Simple Summary
Breast cancer is one of the most prevalent forms of cancer diagnosed in women worldwide. Since the estradiol receptor (ER) is overexpressed in 75% of breast tumors, it is a reasonable target for tumor diagnosis and therapy. This study focuses on the development and preclinical evaluation of readily synthesized 18F-labeled estradiol derivatives with different lipophilicity. The least hydrophilic derivative, 18F-TA-Glyco-EE, showed the highest cellular uptake in ER-positive breast cancer cells. The in vivo PET imaging of breast tumor-bearing mice demonstrated the desired rapid clearance of the tracer from the excretory organ through the liver. The in vitro autoradiography of ER-positive tumor sections confirmed the high specific binding of 18F-TA-Glyco-EE. In conclusion, 18F-TA-Glyco-EE may be a promising candidate for imaging of ER-positive breast cancer.
Abstract
About 75% of breast tumors show an overexpression of the estradiol receptor (ER), making it a valuable target for tumor diagnosis and therapy. To date, 16α-[18F]fluoroestradiol (FES) is the only FDA-approved imaging probe for the positron emission tomography (PET) imaging of ER-positive (ER+) breast cancer. However, FES has the drawback of a high retention in the liver. Therefore, the aim of this study was the development and preclinical evaluation of estradiol (E2) derivatives with different lipophilicity. Three 18F-labeled prosthetic groups (two glycosyl and one PEG azide) were chosen for conjugation with ethinyl estradiol (EE) by 18F-CuAAC (Cu-catalyzed azide-alkyne cycloaddition). The cellular uptake in ER+ MCF-7 tumor cells was highest for the less hydrophilic derivative (18F-TA-Glyco-EE). In nude mice bearing different breast tumors (ER+ MCF-7 and T47D versus ER− MDA-MB-231), 18F-TA-Glyco-EE revealed a high uptake in the liver (13%ID/g, 30 min p.i.), which decreased over 90 min to 1.2%ID/g, indicating fast hepatobiliary clearance. The statistically significant difference of 18F-TA-Glyco-EE uptake in T47D compared to MDA-MB-231 tumors at 60–90 min p.i. indicated ER-specific uptake, whereas in vivo PET imaging did not provide evidence for specific uptake of 18F-TA-Glyco-EE in MCF-7 tumors, probably due to ER occupation by E2 after E2-dependent MCF-7 tumor growth in mice. However, in vitro autoradiography revealed a high specific binding of 18F-TA-Glyco-EE to ER+ tumor slices. We conclude that 18F-TA-Glyco-EE, with its increased hydrophilicity after deacetylation in the blood and thus rapid washout from non-target tissues, may be a viable alternative to FES for the PET imaging of breast cancer.
1. Introduction
Breast cancer is one of the most common types of cancer in the world [1]. At diagnosis, 75% of breast tumors have an overexpression of the estradiol receptor (ER), thus being classified as ER-positive (ER+) [2]. These tumors can respond to hormonal therapy, whereas ER-negative (ER−) tumors require other therapeutic methods like chemotherapy or surgery. The ER is mainly expressed as a nuclear receptor in two isoforms, ERα and ERβ, or as a membrane receptor. These subtypes show distinct tissue distribution; however, in the following, we refer to ERα as ER, since the nuclear ERα is predominantly expressed in breast cancer [3]. The steroid hormone 17β-estradiol (E2), as the endogenous ligand, activates the nuclear ER leading to receptor dimerization, followed by transcriptional activities and gene expression, which in turn affects the development of breast tissue [4]. The status detection of ER is routinely performed by biopsy, followed by immunohistochemistry. However, this invasive method has a low accuracy for the detection of tumor metastasis [5]. To overcome these limitations, non-invasive positron emission tomography (PET) is a viable technique to visualize the complete tumor heterogeneity by the imaging of ER+ lesions [5]. Various PET and single-photon emission computer tomography (SPECT) tracers labeled with, e.g., F-18 [6,7,8,9], Tc-99m [10,11] or I-131 [12,13] as a radionuclide are reported in the literature, but so far, 16α-[18F]fluoroestradiol (FES) is the only FDA-approved imaging probe for PET imaging of ER+ breast cancer [14]. A comprehensive review was published in 2021 that describes the long journey, starting in 1984, of the conception and design that finally led to the discovery of FES by John A. Katzenellenbogen [15].
FES uptake detected by PET correlates with ER expression, such that FES is used in clinics to predict the response of breast cancer tissue to endocrine therapy by evaluating the ER status [15]. However, FES has the drawback of having a high liver uptake and long retention in the liver, due to FES metabolism in the liver and its lipophilicity [16]. Additionally, FES and its so-far reported derivatives suffer from a fast blood clearance, resulting in low tumor uptake [17].
