1012. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Brasan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for K3K227M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mahdi, J.; Ramakrishna, S.; Patel, S.; Chinna Samy, H.; Yeom, K.; Schultz, L.; Barsan, V.; Richards, R.; Conjar, C.; et al. Major tumor regressions in H3K27M-mutated diffuse midline glioma (DNG) following sequential intravenous (IV) and intracerebroventricular (ICV) delivery of GD2-CAR T cells. Cancer Res. 2022, 81 (Suppl. 7), CT001. [Google Scholar] [CrossRef]
- Lin, F.Y.; Stuckert, A.; Tat, C.; White, M.; Ruggieri, L.; Zhang, H.; Mehta, B.; Lapteva, N.; Mei, Z.; Major, A.; et al. Phase I trial of GD2.CART cells augmented with constitutive interleukin-7 receptor for treatment of high-grade pediatric CNS tumors. J. Clin. Oncol. 2024; in press. [Google Scholar]
- Liu, Z.; Zhou, J.; Yang, X.; Liu, Y.; Zou, C.; Lv, W.; Chen, C.; Cheng, K.K.; Chen, T.; Chang, L.J.; et al. Safety and antitumor activity of GD2-specific 4SCAR-T cells in patients with glioblastoma. Mol. Cancer 2023, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, F.; De Angelis, B.; Cornana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART001 for relapsed or refractory high-risk neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Heczey, A.; Xu, X.; Courtney, A.N.; Tian, G.; Barragan, G.A.; Guo, L.; Amador, C.M.; Ghatwai, N.; Rathi, P.; Wood, M.S.; et al. Anti-GD2 CAR-NKT cells in relapsed or refractory neuroblastoma: Updated phase 1 trial interim results. Nat. Med. 2023, 29, 1379–1388. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Meyrs, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Che-Hsing, L.; Sharma, S.; Heczey, A.A.; Steffin, D.; Louis, C.U.; Grilley, B.J.; Thakkar, S.G.; Wu, M.; Wang, T.; Rooney, C.M.; et al. Eighteen-year survival after GD2-direcetd chimeric antigen receptor-modified immune effector cell treatment for neuroblastoma. Res. Sq. 2024, rs.3.ers-4232549. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Ronsley, R.; Huang, W.; Seidel, K.; Thomsen, A.; Gust, J.; Hauptman, J.; Choe, M.; Crotty, E.; Leary, S.; et al. Intravenous B7-H3 CAR T cells for diffuse intrinsic pontine glioma: Safety and efficacy report from the completed phase 1 trial BrainChild-03. Neuro Oncol. 2024, 26 (Suppl. 4), TRLS-01. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, R.; Chi, X.; Xian, N.; Chen, X.; Huang, N.; Zhang, Y.; Zhang, K.; Zhang, J.; Chen, L.; et al. Safety and efficacy of B7-H3 targeting CAR-T cell therapy for patients with recurrent GBM. J. Clin. Oncol. 2024, 142 (Suppl. 16), 2062. [Google Scholar] [CrossRef]
- Liu, G.; Ying, H.; Zeng, G.; Wheelr, C.J.; Black, K.L.; Yu, J.S. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004, 64, 4980–4986. [Google Scholar] [CrossRef]
- Ramezani, M.; Siami, S.; Rezaei, M.; Khazaei, S.; Sadeghi, M. An immunohistochemical study of HER2 expression in primary brain tumors. Biomedicine 2020, 10, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Mineo, J.F.; Bordron, A.; Baroncini, M.; Maurage, C.A.; Ramirez, C.; Siminsky, R.M.; Brethou, C.; Hieu, P.D. Low HER2-expressing glioblastomas are often secondary to anaplastic transformation of low-grade glioma. J. Neuroncol. 2007, 85, 281–287. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, S.V.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-sepcific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Therapy 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Kunkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targhets HER2+ breast cancer metastasis to the brain. Clin. Cancer Res. 2017, 24, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, L.; Li, W.; Gao, P.; Zhang, N. HER2-targeting CAR-T cells show highly efficient anti-tumor activity against glioblastoma both in vitro and in vivo. Genes Immun. 2024, in press. [CrossRef]
