Somatic Mutation Profile as a Predictor of Treatment Response and Survival in Unresectable Pancreatic Ductal Adenocarcinoma Treated with FOLFIRINOX and Gemcitabine Nab-Paclitaxel

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Characteristics of the Patient Population
3.2. Characteristics of the Pathology Samples
3.3. Overall Survival Analysis
3.4. Progression-Free Survival Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| ECOG | Eastern Cooperative Oncology Group |
| NGS | Next-generation sequencing |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| TMB | Tumor mutation burden |
| MSI-H | Microsatellite instability-high |
| OS | Overall survival |
| PFS | Progression-free survival |
| CC | Cell cycle |
| DDR | DNA damage repair |
| PI3K | Phosphoinositide 3-kinase |
| KIT | KIT proto-oncogene |
| NOTCH | Neurogenic locus notch homolog protein |
| ALK | Anaplastic lymphoma kinase |
| ERBB2 | Erb-B2 receptor tyrosine kinase 2 |
| TP53 | Tumor protein p53 |
| TGFB | Transforming growth factor beta |
| PDGFR | Platelet-derived growth factor receptor |
| RAS | Rat sarcoma |
| FTL3 | Fms-like tyrosine kinase 3 |
| FGFR | Fibroblast growth factor receptor |
| WNT | Wnt signaling pathway |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Gene Mutations Included in the Pathway [63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96] | |||||
|---|---|---|---|---|---|---|
| Cell Cycle mutation | ||||||
| CCND1 | MYC | BCOR | BRD4 | RAD54L | AURKA | |
| CCND2 | STK11 | CIC | BTG2 | PRKCI | AURKB | |
| CCND3 | TERT | DAXX | BCL2L2 | TOP2A | XPO1 | |
| CCNE1 | BCL2L1 | ERG | BCL6 | TERC | PTEN | |
| CDK6 | PPP2R2A | ESR1 | JUN | TET2 | ALK | |
| CDKN2A | PPP2R1A | EWSR1 | IDH1 | HNF1A | ||
| CDKN2B | CASP8 | EZH2 | IDH2 | AR | ||
| RB1 | FAS | CDH1 | HDAC1 | PARP1 | ||
| FAT1 | APC | CDKN2C | GNA13 | ATR | ||
| ATRX | MDM2 | CDKN1B | MPL | RANBP2 | ||
| DNA Damage repair mutations | ||||||
| BLM | XRCC2 | WHSC1 | JUN | POLD1 | KRAS | |
| BRCA2 | FAT3 | CUL4A | H3F3A | RAD51L3 | ALK | |
| MLH1 | BRIP1 | FANCL | GEN1 | TP53BP1 | ABL1 | |
| MSH3 | FANCA | FANCM | FUBP1 | CHEK2 | ||
| PARP2 | FANCC | ERCC4 | EMSY | MSH2 | ||
| PMS2 | FANCD2 | CHEK1 | MPL | AR | ||
| POLE | FANCG | CDH1 | MRE11A | PARP1 | ||
| PRKDC | PALB2 | BRCA1 | MSH6 | ATR | ||
| RAD51C | ATM | BARD1 | MUTYH | SF3B1 | ||
| RAD52 | APC | BCL2L2 | NBN | FH | ||
| NOTCH signaling pathway | ||||||
| CREBBP | GATA6 | |||||
| EP300 | EGFR | |||||
| NCOR1 | CBFB | |||||
| NOTCH1 | FBXW7 | |||||
| NOTCH2 | CDK8 | |||||
| NOTCH3 | IKZF1 | |||||
| NOTCH4 | FLT4 | |||||
| SPEN | FH | |||||
| JAK2 | MDM2 | |||||
| RBM10 | BCOR | |||||
| PI3K/AKT signaling pathway | ||||||
| AKT2 | NTRK2 | IGF1R | ||||
| INPP4B | PDK1 | EZH2 | ||||
| MTOR | PHLPP2 | FGF4 | ||||
| PIK3CA | PIK3C2B | BCL2L2 | ||||
| PIK3R1 | PIK3C2G | RPTOR | ||||
| PPP2R1A | PIK3CB | AKT3 | ||||
| PTEN | PPARG | ERBB2 | ||||
| STK11 | PREX2 | ERBB3 | ||||
| TSC2 | RICTOR | ERBB4 | ||||
| MET | TYRO3 | |||||
| KIT signaling pathway | ||||||
| KIT | ||||||
| ROS1 | ||||||
| FGFR3 | ||||||
| MTOR | ||||||
| SRC | ||||||
| LYN | ||||||
| CBL | ||||||
| FTL3 | ||||||
| RAS/RAF/MAPK signaling pathway | ||||||
| BRAF | MAP2K1 | JUN | FGFR3 | |||
| KRAS | MAP2K4 | QKI | ||||
| MAP2K2 | MAP3K1 | RAF1 | ||||
| MET | MAP3K13 | PTPN11 | ||||
| NF1 | MAPK1 | ARAF | ||||
| NTRK1 | PPP2R1A | CBL | ||||
| NTRK2 | IGF1R | RET | ||||
| NTRK3 | DDR2 | ERBB2 | ||||
| XPO1 | CRKL | ERBB3 | ||||
| FGF4 | HRAS | ERBB4 | ||||
| TGF-beta Receptor | ||||||
| ACVR1B | AR | |||||
| SMAD2 | PARP1 | |||||
| SMAD3 | XPO1 | |||||
| SMAD4 | GATA6 | |||||
| TGFRB2 | ||||||
| APC | ||||||
| MYC | ||||||
| CDK8 | ||||||
| JUN | ||||||
| MEN1 | ||||||
| TP53 Activity Alteration | ||||||
| TP53 | BCL6 | AURKA | ||||
| ATM | MDM4 | AURKB | ||||
| CDK12 | PRDM1 | |||||
| MDM2 | PRKCI | |||||
| BCOR | RAD51L3 | |||||
| EP300 | SGK1 | |||||
| CIC | TAF1 | |||||
| DAXX | RPTOR | |||||
| CHEK12 | CHEK2 | |||||
| BCL2L2 | ATR | |||||
| WNT signaling pathway | ||||||
| APC | TRRAP | |||||
| AXIN1 | WT1 | |||||
| CHD4 | FAM123B | |||||
| CTNNB1 | XPO1 | |||||
| RNF43 | BCORL1 | |||||
| MYC | MED12 | |||||
| PPP2R1A | ||||||
| CDC73 | ||||||
| GSK3B | ||||||
| TNKS | ||||||
| ERBB2 signaling pathway | ||||||
| NF2 | ||||||
| EGFR | ||||||
| ERBB2 | ||||||
| ERBB3 | ||||||
| ERBB4 | ||||||
| CBL | ||||||
| BRAF | ||||||