Derivatization with an ethinyl group in the C17 position of E2 was reported to give 17α-ethinylestradiol (EE), which showed a higher ER affinity compared to E2, together with improved pharmacokinetic properties due to decreased first-pass hepatic extraction [18]. Further derivatization by the introduction of a triazole ring in the C17 position by Cu-catalyzed azide-alkyne cycloaddition (CuAAC; click chemistry) resulted in a varied binding affinity for ER which largely depended on the nature of the 4-substituent of the triazole ring [19]. Xu et al. showed that the derivatization of EE by a triazole moiety maintained the ER affinity and demonstrated a specific binding to ER in the case of [18F]FETE, a PEGylated triazolyl derivative (Figure 1) [8], and [125/131I]ITE2, an E2 dimer [13].
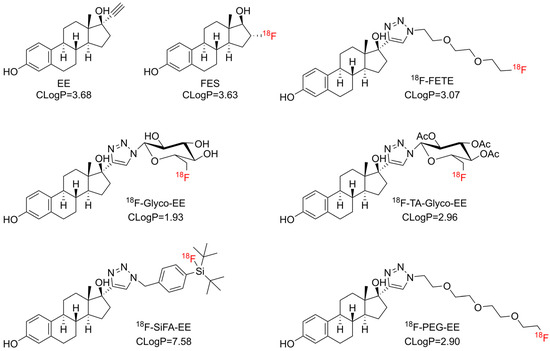
Figure 1.
Chemical structures of FES, 18F-FETE [8], EE and derivatives thereof synthesized in this study aiming at different lipophilicity. CLogP values were calculated using the software ChemDraw, Version 19 (Revvity Signals Software, Revvity Inc., Waltham, MA, USA).
Based on these findings, it appears that EE may be a suitable, readily commercially accessible building block for the synthesis of E2 derivatives by CuAAC with 18F-labeled prosthetic groups. Fluorine-18, with a convenient half-life of 109.8 min, combined with its relatively low positron energy (Emax = 635 keV), is the preferred radioisotope for PET imaging [20].
Therefore, the objective of the present study was to develop an alternative 18F-labeled positron emission tomography (PET) tracer for breast cancer imaging. The tracer was designed to be synthesized using click chemistry, exhibit sufficient ER affinity and display a favorable biodistribution with marked liver clearance. To achieve this objective, three distinct hydrophilic 18F-labeled prosthetic groups (two glycosyl and one PEG azide) were selected for conjugation with EE (Figure 1). Furthermore, a silicon-fluoride acceptor azide (SiFA azide) [21] was selected for comparison (Figure 1), due to its known lipophilicity and easy and fast labeling reaction by isotopic exchange of fluorine-19 [22].
2. Materials and Methods
2.1. General
All chemicals were purchased from commercial sources (ABCR GmbH (Karlsruhe, Germany), Fisher Scientific GmbH (Schwerte, Germany), Alfa Aesar (Haverhill, MA, USA), Carl Roth (Karlsruhe, Germany), Merck KGaA (Darmstadt, Germany), and Sigma Aldrich (Taufkirchen, Germany)) in the highest available quality and used without further purification. NMR spectra were acquired on a Bruker Avance Nanobay V3-I 400 MHz or a Bruker Avance III HD 600 MHz spectrometer. ESI–mass spectra were recorded using LC-MS: Thermo Scientific Dionex Ultimate 3000 UHPLC quaternary pump, autosampler and RS-diode array detector, column: Zorbax-Eclipse XDB-C8 analytical column (3.0 mm × 100 mm, 3.5 μm), flow rate 0.4 mL/min using DAD detection (230 nm; 254 nm), coupled to a Bruker Daltonics Amazon mass spectrometer. High mass accuracy and resolution experiments were performed on a Bruker Daltonics timsTOF Pro spectrometer using electrospray ionization (ESI) as an ionization source. HPLC was performed on an Agilent 1100 system (Agilent Technologies Inc., Santa Clara, CA, USA) with a quaternary pump and variable wavelength detector and radio-HPLC detector HERM LB 500 (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany). No-carrier-added [18F]fluoride was produced through the 18O(p,n)18F reaction on a PETtrace 800 cyclotron (General Electric, Uppsala, Sweden) using H2[18O]O as the target at the Universitätsklinikum Würzburg (Klinik und Poliklinik für Nuklearmedizin, Radiopharmazie/PET-Zentrum, Würzburg, Germany) and transported by road to Erlangen. All radiolabeled derivatives were verified by co-injection with their respective reference compounds on the HPLC system (Figures S1–S4).