- Shabaneh, T.B.; Stevens, A.R.; Stull, S.M.; Shimp, K.R.; Seaton, B.W.; Gad, E.A.; Jaeger-Ruckstuhl, C.A.; Simon, S.; Koehne, A.L.; Price, J.P.; et al. Systemically administered low-affinity HER2 CAR T cells mediate entitumor efficacy without toxicity. J. Immunother. Cancer 2024, 12, e008566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. Erb2/HER2-specific NK cells for targeted therapy of glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef]
- Burger, M.C.; Forster, M.T.; Ramanks, A.; Strabheimer, F.; Macas, J.; Zeiner, P.S.; Steidl, E.; Herky, S.; Weber, K.J.; Sccupp, J.; et al. Intracranial injection of natural killer cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro Oncol. 2023, 25, 2058–2071. [Google Scholar] [CrossRef]
- Yeo, A.T.; Shah, R.; Aliazis, K.; Pal, R.; Xu, T.; Zhang, P.; Rawal, S.; Rose, C.M.; Varn, F.S.; Appleman, V.A.; et al. Driver mutations dictate the immunologic landscape and response to checkpoint immunotherapy of glioblastoma. Cancer Immunol. Res. 2023, 11, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEWs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Tang, O.Y.; Tian, L.; Yoder, T.; Xu, R.; Kulikovskaya, I.; Gupta, M.; Malenhorst, J.J.; Lacey, S.F.; O’Rourke, D.M.; Binder, Z.A. PD1 expression in EGFRvIII-directed CAR T cell infusion product for glioblastoma is associated with clinical response. Front. Immunol. 2022, 13, 872756. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.C.; Iliopoulos, M.; Watson, K.A.; Arcucci, V.; Go, M.; Hughes-Parry, H.E.; Smith, P.; Call, M.J.; Cross, R.S.; Jenkins, M.R. Human EGFRvIII chimeric antigen receptor T cells demonstrate favorable safety profile and curative resonses in orthoptic glioblastoma. Clin. Transl. Immunol. 2023, 12, e1440. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.H.; Plautz, G.E.; Puri, R.K. Interleukin-13 receptor α chain: A novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2020, 60, 1168–1172. [Google Scholar]
- Knudson, K.M.; Hwang, S.; McCann, M.S.; Joshi, B.H.; Husain, S.R. Recent advances in IL-13Rα2-directed cancer immunotherapy. Front. Immunol. 2022, 13, 878365. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4070. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Gholamin, S.; Maka, M.; Aftabizadeh, M.; Xang, W.; Pecoraro, J.R.; Jeppson, J.D.; Wang, D.; Aguilar, B.; et al. IFNγ is critical for CAR T cell-mediated myeloid activation and induction of endogenous immunity. Cancer Discov. 2021, 11, 2248–2265. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro-Oncology 2022, 24, 1318–1330. [Google Scholar] [CrossRef]
- Stern, L.A.; Gholamin, S.; Moraga, I.; Yang, X.; Saravanakumar, S.; Cohen, J.R.; Cohen, J.R.; Starr, R.; Aguilar, B.; Salvary, V.; et al. Engineered IL13 variants direct specificity of IL13Rα2-targeted CAR T cell therapy. Proc. Natl. Acad. Sci. USA 2022, 119, e2112006119. [Google Scholar] [CrossRef]
- Kim, K.; Gwak, H.S.; Han, N.; Hong, E.K.; Choi, B.K.; Lee, S.; Choi, S.; Park, Y.H.; Seok, J.H.; Jeon, Y.; et al. Chimeric antigen receptor T cells with modified interleukin-13 preferentially recognize IL13Rα2 and suppress malignant glioma: A preclinical study. Fron Immunol. 2021, 12, 715000. [Google Scholar] [CrossRef] [PubMed]
- Leland, P.; Degheidy, H.; Lea, A.; Bauer, S.R.; Puri, R.K.; Joshi, B.H. Identification and characterization of novel CAR-T cells to target IL13Rα2 positive human glioma in vitro and in vivo. Clin. Transl. Med. 2024, 14, e1664. [Google Scholar] [CrossRef]
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M. Glioma. Nat. Rev. Dis. Prim. 2024, 10, 33. [Google Scholar]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rierberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-H27M+ diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- Monje, M.; Mahdi, J.; Majzner, R.; Yeom, K.; Schultz, L.M.; Richards, R.M.; Barsan, V.; Song, K.W.; Kamens, J.; Baggott, K.; et al. Sequential intravenous and intracerebroventricular GD2-CAR T-cell tharapy for H3K27M-mutated diffuse midline gliomas. medRxIV, 2024, in press.