| PDGFR signaling pathway | ||||||
| PDGFRA | ||||||
| PDGFRB | ||||||
| KDR | ||||||
| EGFR | ||||||
| IDH1 | ||||||
| LRP1B | ||||||
| AR | ||||||
| FH | ||||||
| KIT | ||||||
| NF1 | ||||||
| FGFR signaling pathway | ||||||
| FGFR1 | FGF6 | |||||
| FGFR3 | BCL2L2 | |||||
| MTOR | SOX2 | |||||
| PPP2R1A | FGFR2 | |||||
| PTEN | CBL | |||||
| ERBB3 | FH | |||||
| FGF19 | ||||||
| FGF23 | ||||||
| FGF3 | ||||||
| FGF4 | ||||||
| FTL3 signaling pathway | ||||||
| FLT3 | ||||||
| CBL | ||||||
| PIM1 | ||||||
| RAF1 | ||||||
| SRC | ||||||
| SYK | ||||||
| FH | ||||||
| PDGFRB | ||||||
| ALK signaling pathway | ||||||
| ALK | ||||||
| STAT3 | ||||||
| LTK | ||||||
| EGFR | ||||||
| ROS1 | ||||||
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49, Erratum in CA Cancer J. Clin. 2024, 74, 203. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Cronin, K.A.; Scott, S.; Firth, A.U.; Sung, H.; Henley, S.J.; Sherman, R.L.; Siegel, R.L.; Anderson, R.N.; Kohler, B.A.; Benard, V.B.; et al. Annual report to the nation on the status of cancer, part 1: National cancer statistics. Cancer 2022, 128, 4251–4284. [Google Scholar] [CrossRef] [PubMed]
- Maloney, S.; Itchins, M.; Arena, J.; Sahni, S.; Howell, V.M.; Hayes, S.A.; Gill, A.J.; Clarke, S.J.; Samra, J.; Mittal, A.; et al. Optimal Upfront Treatment in Surgically Resectable Pancreatic Cancer Candidates: A High-Volume Center Retrospective Analysis. J. Clin. Med. 2021, 10, 2700. [Google Scholar] [CrossRef]
- Chhoda, A.; Vodusek, Z.; Wattamwar, K.; Mukherjee, E.; Gunderson, C.; Grimshaw, A.; Sharma, A.; Ahuja, N.; Kastrinos, F.; Farrell, J.J. Late-stage pancreatic cancer detected during high-risk individual surveillance: A systematic review and meta-analysis. Gastroenterology 2022, 162, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 2021, 11, 688377. [Google Scholar] [CrossRef]
- De Dosso, S.; Siebenhüner, A.R.; Winder, T.; Meisel, A.; Fritsch, R.; Astaras, C.; Szturz, P.; Borner, M. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 2021, 96, 102180. [Google Scholar] [CrossRef] [PubMed]
- Damanakis, A.I.; Gebauer, F.; Bruns, C.J. Molekulare Prädiktoren für den Krankheitsverlauf und die individualisierte Therapie beim Pankreaskarzinom. Chirurg 2020, 91, 642–649. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures with Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Shen, G.Q.; Aleassa, E.M.; Walsh, R.M.; Morris-Stiff, G. Next-Generation Sequencing in Pancreatic Cancer. Pancreas 2019, 48, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Qiu, X.; Lu, C.; Zhu, Y.; Kong, W.; Xu, M.; Jiang, W.; Wang, Y.; Li, Y.; Zhang, W.; et al. Molecular Landscape and Prognostic Biomarker Analysis of Advanced Pancreatic Cancer and Predictors of Treatment Efficacy of AG Chemotherapy. Front. Oncol. 2022, 12, 844527. [Google Scholar] [CrossRef]
- Shaya, J.; Kato, S.; Adashek, J.J.; Patel, H.; Fanta, P.T.; Botta, G.P.; Sicklick, J.K.; Kurzrock, R. Personalized matched targeted therapy in advanced pancreatic cancer: A pilot cohort analysis. NPJ Genom. Med. 2023, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, K.Y.; Bentrem, D.J.; Ko, C.Y.; Ritchey, J.; Stewart, A.K.; Winchester, D.P.; Talamonti, M.S.; Jennings, G.; Bhayani, N.H.; Stamos, M.J. Validation of the 6th Edition AJCC Pancreatic Cancer Staging System: Report from the National Cancer Database. Cancer 2007, 110, 738–744. [Google Scholar] [CrossRef]
- Referenced with Permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Pancreatic Adenocarcinoma V.1.2022. National Comprehensive Cancer Network, Inc. 2022. Available online: www.NCCN.org (accessed on 6 June 2023).
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Milbury, C.A.; Creeden, J.; Yip, W.-K.; Smith, D.L.; Pattani, V.; Maxwell, K.; Sawchyn, B.; Gjoerup, O.; Meng, W.; Skoletsky, J.; et al. Clinical and analytical validation of FoundationOne® CDx, a comprehensive genomic profiling assay for solid tumors. PLoS ONE 2022, 17, e0264138. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T.; Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; et al. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019, 95, 103208. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 6 June 2023).
- Therneau, T. A Package for Survival Analysis in R. R Package Version 3.4-0. Available online: https://CRAN.R-project.org/package=survival (accessed on 6 June 2023).