2.2. Syntheses
2.2.1. Synthesis of Glyco-EE
17α-Ethinylestradiol (29.6 mg, 100 µmol, 1 eq) and 6-deoxy-6-fluoro-β-D-glucopyranosyl-1-azide [23] (Glyco-N3, 20.7 mg, 100 µmol, 1 eq) were dissolved in water (1.3 mL) and ethanol (500 µL). Sodium ascorbate (100 mM, 20 µL), Cu(OAc)2 (4 mM, 20 µL) and tris(3-hydroxypropyltriazolylmethyl)amine (THPTA, 20 mM, 20 µL) were added, and the reaction was stirred for 30 min at room temperature. The crude product was purified by HPLC (Kromasil C8, 125 × 8 mm, 20–50% acetonitrile (with 0.1% TFA) in water (with 0.1% TFA) over 20 min, 4 mL/min, tR = 8.30 min), and Glyco-EE was obtained after lyophilization in a yield of 3% (1.66 mg, 3.3 µmol). Purity (HPLC): >99% (Chromolith RP18e 100 × 4.6 mm, 4 mL/min, 10–100% acetonitrile in water + 0.1% TFA, tR = 2.01 min), ESI-MS: m/z 504.2 [M + H]+.
2.2.2. Synthesis of TA-Glyco-EE
17α-Ethinylestradiol (21.0 mg, 70.8 µmol, 1 eq) and 2,3,4-tri-O-acetyl-6-deoxy-6-fluoro-β-D-glucopyranosyl-1-azide [23] (TA-Glyco-N3, 23.6 mg, 70.8 µmol, 1 eq) were dissolved in DMSO (500 µL). A solution of Cu(II) sulfate (0.2 M, 40 µL) and sodium ascorbate (0.6 M, 40 µL) was added, and the reaction was stirred at 40 °C overnight. The crude product was prepurified by column chromatography (Silica gel 60, n-hexane/ethyl acetate = 3/2), followed by purification with flash column chromatography (Büchi Ecoflex C18, 50 µm, spherical, 20 g, 25–50% acetonitrile in water + 0.1% TFA). After lyophilization of the product fraction, TA-Glyco-EE was obtained as a white solid in a yield of 65% (29.2 mg, 46.4 µmol). Purity (HPLC): >99% (Chromolith RP18e 100 × 4.6 mm, 4 mL/min, 10–100% acetonitrile in water + 0.1% TFA, tR = 2.69 min), HR-MS m/z calculated for C32H40FN3O9 630.2821 [M + H]+, found 630.2827 [M + H]+. 1H NMR (600 MHz, DMSO-d6) δ 8.94 (s, 1H), 8.11 (s, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.46 (dd, J = 8.4, 2.6 Hz, 1H), 6.41 (d, J = 2.6 Hz, 1H), 6.35 (d, J = 9.3 Hz, 1H), 5.68 (t, J = 9.5 Hz, 1H), 5.56 (t, J = 9.5 Hz, 1H), 5.21 (t, J = 9.8 Hz, 1H), 4.62–4.58 (m, 0.5 H), 4.55–4.49 (m, 1H), 4.45–4.40 (m, 1H), 4.39–4.36 (m, 0.5 H), 2.75–2.64 (m, 2H), 2.43–2.37 (m, 1H), 2.05 (s, 3H), 1.97 (s, 3H), 1.92 (ddd, J = 13.3, 11.7, 3.8 Hz, 1H), 1.86–1.78 (m, 2H), 1.74 (s, 3H), 1.73–1.70 (m, 1H), 1.53–1.47 (m, 1H), 1.46–1.38 (m, 2H), 1.35–1.14 (m, 4H), 0.91 (s, 3H), 0.43 (td, J = 13.0, 4.2 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 169.5, 169.3, 168.2, 154.8, 154.8, 137.1, 130.3, 125.9, 121.3, 114.9, 112.6, 83.7, 81.2 (d, J = 119.4 Hz), 80.5, 73.9 (d, J = 18.2 Hz), 72.2, 70.1, 66.7 (d, J = 7.2 Hz), 47.5, 46.6, 43.2, 39.2, 36.8, 32.5, 29.2, 27.2, 26.1, 23.6, 20.4, 20.2, 19.8, 14.2.