- Ramakrishna, S.; Good, Z.; Desai, M.; Zamler, D.; Mancusi, R.; Mahdi, J.; Majzner, R.; Schulz, L.; Richards, R.; Kamen, J.; et al. Immune signatures of GD2 CAR T cell activity in H3K27M+ diffuse midline glioma patients. Cancer Res. 2023, 23 (Suppl. 7), 959. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Oarihar, R.; et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Chiavelli, C.; Prapa, M.; Rovesti, C.; Slingardi, M.; Neri, G.; Pugliese, G.; Trudu, L.; Dall’Ora, M.; Golinelli, G.; Griseni, G.; et al. Autologous anti-GD2 CART cells efficiently target primay human glioblastoma. NPJ Precis. Oncol. 2024, 8, 26. [Google Scholar] [CrossRef]
- Gargett, T.; Ebert, L.M.; Truong, N.; Kollis, P.M.; Sedivakova, K.; Yu, W.; Yeo, E.; Wittver, N.L.; Gliddon, B.L.; Tea, M.N.; et al. GD2-targheting CAR-T cells enhanced by transgenic IL-15 expression are an effective and clinically feasible therapy for glioblastoma. Immunother. Cancer 2022, 10, e005187. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukemi-2, and isotretinoin for neuroblastoma. N. Eng. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.K.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in pateints with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J. Cancer Res. Clin. Oncol. 2022, 148, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatway, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: An interim analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef]
- Paret, C.; Ustjanzew, A.; Ersali, S.; Seidmann, L.; Jennemann, R.; Ziegler, N.; El Malki, K.; Russo, A.; Wingerter, A.; Ortmuller, F.; et al. GHD2 expression in medulloblastoma and neuroblastoma for personalized immunotherapy: A matter of subtype. Cancers 2022, 14, 6059. [Google Scholar] [CrossRef]
- Ciccone, R.; Quintarelli, C.; Camera, A.; Pezzella, M.; Caruso, S.; Manni, S.; Ottaviani, A.; Guercio, M.; Del Bufalo, F.; Quadraccia, M.C.; et al. GD2-targeting CAR T-cell therapy for patients with GD2+ medulloblastoma. Clin. Cancer Res. 2024, 30, 2545–2557. [Google Scholar] [CrossRef]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Odé, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumots: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro Oncol. 2021, 23, 991–1101. [Google Scholar] [CrossRef]
- Nehama, D.; Di Ianni, N.; Musio, S.; Du, H.; Patané, M.; Pollo, B.; Finocchiaro, G.; Park, J.; Dunn, D.E.; Edwards, D.S.; et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neuropspheres. E BioMed. 2019, 47, 33–43. [Google Scholar]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol. Ther. Oncolytics 2019, 14, 279–285. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Wilson, A.L.; Huang, W.; Seidel, K.; Brown, C.; Gustafson, J.A.; Yokoyama, J.K.; Johnson, A.J.; Baxter, B.A.; Koning, R.W.; et al. Intraventricular b7-H3 CART cells for diffuse intrinsic pontine glioma: Preliminary first-in-human bioactivity and safety. Cancer Discov. 2023, 13, 114–131. [Google Scholar] [CrossRef]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregional administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, Y.; Huang, J.; Zhang, Z.; Liu, F.; Xu, J.; Guo, G.; Wang, W.; Tong, A.; Zhou, L. Administration of B7-H3-targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transd Targetes Ther. 2021, 6, 125. [Google Scholar] [CrossRef]
- Shang, X. T-MAXIMUM Pharmaceutical. An exploratory clinical trial on intra-lumbar injection of B7-H3-specific allogeneic universal CAR-T cells in patients with recurrent high-grade gliomas. J. Clin. Oncol. 2023, 141 (Suppl. 16), 2043. [Google Scholar]