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Ghidini, M.; Lampis, A.; Mirchev, M.B.; Okuducu, A.F.; Ratti, M.; Valeri, N.; Hahne, J.C. Immune-Based Therapies and the Role of Microsatellite Instability in Pancreatic Cancer. Genes 2020, 12, 33. [Google Scholar] [CrossRef]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef]
- Rosenberg, A.; Mahalingam, D. Immunotherapy in pancreatic adenocarcinoma-overcoming barriers to response. J. Gastrointest. Oncol. 2018, 9, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Mullendore, M.E.; Koorstra, J.B.; Li, Y.M.; Offerhaus, G.J.; Fan, X.; Henderson, C.M.; Matsui, W.; Eberhart, C.G.; Maitra, A.; Feldmann, G.; et al. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 2291–2301. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [PubMed]
- Graziani, I.; Eliasz, S.; De Marco, M.A.; Chen, Y.; Pass, H.I.; De May, R.M.; Strack, P.R.; Miele, L.; Bocchetta, M. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 2008, 68, 9678–9685. [Google Scholar] [CrossRef]
- De La, O.J.P.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef]
- Misiorek, J.O.; Przybyszewska-Podstawka, A.; Kałafut, J.; Paziewska, B.; Rolle, K.; Rivero-Müller, A.; Nees, M. Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis. Cells 2021, 10, 94. [Google Scholar] [CrossRef]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Böttinger, E.P. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef]
- Lombardo, Y.; Faronato, M.; Filipovic, A.; Vircillo, V.; Magnani, L.; Coombes, R.C. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells. Breast Cancer Res. 2014, 16, R62. [Google Scholar] [CrossRef]
- Jia, Y.; Xie, J. Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes Dis. 2015, 2, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Li, J.; Sun, H.; Liu, S.; Cui, Y.; Li, F. HES 1 is essential for chemoresistance induced by stellate cells and is associated with poor prognosis in pancreatic cancer. Oncol. Rep. 2015, 33, 1883–1889. [Google Scholar] [CrossRef]
- Chanrion, M.; Kuperstein, I.; Barrière, C.; El Marjou, F.; Cohen, D.; Vignjevic, D.; Stimmer, L.; Paul-Gilloteaux, P.; Bièche, I.; Tavares Sdos, R.; et al. Concomitant Notch activation and p53 deletion trigger epithelial-to-mesenchymal transition and metastasis in mouse gut. Nat. Commun. 2014, 5, 5005. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat. Rev. Cancer 2009, 9, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Li, X.; Feng, G.; Hu, S.; Bai, Y. The Impact of NOTCH Pathway Alteration on Tumor Microenvironment and Clinical Survival of Immune Checkpoint Inhibitors in NSCLC. Front. Immunol. 2021, 12, 638763. [Google Scholar] [CrossRef] [PubMed]
- Takam Kamga, P.; Collo, G.D.; Resci, F.; Bazzoni, R.; Mercuri, A.; Quaglia, F.M.; Tanasi, I.; Delfino, P.; Visco, C.; Bonifacio, M.; et al. Notch Signaling Molecules as Prognostic Biomarkers for Acute Myeloid Leukemia. Cancers 2019, 11, 1958. [Google Scholar] [CrossRef] [PubMed]