2.2.3. Synthesis of PEG-EE
17α-Ethinylestradiol (35.2 mg, 118.8 µmol, 1 eq) and 1-azido-2-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)ethan [24] (PEG-N3, 26.9 mg, 121.4 µmol, 1.02 eq) were dissolved in DMSO (700 µL). A solution of Cu(II) sulfate (0.2 M, 80 µL) and sodium ascorbate (0.6 M, 80 µL) was added, and the reaction was stirred at 45 °C for 5.5 h. The crude product was purified by flash column chromatography (Büchi Ecoflex C18, 50 µm, spherical, 40 g; 15–50% acetonitrile in water + 0.1% TFA). After lyophilization, PEG-EE was obtained as a yellow-brown solid in a yield of 89% (54.8 mg, 105.9 µmol). Purity (HPLC): >99% (Chromolith RP18e 100 × 4.6 mm, 4 mL/min, 10–100% acetonitrile in water + 0.1% TFA, tR = 2.24 min), HR-MS m/z calculated for C28H40FN3O5 518.3025 [M + H]+, found 518.3029 [M + H]+. 1H NMR (600 MHz, DMSO-d6) δ 8.94 (br s, 1H), 7.82 (s, 1H), 6.95 (d, J = 8.6 Hz, 1H), 6.46 (dd, J = 8.4, 2.6 Hz, 1H), 6.41 (d, J = 2.6 Hz, 1H), 4.53 (t, J = 4.0 Hz, 1H), 4.49 (t, J = 5.3 Hz, 2H), 4.45 (t, J = 4.0 Hz, 1H), 3.82 (t, J = 5.3 Hz, 2H), 3.64 (t, J = 4.1 Hz, 1H), 3.59 (t, J = 4.1 Hz, 1H), 3.52 (dt, J = 6.3, 2.6 Hz, 4H), 3.51–3.47 (m, 4H), 2.77–2.64 (m, 2H), 2.40–2.31 (m, 1H), 2.11–2.04 (m, 1H), 1.93 (ddd, J = 13.3, 11.8, 3.7 Hz, 1H), 1.87–1.71 (m, 3H), 1.61 (td, J = 11.4, 7.5 Hz, 1H), 1.50–1.37 (m, 2H), 1.33 (qd, J = 11.0, 2.6 Hz, 1H), 1.29–1.16 (m, 2H), 0.92 (s, 3H), 0.59 (td, J = 13.0, 4.2 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 154.8, 153.9, 137.1, 130.4, 125.9, 123.0, 114.9, 112.6, 83.0 (d, J = 165.6 Hz), 81.0, 69.8 (2C), 69.7, 69.7, 69.6, 68.9, 49.2, 47.5, 46.7, 43.2, 39.3, 37.1, 32.6, 29.2, 27.2, 26.1, 23.5, 14.4.
2.2.4. Synthesis of SiFA-EE
17α-Ethinylestradiol (25.0 mg, 84.3 µmol, 1 eq) and 4-(di-tert-butylfluorosilyl)benzylazide [21] (SiFA azide, 25.2 mg, 85.9 µmol, 1.02 eq) were dissolved in DMSO (440 µL). A solution of Cu(II) sulfate (0.2 M, 80 µL) and sodium ascorbate (0.6 M, 80 µL) was added, and the reaction was stirred at 45 °C for 2 d. The crude product was purified by flash column chromatography (Büchi Select C18, 30 µm, spherical, 4 g; 55–70% acetonitrile in water + 0.1% TFA). After lyophilization, SiFA-EE was obtained as a white solid in a yield of 46% (22.7 mg, 38.6 µmol). Purity (HPLC): 97%, (Chromolith RP18e 100 × 4.6 mm, 4 mL/min, 10–100% acetonitrile in water + 0.1% TFA, tR = 3.91 min) HR-MS m/z calculated for C35H48FN3O2Si 590.3573 [M + H]+, found 590.3576 [M + H]+. 1H NMR (600 MHz, CDCl3) δ 7.61 (d, J = 8.0 Hz, 2H), 7.39 (s, 1H), 7.26 (s, overlap with CHCl3), 7.25 (s, 1H), 7.05 (dd, J = 8.5, 1.0 Hz, 1H), 6.59 (dd, J = 8.5, 2.8 Hz, 1H), 6.54 (d, J = 2.7 Hz, 1H), 5.56 (s, 2H), 2.88–2.71 (m, 3H), 2.40–2.31 (m, 1H), 2.15–2.11 (m, 2H), 1.92–1.87 (m, 3H), 1.68–1.61 (m, 1H), 1.58–1.36 (m, 5H), 1.32–1.24 (m, 2H), 1.03 (dd, J = 5.5, 1.2 Hz, 21H).