- Mainprize, T.; Lipman, N.; Huang, Y.; Mang, Y.; Bethuna, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-brain barrier opening in primary brain tumors with non-invasive MR-guided focused ultrasound: A clinical safety and feasibility study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Gandhi, D.; Melhem, E.R.; Frenkel, V. MTI guided focused ultrasound-mediated delivery of therapeutic cells to the brain: A review of the state-of-art methodology and future applications. Front. Neurol. 2021, 12, 669449. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]

{kind=link}
| Author | Trial and Phase | Target Antigen | Dose and Route of Administration | Number of Patients (Age) | Clinical Results | Adverse Events |
|---|---|---|---|---|---|---|
| Ahmed et al. 2017 [23] | NCT 01109059 Phase I | HER2 | 1 × 106 to 1 × 108 Intravenous without prior lymphodepletion | Pediatric 7 (10–17 yr) Adult 10 (30–69 yr) Recurrent GBM | 1 partial response; 7 stable disease; 8 disease progression In adult pts: mOS 9.4 mo | No severe adverse events related to treatment |
| O’Rourke et al. 2017 [24] | NCT 02209376 Phase I | EGFRvIII | 1.75 × 108 to 5 × 108 Intravenous after lymphodepletion | 11 Adult recurrent GBM | 1 SD; 10 no response Evidence of CAR-T cell trafficking to the tumor Reduction in target antigen | No off-tumor toxicity No CRS |
| Goff et al. 2019 [25] | NCT 01454596 Phase I | EGFRvIII | 4 pts 1 × 107; 3 pts 1 × 108 5 pts 1 × 109; 5 pts 1 × 1010 Intravenous after lymphodepletion | 18 Adult recurrent GBM | No objective response mPFS: 1.2 mo mOS: 6.9 mo | Severe adverse events and dose-limiting toxicities in the group at 1 × 1010 CAR-T cells |
| Choi et al. 2024 [26] | NCT 05660369 Phase I/Pilot | EGFRvIII | Patient 1: 2 infusions of 10 × 106 CAR-T cells Patients 2 and 3: 1 infusion of 10×106 CAR-T cells | 3 Adult recurrent GBM | All patients displayed rapid and dramatic radiographic tumor regression. In 2/3 pts, this response was transient | No dose-limiting toxicity No adverse events greater than 3 |
| Bagley et al. 2024 [27] | NCT 03726515 Phase I | EGFRvIII | 4.65 × 107 to 2 × 108 1–3 cycles of CAR-T cells 1 cycle of Pembrolizumab | 7 (56–76 yr) Adult recurrent GBM | No objective responses mPFS: 5.2 mo mOD: 11.8 mo | No dose-limiting toxicity |
| Bagley et al. 2024 [28] | NCT 05168423 Phase I | EGFRvIII—IL-13Rα2 (bicistronic lentiviral vector) | 3 pts (1 × 107 cells/m2) 3 pts (2.5 × 107 cells/m2) Intrathecal | 6 (33–71 yr) Adult recurrent GBM | Significant reduction in tumor size at MRI, but none with radiographic objective response | Low-grade CRS Early moderate–severe neurotoxicity |
| Brown et al. 2024 [29] | NCT 02208362 Phase I | IL-13Rα2 | 57 pts From 2 to 200 × 106 IL13-CAR-T cells ICT or ICV or ICT and ICV infusions | 57 pts (16–71 yr) 41 pts GBM 2 pts DMG 7 pts gr4 Astrocytoma 7 pts gr3 Glioma | SD: 50% PR: 2 pts CR: 2 pts mOS: 7.7 mo GBM(all) mOS: 10.2 mo GBM(ICT and ICV) | No dose-limiting toxicity 2 pts grade3 neurologic events |
| Majzner et al. 2022 [30] | NCT 04196413 Phase I | GD2 | IV (1 × 106 cells/Kg) Optional ICV infusions in responding patients | 4 (5–25 yr) DMG H3K2M7-mutated | 75% patients clinical and radiographic response | Manageable toxicity On target, off-tumor toxicity not observed |
| Majzner et al. 2022 [31] | NCT 04196413 Phase I | GD2 | 4 pts IV (1 × 106 cells/Kg) 9 pts IV (3 × 106 cells/Kg) | 13 (2–30 yr) DMG H3K2M7-mutated | 90% pts clinical and radiographic response. 2 pts with complete response. | Grade 4 CRS in 3 pts at 3 × 106 cells/Kg. Transient tumor inflammation neurotoxicity |