- Maraver, A.; Fernández-Marcos, P.J.; Herranz, D.; Muñoz-Martin, M.; Gomez-Lopez, G.; Cañamero, M.; Mulero, F.; Megías, D.; Sanchez-Carbayo, M.; Shen, J.; et al. Therapeutic effect of γ-secretase inhibition in KrasG12V-driven non-small cell lung carcinoma by derepression of DUSP1 and inhibition of ERK. Cancer Cell 2012, 22, 222–234. [Google Scholar] [CrossRef]
- Massard, C.; Azaro, A.; Soria, J.C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.K.R.; et al. First-in-human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Aste-Amézaga, M.; Zhang, N.; Lineberger, J.E.; Arnold, B.A.; Toner, T.J.; Gu, M.; Huang, L.; Vitelli, S.; Vo, K.T.; Haytko, P.; et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS ONE 2010, 5, e9094. [Google Scholar] [CrossRef]
- Hu, Z.I.; Bendell, J.C.; Bullock, A.; LoConte, N.K.; Hatoum, H.; Ritch, P.; Hool, H.; Leach, J.W.; Sanchez, J.; Sohal, D.P.S.; et al. A randomized phase II trial of nab-paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019, 8, 5148–5157. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dias-Santagata, D.; Selim, M.A.; Su, Y.; Peng, Y.; Vollmer, R.; Chłopik, A.; Tell-Marti, G.; Paral, K.M.; Shalin, S.C.; Shea, C.R.; et al. KIT mutations and CD117 overexpression are markers of better progression-free survival in vulvar melanomas. Br. J. Dermatol. 2017, 177, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Chi, S.; Minami, Y.; Fukushima, K.; Shibayama, H.; Hosono, N.; Yamauchi, T.; Morishita, T.; Kondo, T.; Yanada, M.; et al. Mutated KIT Tyrosine Kinase as a Novel Molecular Target in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 4694. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Jiang, Z.; Zhang, J.; Li, Z.; Liu, Y.; Wang, D. A meta-analysis of prognostic value of KIT mutation status in gastrointestinal stromal tumors. OncoTargets Ther. 2016, 9, 3387–3398. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Qian, Y.; Wang, Y.; Fang, F.; Wu, G. Prognostic value of Beclin 1, EGFR and ALK in non-squamous non-small cell lung cancer. Discov. Oncol. 2022, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Devasthali, V.; Cheng, J.H.; Castillo, J.; Metcalfe, C.; Clermont, A.C.; Otter, D.D.; Chan, E.; Bou-Reslan, H.; Cao, T.; et al. Modeling targeted inhibition of MEK and PI3 kinase in human pancreatic cancer. Mol. Cancer Ther. 2015, 14, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.C.; Hannan, L.M.; Zhen, D.B.; Coveler, A.L.; King, G.; Cohen, S.A.; Harris, W.P.; Shankaran, V.; Wong, K.M.; Green, S.; et al. KRAS Mutation Variants and Co-occurring PI3K Pathway Alterations Impact Survival for Patients with Pancreatic Ductal Adenocarcinomas. Oncologist 2022, 27, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Jiang, Z.; Yang, Y.; Zhang, C.; Jiang, M.; Zhu, J.; Xu, L.; Xie, A.; Yan, M.; Zhang, Y.; et al. Combined homologous recombination repair deficiency and immune activation analysis for predicting intensified responses of anthracycline, cyclophosphamide and taxane chemotherapy in triple-negative breast cancer. BMC Med. 2021, 19, 190. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D.; et al. PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight 2020, 5, e135544. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Marin-Garcia, P.; Ping, P.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome diagram viewer: Data structures and strategies to boost performance. Bioinformatics 2018, 34, 1208–1214. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stacey, D.W. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr. Opin. Cell Biol. 2003, 15, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Yu, M.; Kumarasiri, M.; Le, B.T.; Wang, S. Targeting CDK6 in cancer: State of the art and new insights. Cell Cycle 2015, 14, 3220–3230. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agarwal, P.; Sandey, M.; DeInnocentes, P.; Bird, R.C. Tumor suppressor gene p16/INK4A/CDKN2A-dependent regulation into and out of the cell cycle in a spontaneous canine model of breast cancer. J. Cell Biochem. 2013, 114, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Lim, D.; Qi, C.; Zhang, Z.; Wang, J.; Zhang, F.; Dong, C.; Feng, Z. VPA mediates bidirectional regulation of cell cycle progression through the PPP2R2A-Chk1 signaling axis in response to HU. Cell Death Dis. 2023, 14, 114. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de Miranda, N.F.; Peng, R.; Georgiou, K.; Wu, C.; Falk Sörqvist, E.; Berglund, M.; Chen, L.; Gao, Z.; Lagerstedt, K.; Lisboa, S.; et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J. Exp. Med. 2013, 210, 1729–1742. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ronen, A.; Glickman, B.W. Human DNA repair genes. Environ. Mol. Mutagen. 2001, 37, 241–283. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.; Bernstein, H.; Payne, C.M.; Garewal, H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: Fail-safe protection against carcinogenesis. Mutat. Res. 2002, 511, 145–178. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.; Ngeow, J. Homologous recombination deficiency: Cancer predispositions and treatment implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mekonnen, N.; Yang, H.; Shin, Y.K. Homologous recombination deficiency in ovarian, breast, colorectal, pancreatic, non-small cell lung and prostate cancers, and the mechanisms of resistance to PARP inhibitors. Front. Oncol. 2022, 12, 880643. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ohashi, S.; Natsuizaka, M.; Yashiro-Ohtani, Y.; Kalman, R.A.; Nakagawa, M.; Wu, L.; Klein-Szanto, A.J.; Herlyn, M.; Diehl, J.A.; Katz, J.P.; et al. NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology 2010, 139, 2113–2123. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ho, D.M.; Guruharsha, K.G.; Artavanis-Tsakonas, S. The Notch Interactome: Complexity in Signaling Circuitry. Adv. Exp. Med. Biol. 2018, 1066, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Knobbe, C.B.; Reifenberger, G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003, 13, 507–518. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bonin, S.; Pracella, D.; Barbazza, R.; Dotti, I.; Boffo, S.; Stanta, G. PI3K/AKT Signaling in Breast Cancer Molecular Subtyping and Lymph Node Involvement. Dis. Markers 2019, 2019, 7832376. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Phung, B.; Sun, J.; Schepsky, A.; Steingrimsson, E.; Rönnstrand, L. C-KIT signaling depends on microphthalmia-associated transcription factor for effects on cell proliferation. PLoS ONE 2011, 6, e24064. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bosbach, B.; Rossi, F.; Yozgat, Y.; Loo, J.; Zhang, J.Q.; Berrozpe, G.; Warpinski, K.; Ehlers, I.; Veach, D.; Kwok, A.; et al. Direct engagement of the PI3K pathway by mutant KIT dominates oncogenic signaling in gastrointestinal stromal tumor. Proc. Natl. Acad. Sci. USA 2017, 114, E8448–E8457. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rocca, A.; Braga, L.; Volpe, M.C.; Maiocchi, S.; Generali, D. The Predictive and Prognostic Role of RAS-RAF-MEK-ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets. Cancers 2022, 14, 5306. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gomes, Á.N.M.; Oliveira, K.K.; Marchi, F.A.; Bettim, B.B.; Germano, J.N.; Gonçalves Filho, J.; Pinto, C.A.L.; Lourenço, S.V.; Coutinho-Camillo, C.M. TGFβ signaling pathway in salivary gland tumors. Arch. Oral Biol. 2024, 162, 105943. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Murphy, M.E. Genetic Modifiers of the p53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026302. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yoon, H.; Liyanarachchi, S.; Wright, F.A.; Davuluri, R.; Lockman, J.C.; de la Chapelle, A.; Pellegata, N.S. Gene expression profiling of isogenic cells with different TP53 gene dosage reveals numerous genes that are affected by TP53 dosage and identifies CSPG2 as a direct target of p53. Proc. Natl. Acad. Sci. USA 2002, 99, 15632–15637. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Zhang, X.; Han, C.; Wan, G.; Huang, X.; Ivan, C.; Jiang, D.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Rao, P.H.; et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 2015, 520, 697–701, Erratum in Nature 2021, 597, E6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aghabozorgi, A.S.; Ebrahimi, R.; Bahiraee, A.; Tehrani, S.S.; Nabizadeh, F.; Setayesh, L.; Jafarzadeh-Esfehani, R.; Ferns, G.A.; Avan, A.; Rashidi, Z. The genetic factors associated with Wnt signaling pathway in colorectal cancer. Life Sci. 2020, 256, 118006. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.A.; Michalski, M.N.; Stevens, P.D.; Sall, E.A.; Williams, B.O. Regulation of Wnt receptor activity: Implications for therapeutic development in colon cancer. J. Biol. Chem. 2021, 296, 100782. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kikuchi, A.; Kishida, S.; Yamamoto, H. Regulation of Wnt signaling by protein-protein interaction and post-translational modifications. Exp. Mol. Med. 2006, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 2007, 25, 118–145. [Google Scholar] [CrossRef] [PubMed]