2.3. Radiosyntheses
2.3.1. Radiosynthesis of 18F-TA-Glyco-EE and 18F-Glyco-EE
The prosthetic group 2,3,4-tri-O-acetyl-6-deoxy-6-[18F]fluoroglucopyranosyl azide (TA-Glyco-N3) was synthesized, purified by HPLC and isolated using a C18 cartridge (SepPak light, Waters GmbH, Eschborn, Germany) as described previously [23]. TA-Glyco-N3 was eluted from the cartridge with 600 µL ethanol into an Eppendorf vial, containing CuSO4 (0.2 M, 20 µL), sodium ascorbate (0.6 M, 20 µL) and 17α-ethinylestradiol (10 mM, 40 µL). The reaction mixture was heated at 60 °C for 10 min, and the solvent was continuously reduced under a stream of helium. For the radiosynthesis of 18F-TA-Glyco-EE, water (600 µL, 0.1% TFA) and acetonitrile (100 µL) were added to adjust the reaction volume to 1 mL, and the product was subsequently purified by semi-preparative HPLC (Chromolith RP-18e, 100 × 10 mm, 30–75% acetonitrile (with 0.1% TFA) in water (with 0.1% TFA) over 30 min, 4 mL/min, tR = 11.11 min). The product fraction containing 18F-TA-Glyco-EE was diluted with water (16 mL) and passed through a preconditioned (acetonitrile followed by water, each 5 mL) SepPak light C18 cartridge (Waters GmbH, Eschborn, Germany). 18F-TA-Glyco-EE was eluted from the cartridge with ethanol (1 mL). and was obtained in an activity yield (AY) of 25% (referred to [18F]fluoride) after 77 min and identified by co-injection with TA-Glyco-EE by HPLC.
For the radiosynthesis of 18F-Glyco-EE, 18F-TA-Glyco-EE was deacetylated after the click reaction described above by the addition of NaOH (0.5 M, 100 µL) for 5 min at 60 °C. Afterwards, the reaction mixture was neutralized with HCl (0.5 M, 100 µL), the reaction volume was adjusted to 1 mL with water, and the 18F-Glyco-EE was purified by semi-preparative HPLC (Kromasil C8, 125 × 8 mm, 20–50% acetonitrile (with 0.1% TFA) in water (with 0.1% TFA) over 20 min, 4 mL/min, tR = 8.40 min). The product fraction was diluted with water (16 mL) and passed through a preconditioned (acetonitrile followed by water, each 5 mL) SepPak light C18 cartridge (Waters GmbH, Eschborn, Germany). The cartridge was eluted with ethanol (1 mL). 18F-Glyco-EE was obtained in an AY of 22% (referred to [18F]fluoride) after 90 min and identified by co-injection with Glyco-EE by HPLC.
2.3.2. Radiosynthesis of 18F-PEG-EE
[18F]Fluoride was trapped on a QMA cartridge (Waters GmbH, Eschborn, Germany) and eluted with a solution of Kryptofix 2.2.2 (15 mg) and K2CO3 (1 M, 15 µL) in acetonitrile (900 µL) and water (100 µL) into a reaction vial. The solvent was evaporated under a stream of helium at 85 °C and azeotropically dried with anhydrous acetonitrile (3× 500 µL). The labeling precursor 2-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)ethyl methanesulfonate [24] (8 mg, pre-dried under vacuum overnight) in acetonitrile (400 µL) was added, and the reaction was stirred at 85 °C for 5 min. By adding water (600 µL, 0.1% TFA), the reaction volume was adjusted to 1 mL, and the reaction solution was subjected to semi-preparative HPLC (Kromasil C8, 125 × 10 mm, 10–55% acetonitrile (with 0.1% TFA) in water (with 0.1% TFA) over 20 min, 4 mL/min, tR = 7.07 min). The product fraction containing 1-azido-2-(2-(2-(2-[18F]fluoroethoxy)ethoxy)ethoxy)ethan (PEG-N3) was diluted with water (16 mL) and passed through a preconditioned (acetonitrile followed by water, each 5 mL) StrataX C18 (60 mg) cartridge (Phenomenex, Torrance, CA, USA). The cartridge was eluted with 600 µL DMSO into a reaction vial, containing CuSO4 (0.2 M, 20 µL), sodium ascorbate (0.6 M, 20 µL) and 17α-ethinylestradiol (10 mM, 40 µL). The reaction mixture was heated in a heating block for 15 min at 60 °C. By adding water (400 µL, 0.1% TFA), the reaction volume was adjusted to 1 mL, and 18F-PEG-EE was subsequently purified by semi-preparative HPLC (Kromasil C8, 125 × 10 mm, 30–75% acetonitrile (with 0.1% TFA) in water (with 0.1% TFA) over 30 min, 4 mL/min, tR = 9.5 min). The product fraction was diluted with water (16 mL) and passed through a preconditioned (acetonitrile followed by water, each 5 mL) SepPak light C18 cartridge (Waters GmbH, Eschborn, Germany). 18F-PEG-EE was eluted from the cartridge with ethanol (1 mL) and was obtained in an AY of 19% (referred to [18F]fluoride) after 81 min and identified by co-injection with PEG-EE by HPLC.