| Lin et al. 2024 [32] | NCT 04099797 | GD2—IL-7R | 3 pts: IV GD2-CAR-T cells 1 × 107 cells/m2 Ppts: IV CR7-GD2-CAR-T cells 1 × 107 cells/m2 | 11 (4–18 yr) 8 DMG H3K27M-mut 2 recurrent MB 1 ATRT (atypical teratoid rhabdoid tumor) | 3 pts: GD2-CAR-T group: PD 8 pts CR7-GD2-CAR-T group: 2 pts PR; 6 pts SD | CRS grade 1 75% Tumor inflammation-associated toxicity grade 1 88% |
| Liu et al. 2023 [33] | NCT 03170141 Phase I | GD2 | 8 pts IV: 3 × 107–2.1 × 108 3 pts IC: 2.6 × 106–6.4 × 106 4SCAR-T cells | 8 (3–63 yr) Recurrent GBM | 3 pts: PD; 4 pts: PR; 1 pt: SD mOS from diagnosis: 19.7 mo; mOS from infusions: 11.5 mo | Grade 2 or 3 neurologic events: 2 pts |
| Del Bufalo et al. 2023 [34] | NCT 03373097 Phase I/II | GD2 | 17 pts single infusion; 11 pts multiple infusions 3 × 106, 6 × 106, 10 × 106 GD2-CART01 cells/Kg | 27 (2.7–18.6 yr) Refractory/relapsed Neuroblastoma | CR: 33%; PR: 30%; SD: 19%; NR: 19% At 3 yrs: OS 67% LDB vs. 0% HDB; EFS 58% LDB vs. 0% HDB | No dose-limiting toxicities Grade 1–2 CRS: 70% Grade 3 CRS: 4% Neurologic toxicity grade 1–2: 22% |
| Heczey et al. 2023 [35] | NCT 03294954 Phase I | GD2 | 3 pts 3 × 106, 3 pts 1 × 107, 3pta 3 × 107, 3 pts 1 × 108 GD2-CAR-NKT cells/m2 8 pts single infusion, 4 pts double infusion | 12 (2–12 yr) Refractory/relapsed Neuroblastoma | After first infusion: PR: 3pta; SD: 4 pts; PD: 5 pts After second infusion: CR: 1 pt; PR: 1 pt; PD: 2 pts. | |
| Pule et al. 2008 Louise et al. 2011 Che-Hsing et al. 2024 [36,37,38] | NCT 00085930 Phase I | GD2 | 19 pts IV infusion 1.2 × 107 cells/m2 5 × 107 cells/m2 3.1 × 108 cells/m2 | 19 pts with R/R Neuroblastoma 11 with active disease 8 in remission | After a follow-up of 8–14 yrs survived: 5/8 in remission 2/11 with active disease | No dose-limiting toxicities |
| Brown et al. 2024 [29] | NCT 02208362 Phase I | IL-13Rα2 | From 2 × 106 to 200 × 106 IL13-CAR-T cells ICT or ICV or ICT and ICV infusions | 57 pts 41 pts GBM 2pta DMG 7 pts Astrocytoma 7 pts Glioma | SD: 50% PR: 2 pts CR: 2 pts mOS: 7.7 mo (GBM); 10.2 mo (GBM ICT and ICV) | No dose-limiting toxicity 2 pts with grade 3 neurologic events |
| Vitanza et al. 2024 [39] | NCT 04185038 Phase I Brain Child 03- Arm C | B7-H3 | ICV infusions of 10 × 107 B7-H3 CAR-T cells Multiple infusions (median 7) | 21 pediatric DIPG | mOS for pts after progression: 9.7 mo, before progression: 16.9 mo 3 pts alive 3 yrs from diagnosis | No dose-limiting toxicity Common neurologic events: headache, nausea, vomiting, fever |
| Zhang et al. 2024 [40] | NCT 05241392 Phase I | B7-H3 | ICT or ICV infusions of B7-H3 CAR-T cells 3 × 107 cells (3 pts) 6 × 107 cells (4 pts) 15 × 107 cells (6 pts) | 13 adult R/R GBM patients | At 12 mo: 83% OS mOS: 20.3 mo 1 pt: PR 1 pt: CR | No dose-limiting toxicity 2 pts neurologic events gr.3 some pts CRS gr.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Castelli, G.; Pelosi, E. CAR-T Cells in the Treatment of Nervous System Tumors. Cancers 2024, 16, 2913. https://doi.org/10.3390/cancers16162913
Testa U, Castelli G, Pelosi E. CAR-T Cells in the Treatment of Nervous System Tumors. Cancers. 2024; 16(16):2913. https://doi.org/10.3390/cancers16162913
Chicago/Turabian StyleTesta, Ugo, Germana Castelli, and Elvira Pelosi. 2024. "CAR-T Cells in the Treatment of Nervous System Tumors" Cancers 16, no. 16: 2913. https://doi.org/10.3390/cancers16162913
APA StyleTesta, U., Castelli, G., & Pelosi, E. (2024). CAR-T Cells in the Treatment of Nervous System Tumors. Cancers, 16(16), 2913. https://doi.org/10.3390/cancers16162913