- Black, L.E.; Longo, J.F.; Carroll, S.L. Mechanisms of Receptor Tyrosine-Protein Kinase ErbB-3 (ERBB3) Action in Human Neoplasia. Am. J. Pathol. 2019, 189, 1898–1912. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Funa, K.; Sasahara, M. The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J. Neuroimmune Pharmacol. 2014, 9, 168–181. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rogers, M.A.; Campaña, M.B.; Long, R.; Fantauzzo, K.A. PDGFR dimer-specific activation, trafficking and downstream signaling dynamics. J. Cell Sci. 2022, 135, jcs259686. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ho, B.B.; Bergwitz, C. FGF23 signalling and physiology. J. Mol. Endocrinol. 2021, 66, R23–R32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, K.; Ji, W.; Yu, Y.; Li, Z.; Niu, X.; Xia, W.; Lu, S. FGFR1-ERK1/2-SOX2 axis promotes cell proliferation, epithelial-mesenchymal transition, and metastasis in FGFR1-amplified lung cancer. Oncogene 2018, 37, 5340–5354, Erratum in Oncogene 2020, 39, 6619–6620. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.J.; Xue, J.; Corey, S.J. Src family tyrosine kinases are activated by Flt3 and are involved in the proliferative effects of leukemia-associated Flt3 mutations. Exp. Hematol. 2005, 33, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Reindl, C.; Quentmeier, H.; Petropoulos, K.; Greif, P.A.; Benthaus, T.; Argiropoulos, B.; Mellert, G.; Vempati, S.; Duyster, J.; Buske, C.; et al. CBL exon 8/9 mutants activate the FLT3 pathway and cluster in core binding factor/11q deletion acute myeloid leukemia/myelodysplastic syndrome subtypes. Clin. Cancer Res. 2009, 15, 2238–2247. [Google Scholar] [CrossRef] [PubMed]

| Patient Characteristics | |
|---|---|
| Age, years (median, IQR) | 66 (59–72) |
| Gender | |
| Male | 70 (49%) |
| Female | 72 (51%) |
| ECOG | |
| 0 | 83 (58%) |
| 1 | 43 (30%) |
| 2 | 10 (8%) |
| 3 | 3 (2%) |
| Unknown | 3 (2%) |
| Stage | |
| III | 52 (37%) |
| IV | 90 (63%) |
| First-line Chemotherapy | |
| FOLFIRINOX | 62 (44%) |
| Gemcitabine nab-paclitaxel | 62 (44%) |
| FOLFOX | 8 (6%) |
| Gemcitabine monotherapy | 6 (4%) |
| Gemcitabine plus Cisplatin | 2 (1%) |
| Other cytotoxic chemotherapy | 2 (1%) |
| Sample Characteristics | |
| Source | |
| Primary | 68 (48%) |
| Metastasis | 68 (48%) |
| Blood | 6 (4%) |
| TMB (median, IQR) | 2.5 (1.3–3.8) |
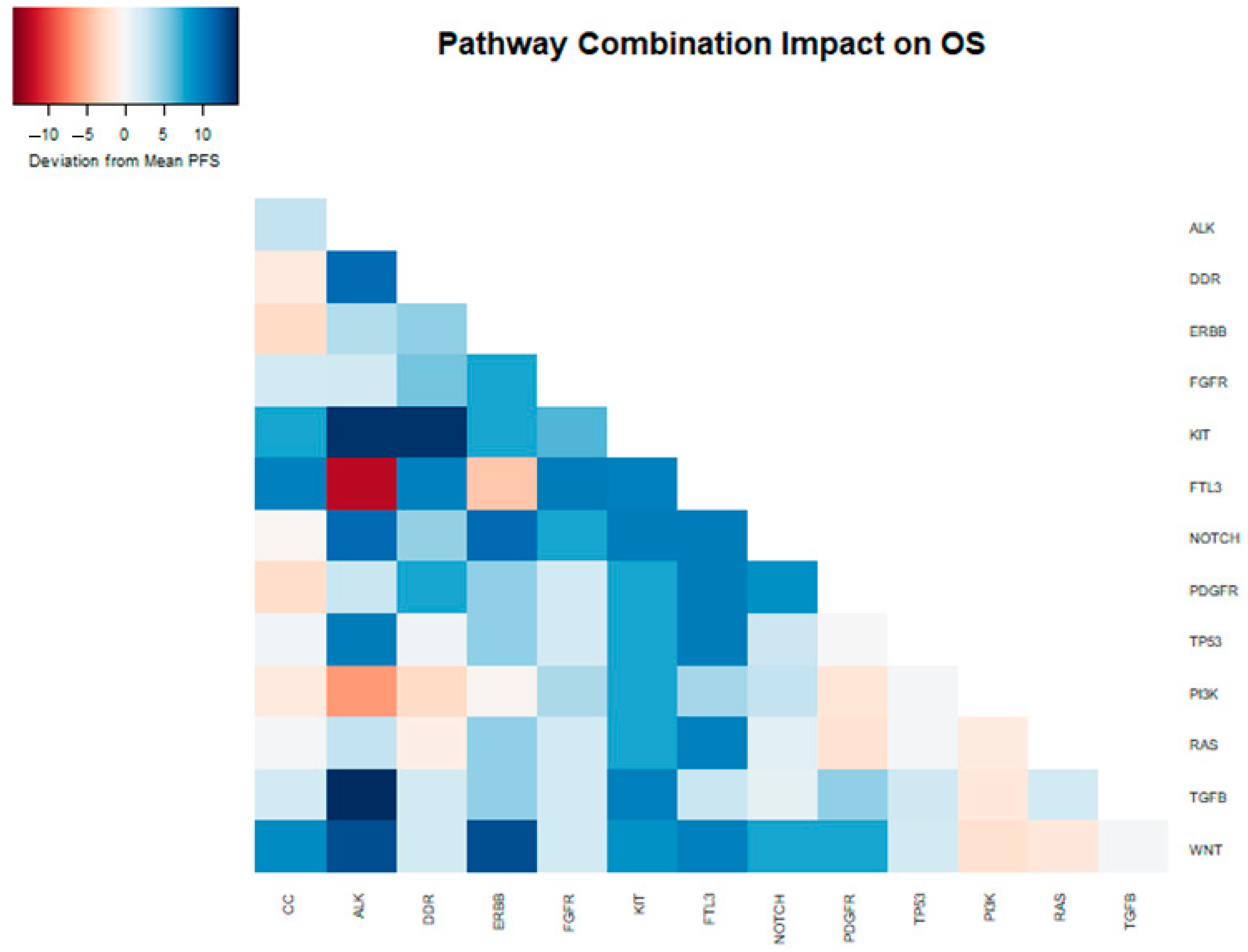
| Main Pathway | Mutation Combination | n | Median OS (months) | HR | 95% CI | p-Value (HR) |
|---|---|---|---|---|---|---|
| Overall | 142 | 13.6 | ||||
| NOTCH | 51 | 15.0 | 0.57 | 0.38–0.86 | 0.008 | |
| NOTCH + CC | 38 | 13.3 | 0.59 | 0.38–0.91 | 0.017 | |
| NOTCH + PI3K | 18 | 16.7 | 0.57 | 0.32–1 | 0.052 | |
| NOTCH + ALK | 11 | 24.9 | 0.39 | 0.19–0.81 | 0.011 | |
| NOTCH + KIT | 14 | 23.8 | 0.43 | 0.23–0.81 | 0.009 | |
| NOTCH + DDR | 29 | 18.4 | 0.60 | 0.38–0.96 | 0.030 | |
| NOTCH + ERBB2 | 11 | 24.9 | 0.42 | 0.2–0.86 | 0.018 | |
| NOTCH + PDGFR | 14 | 22.5 | 0.41 | 0.21–0.8 | 0.009 | |
| NOTCH + TP53 | 39 | 16.4 | 0.53 | 0.34–0.82 | 0.004 | |
| NOTCH + RAS | 42 | 15.0 | 0.66 | 0.45–0.99 | 0.044 | |
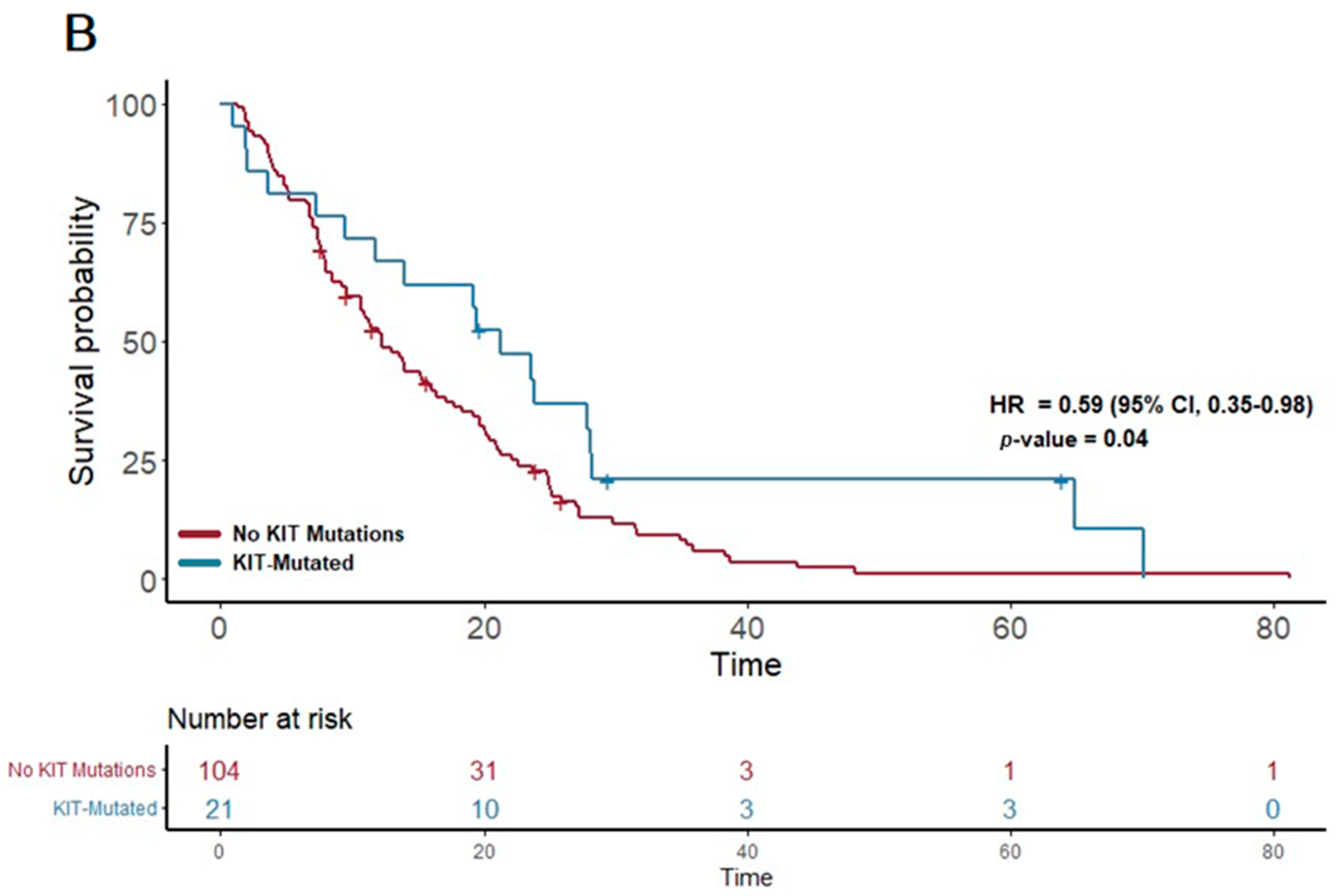
| KIT | 23 | 21.3 | 0.59 | 0.35–0.98 | 0.043 | |
| KIT + CC | 19 | 21.3 | 0.56 | 0.33–0.96 | 0.035 | |
| KIT+ DDR | 13 | 27.8 | 0.49 | 0.26–0.92 | 0.028 | |
| KIT + TP53 | 20 | 21.3 | 0.57 | 0.33–0.96 | 0.033 | |