2.3.3. Radiosynthesis of 18F-SiFA-EE
[18F]Fluoride was trapped on a QMA cartridge and eluted with a solution of Kryptofix 2.2.2 (10 mg), K2CO3 (0.1 M, 17.5 µL) and KH2PO4 (0.1 M, 17.5 µL) in acetonitrile (800 µL) and water (165 µL) into a reaction vial. The solvent was evaporated under a stream of helium at 85 °C and azeotropically dried with anhydrous acetonitrile (3 × 500 µL). The labeling precursor SiFA-EE [21] (10 nmol) was added to 300 µL dry acetonitrile, and the reaction was stirred for 5 min at room temperature. The reaction was quenched with NaH2PO4 (0.1 M, pH = 4–5, 10 mL) and passed through a preconditioned (acetonitrile followed by water, each 5 mL) SepPak light C18 cartridge (Waters GmbH, Eschborn, Germany). The cartridge was washed with water (5 mL), 18F-SiFA-EE was eluted from the cartridge with ethanol (1 mL), and the solvent was evaporated under reduced pressure. The product was redissolved in Hanks′ Balanced Salt Solution (HBSS) (pH = 6.4) for further use. 18F-SiFA-EE was obtained in an AY of 44% after 27 min and identified by co-injection with SiFA-EE by HPLC.
2.4. In Vitro Studies
2.4.1. Determination of logD7.4
For the determination of the octanol–water coefficient, an aliquot of the radiotracer was added to PBS (500 µL, pH 7.4) and n-octanol (500 µL) in an Eppendorf tube. The mixture was vortexed and subsequently centrifuged, each for 1 min. Three samples (each 100 µL) were taken from both the PBS and the n-octanol layer and measured using a γ-counter (Wallac Wizard, PerkinElmer, Waltham, MA, USA). The logD7.4 was expressed as log(cpmoctanol/cpmPBS) and reported as mean ± SD. The determination of logD7.4 was performed in three independent experiments, each performed in triplicate.
2.4.2. Stability in Serum and Plasma In Vitro
An aliquot of the formulated tracer solution (40 µL containing approx. 10 MBq in 0.9% NaCl) was incubated with 200 µL of human plasma (Hölzel Diagnostika Handels GmbH, from CosmoBio USA, Carlsbad, CA, USA) or human serum (Biochrom GmbH, Merck KGaA, Darmstadt, Germany), respectively, at 37 °C. After various time points (5–120 min), aliquots (30 µL) were taken and quenched in methanol or 10% TFA (30 µL). The samples were centrifuged for 3 min, and subsequently the supernatants were analyzed by radio-HPLC (Chromolith RP-18e, 100 × 4.6 mm, 10–100% acetonitrile (with 0.1% TFA) in water (with 0.1% TFA) over 5 min, 4 mL/min).
2.4.3. Plasma Protein Binding
MicroSPin G-50 columns (Cytiva, Merck KGaA, Darmstadt, Germany) were preconditioned according to the manufacturer’s protocol and the experiment was performed as follows. In brief, an aliquot of the tracer (1 µL, approx. 250 kBq) was added to NaCl (0.9%, 200 µL, negative control) or human plasma (200 µL) and incubated for 10 min at 37 °C. A sample from the plasma or negative control (40 µL) was carefully added to the resin, and the column was placed in an Eppendorf tube. After centrifugation (2 min, 2000× g), the eluate and the resin were collected separately. Both were measured for radioactivity using a γ-counter (Wallac Wizard, PerkinElmer, Waltham, MA, USA), and the radioactivity eluted from the resin was calculated as a percentage of the total amount of radioactivity. The determination of plasma protein binding was performed twice in duplicate.
2.4.4. Cell Lines
The human breast cancer cell lines MCF-7 and T47D were used as positive controls (ER+), whereas the human breast cancer cell line MDA-MB-231 was used as a negative control (ER−) [25]. MCF-7 cells were obtained from DSMZ (Leibniz Institute, DSMZ—German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany); MDA-MB-231 and T47D cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). MCF-7 cells were cultured in DMEM/Ham F-12 containing 10% FBS and 1% L-glutamine. MDA-MB-231 cells were cultured in DMEM containing 10% FBS and 1% L-glutamine, and T47D cells were cultured in RPMI 1640 containing 10% FBS and 1% L-glutamine. All cells were cultured under sterile conditions in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were routinely passaged twice a week.