| KIT + PI3K | 14 | 21.3 | 0.46 | 0.24–0.87 | 0.017 | |
| KIT + RAS | 21 | 21.3 | 0.59 | 0.35–0.98 | 0.043 | |
| ALK | 19 | 16.7 | 0.57 | 0.32–1.03 | 0.064 | |
| ALK + CC | 14 | 16.7 | 0.53 | 0.28–1 | 0.050 | |
| ALK + TGFB | 7 | 28.2 | 0.34 | 0.14–0.85 | 0.021 | |
| ALK + TP53 | 15 | 23.8 | 0.52 | 0.28–0.96 | 0.037 |
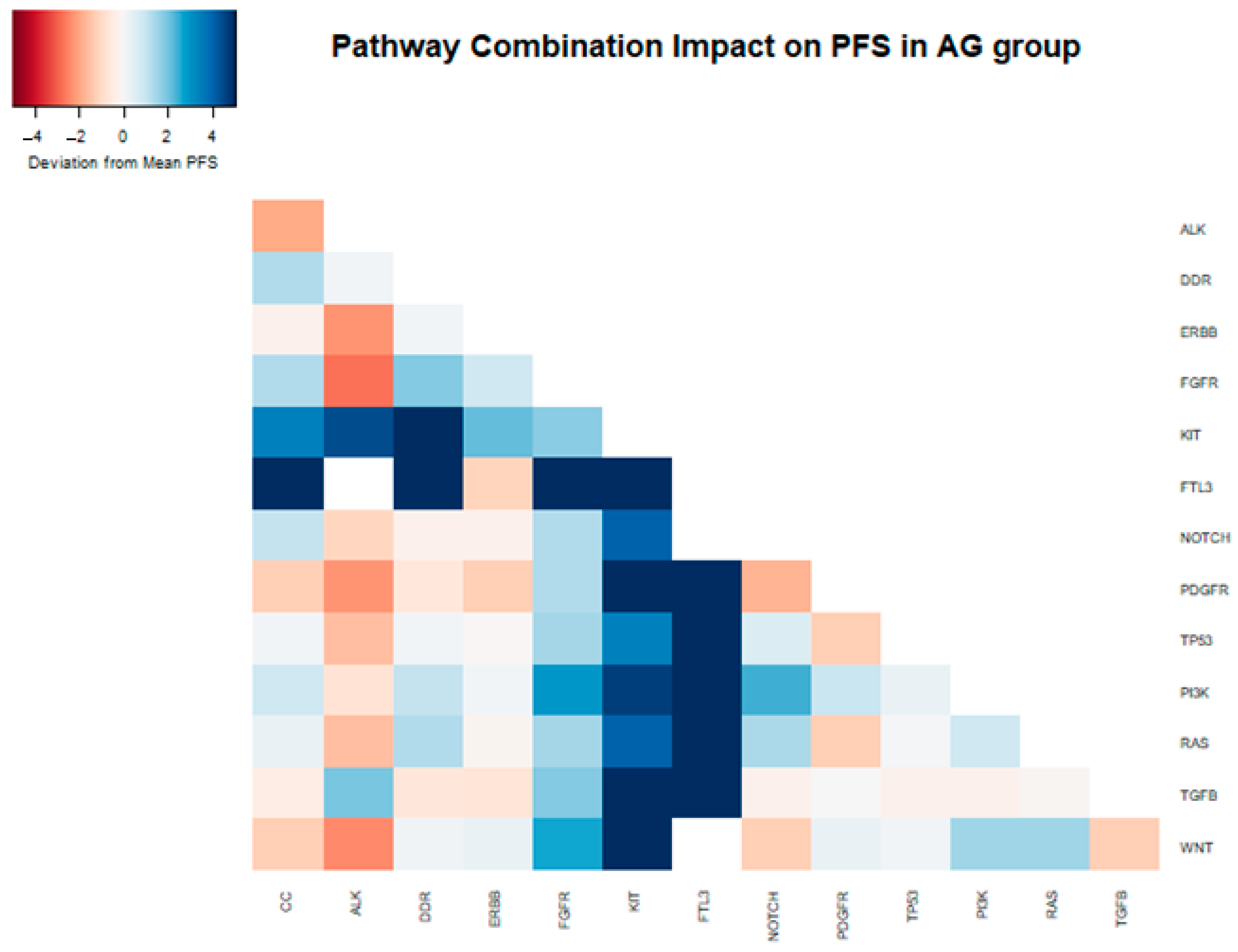
| Main Pathway | Mutation Combination | n | Median PFS (months) | HR | 95% CI | p-Value (HR) |
|---|---|---|---|---|---|---|
| Overall | 62 | 6.3 | ||||
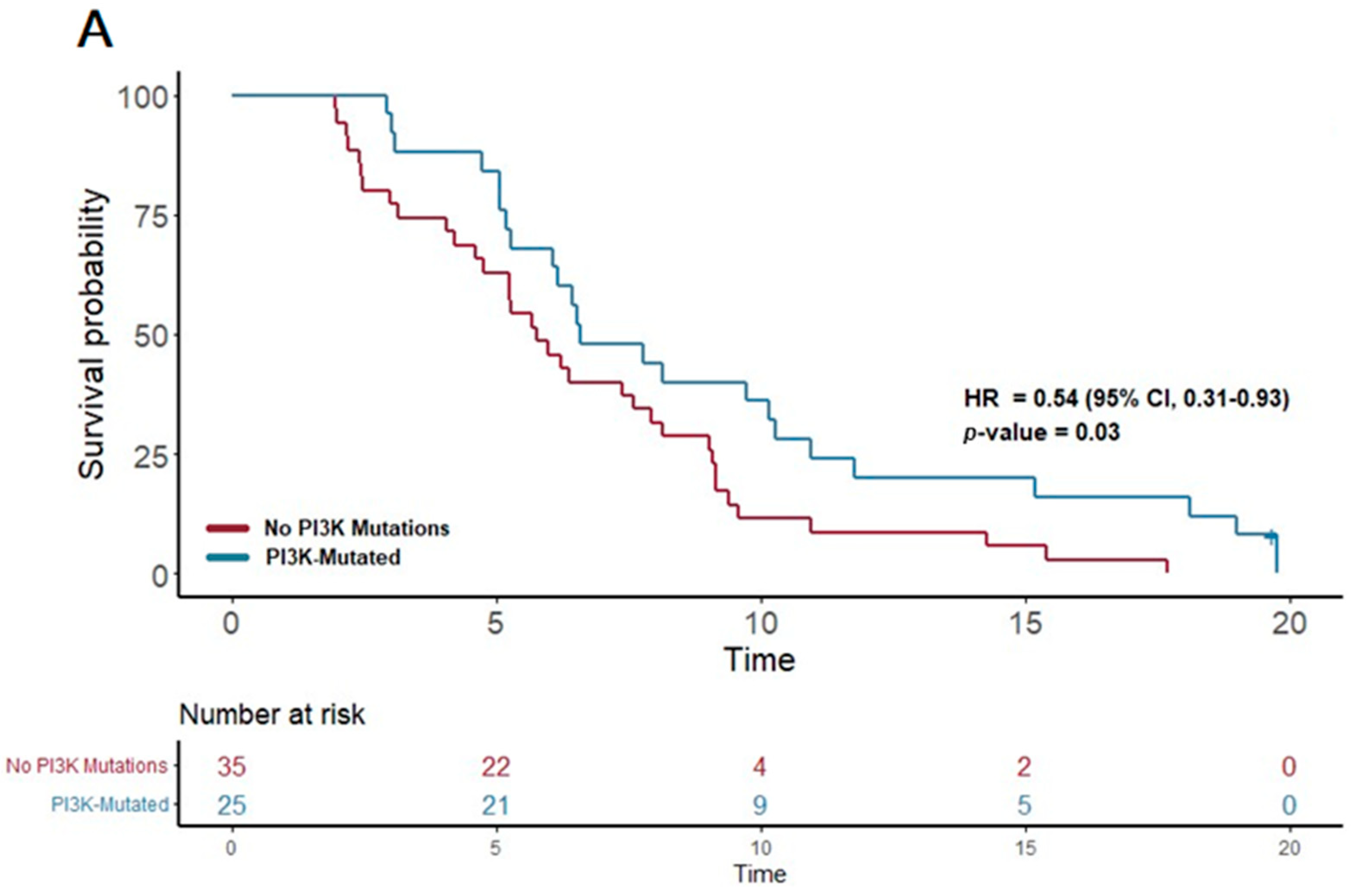
| PI3K | 25 | 6.6 | 0.54 | 0.31–0.93 | 0.027 | |
| PI3K + CC | 20 | 7.2 | 0.53 | 0.29–0.95 | 0.034 | |
| PI3K + DDR | 16 | 7.4 | 0.47 | 0.24–0.9 | 0.023 | |
| PI3K + FGFR | 12 | 9.2 | 0.45 | 0.22–0.9 | 0.024 | |
| PI3K + KIT | 7 | 10.9 | 0.32 | 0.13–0.83 | 0.018 | |
| PI3K + FTL3 | 4 | 13.1 | 0.21 | 0.05–0.89 | 0.035 | |
| PI3K + TP53 | 23 | 6.6 | 0.55 | 0.31–0.97 | 0.037 | |
| PI3K + RAS | 24 | 7.2 | 0.52 | 0.3–0.91 | 0.022 | |
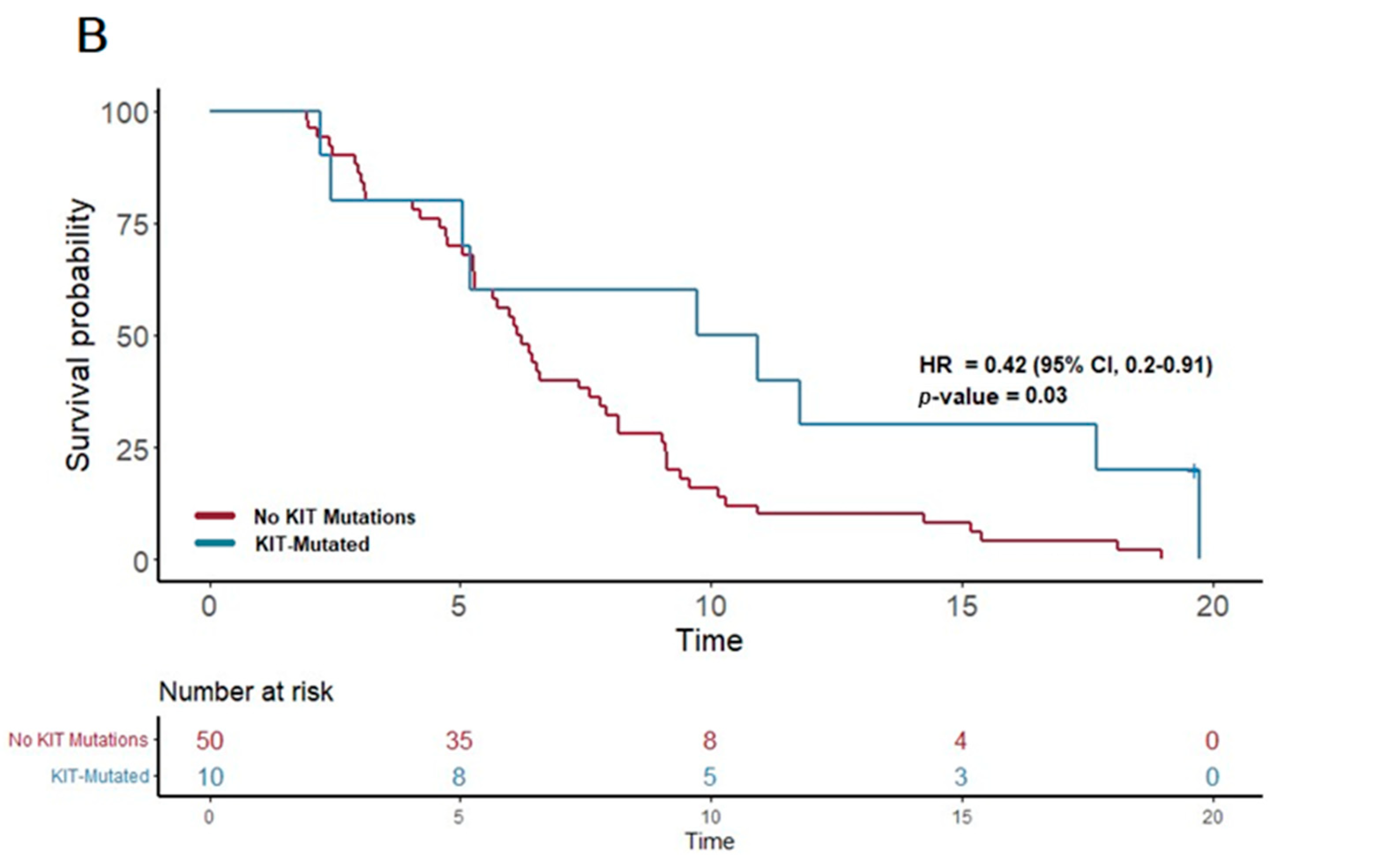
| KIT | 10 | 10.3 | 0.42 | 0.2–0.91 | 0.028 | |
| KIT + CC | 9 | 9.7 | 0.43 | 0.19–0.98 | 0.046 | |
| KIT + DDR | 6 | 14.7 | 0.26 | 0.09–0.74 | 0.012 | |
| KIT + FTL3 | 3 | 19.7 | 0.11 | 0.01–0.83 | 0.032 | |
| KIT + TP53 | 9 | 9.7 | 0.43 | 0.19–0.97 | 0.043 | |
| KIT + RAS | 10 | 10.3 | 0.42 | 0.2–0.91 | 0.028 | |
| FTL3 | 4 | 13.1 | 0.21 | 0.05–0.89 | 0.035 | |
| FTL3 + CC | 4 | 13.3 | 0.21 | 0.05–0.89 | 0.035 | |
| FTL3 + DDR | 3 | 19.7 | 0.11 | 0.01–0.83 | 0.032 | |
| FTL3 + FGFR | 3 | 19.7 | 0.11 | 0.01–0.83 | 0.032 | |
| FTL3 + TP53 | 4 | 13.1 | 0.21 | 0.05–0.89 | 0.035 | |
| FTL3 + RAS | 4 | 13.3 | 0.21 | 0.05–0.89 | 0.035 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paredes de la Fuente, R.; Sucre, S.; Ponce, C.; Rattani, A.A.A.; Peters, M.L.B. Somatic Mutation Profile as a Predictor of Treatment Response and Survival in Unresectable Pancreatic Ductal Adenocarcinoma Treated with FOLFIRINOX and Gemcitabine Nab-Paclitaxel. Cancers 2024, 16, 2734. https://doi.org/10.3390/cancers16152734
Paredes de la Fuente R, Sucre S, Ponce C, Rattani AAA, Peters MLB. Somatic Mutation Profile as a Predictor of Treatment Response and Survival in Unresectable Pancreatic Ductal Adenocarcinoma Treated with FOLFIRINOX and Gemcitabine Nab-Paclitaxel. Cancers. 2024; 16(15):2734. https://doi.org/10.3390/cancers16152734
Chicago/Turabian StyleParedes de la Fuente, Rodrigo, Santiago Sucre, Cristina Ponce, Ahmed Anwer Ali Rattani, and Mary Linton B. Peters. 2024. "Somatic Mutation Profile as a Predictor of Treatment Response and Survival in Unresectable Pancreatic Ductal Adenocarcinoma Treated with FOLFIRINOX and Gemcitabine Nab-Paclitaxel" Cancers 16, no. 15: 2734. https://doi.org/10.3390/cancers16152734
APA StyleParedes de la Fuente, R., Sucre, S., Ponce, C., Rattani, A. A. A., & Peters, M. L. B. (2024). Somatic Mutation Profile as a Predictor of Treatment Response and Survival in Unresectable Pancreatic Ductal Adenocarcinoma Treated with FOLFIRINOX and Gemcitabine Nab-Paclitaxel. Cancers, 16(15), 2734. https://doi.org/10.3390/cancers16152734