2.4.5. Stability in Cells
18F-TA-Glyco-EE was incubated for 90 min at 37 °C with MCF-7 cells (350,000 cells/well, seeded one day prior to the experiment in 24-well plates) in cell culture medium. After washing twice with ice-cold PBS, the cells were scraped off, and 200 µL PBS was added for resuspension. The cell suspension was transferred to Eppendorf tubes, homogenized using ultrasound (Bandelin sonoplus HD2070 ultrasonic homogenizer, room temperature, BANDELIN electronic GmbH, Berlin, Germany) and then centrifuged. The supernatant was quenched in 10% TFA, and after centrifugation the resulting supernatant was analyzed by radio-HPLC (Chromolith RP-18e, 100 × 4.6 mm, 10–100% acetonitrile (containing 0.1% TFA) in water (containing 0.1% TFA) over 5 min, 4 mL/min).
2.4.6. Cellular Uptake Assays
One day prior to the experiment, MCF-7 and MDA-MB-231 cells were seeded in 24-well plates (350,000 cells/well). The cells were washed once with cell medium. The cells were incubated with the respective radiotracer in cell medium (510 µL) for 10, 20, 30, 60, 90, 120 min at 37 °C in the absence and presence of estradiol (E2, final concentration of 72 µM). After incubation, the plates were set on ice and washed with ice-cold PBS twice, following cell lysis and detachment with NaOH (0.1 M, 750 µL, 37 °C). The cells were transferred to counting tubes, and the radioactivity was measured using a γ-counter (Wallac Wizard, PerkinElmer, Waltham, MA, USA). The protein concentration of each well was determined in duplicate using Bradford reagent (Bio-Rad Laboratories GmbH, Feldkirchen, Germany). The cellular uptake assay was performed in 2–4 independent experiments, each performed in quadruplicate for each time point.
2.4.7. Competitive Cellular Uptake
Competitive uptake experiments were performed using 18F-TA-Glyco-EE as a radioligand. One day prior to the experiment, MCF-7 cells were seeded in 96-well plates (50,000 cells/well). The cell medium was removed, and the respective estradiol derivative, as a competitor of the radioligand, was added in concentrations from 0 to 500 µM in 90 µL cell culture medium each. After 18F-TA Glyco-EE (10 µL, approx. 1 MBq/well) was added, the incubation proceeded for 60 min at 37 °C. The plate was set on ice and washed with ice-cold PBS, followed by cell lysis and detachment with NaOH (0.1 M, 200 µL, 37 °C). The cells were transferred into counting tubes, and the radioactivity was measured using a γ-counter (Wallac Wizard, PerkinElmer, Waltham, MA, USA). One single experiment was performed in triplicate.
2.5. In Vivo Studies
2.5.1. Animal Models
All mouse experiments were approved by the local animal protection authorities (Government of Lower Franconia, Germany, No. 55.2 2532-2-279) and performed at the FAU in accordance with the relevant E.U. guidelines and regulations.
Female NMRI nude mice (Crl:NMRI-Foxn1nu) were purchased from Charles River (Charles River Laboratories Inc., Wilmington, MA, USA) at the age of four weeks and kept in groups of four to five animals in individually ventilated cages in a twelve hours dark/light cycle, with unlimited access to water and standard chow. At the age of nine to ten weeks, one 17β-estradiol pellet per animal (0.72 mg per pellet, 3 mm diameter) with a 60-day release time (Innovative Research of America, Sarasota, FL, USA) was subcutaneously implanted on the back under isoflurane anesthesia. After three days, approximately 2–3 × 106 MCF-7, T47D or MDA-MB-231 tumor cells (in 50 µL PBS) were mixed with Matrigel (50 µL, BD Biosciences, Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and injected subcutaneously to the back of the mice. Tumor diameters and weight of the animals were recorded five times a week. Biodistribution or imaging studies were performed three–four weeks after the inoculation of the cells.
2.5.2. Biodistribution
For biodistribution experiments, nude mice bearing T47D and MDA-MB-231 tumors (n = 3, per group) were injected with 18F-TA-Glyco-EE (2–4 MBq) under isoflurane anesthesia. After 30, 60 or 90 min, the mice were sacrificed by cervical dislocation. Blood, as well as organs and tissue from the lung, liver, kidneys, heart, spleen, brain, muscle, femur and tumors, were harvested and measured for radioactivity in the γ-counter (Wallac Wizard, PerkinElmer, Waltham, MA, USA). The samples were weighed, and the radioactivity of each tissue sample was calculated as a percentage of the total injected dose per gram tissue (%ID/g).
2.5.3. PET Imaging
Small animal PET scans were performed on an Inveon™ microPET scanner (Siemens Healthineers AG, Erlangen, Germany) under isoflurane anesthesia (3%) using four MCF-7 tumor-bearing mice. 18F-TA-Glyco-EE (2–4 MBq in about 100 µL of saline) was intravenously injected into the tail vein under isoflurane anesthesia (3–4%). Dynamic images were acquired for 60 min, starting with the injection of the radiotracer. For displacement studies, the same mice were injected with fulvestrant (0.5 mg, s.c.) directly after the first PET scan, and two days later, 18F-TA-Glyco-EE (2–4 MBq in about 100 µL of saline) was injected, and a second dynamic PET scan was performed. Images were corrected for decay and attenuation, and MAP (iterative maximum a posteriori) images were reconstructed (12 × 10 s, 3 × 1 min, 5 × 5 min, 3 × 10 min, a total of 23 frames) using the built-in software of the PET scanner. An evaluation of the MAP images was conducted using the software Pmod (Version 3.6, PMOD Technologies LLC, Fällanden, Switzerland, Part of Bruker’s Preclinical Imaging Division). Regions of interest (ROIs) were drawn over the tumor, and the mean radioactivity concentration within these regions was obtained from the mean values and calculated as a percentage of the total injected dose per gram tissue (%ID/g).
2.6. In Vitro Autoradiography
MCF-7, MDA-MB231 and T47D tumors grown in nude mice were collected and frozen in a hexane/dry ice bath (−70 °C). Tumor slices (14 µm) were prepared on a cryostat microtome HM 500 O (Microm, Walldorf, Germany), thaw-mounted on HistoBond® adhesive glass slides (Marienfeld, Lauda-Königshofen, Germany) and stored at −80 °C. For in vitro autoradiography studies, the tumor tissue sections were thawed and pre-incubated in incubation buffer (50 mM Tris HCl, 5 mM MgCl2, 0.2% bovine serum albumin (BSA), pH 7.4) at room temperature for 15 min. Then, the sections were transferred to incubation buffer containing 18F-TA-Glyco-EE (3–5 MBq) in a total volume of 70 mL. For the determination of non-specific binding, separate sections were incubated with 18F-TA-Glyco-EE in the presence of E2 (72 µM). After an incubation time of 60 min, the sections were washed three times with incubation buffer, followed by a short dip in ice-cold distilled water. The slices were carefully dried in a stream of warm air and finally placed on a phosphor imager screen (Fuji Imaging Plate BAS-IP SR 2025 E) overnight prior to readout (30 µm resolution) using the HD-CR-35 Bio scanner (Elysia-Raytest, Straubenhardt, Germany). The image analysis was performed using the software AIDA (version 4.21, Elysia-Raytest, Straubenhardt, Germany). Regions of interest were analyzed from 3–4 tumor slices from the MCF-7, MDA-MB231 and T47D tumors, and mean values of the intensity per area were calculated.
3. Results
3.1. Chemistry and Radiochemistry
The reference compounds SiFA-EE, PEG-EE, TA-Glyco-EE and Glyco-EE were obtained, applying a straightforward CuAAC using commercially available EE together with the respective azides (Figure 2). The CuAAC was performed in DMSO or ethanol as solvents in the presence of CuSO4 as a catalyst and sodium ascorbate as a reducing agent. All compounds were purified by semi-preparative HPLC or flash column chromatography and obtained in a high chemical purity of at least 97%, as determined by HPLC.
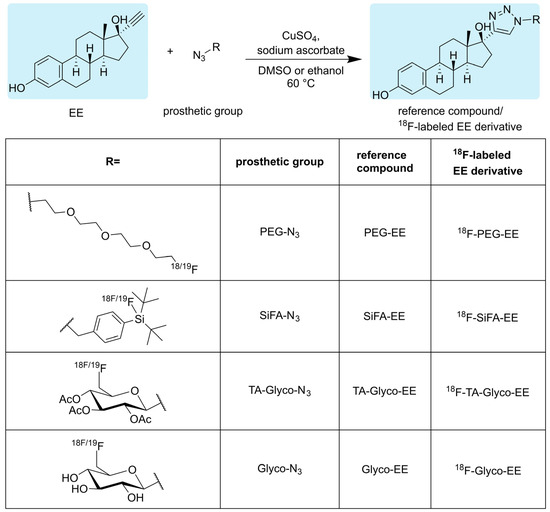
Figure 2.
Click chemistry-based synthesis of triazolyl derivatives of estradiol with different fluorine-containing substituents (R) in the 4-position of the triazole ring and the respective radiosynthesis of their 18F-labeled analogs using 18F-labeled azides as prosthetic groups.
Applying a similar straightforward approach to the radiosynthesis of the series of 18F-labeled triazolyl conjugated analogs of estradiol (Figure 2; 18F-TA-Glyco-EE, 18F-Glyco-EE, 18F-PEG-EE and 18F-SiFA-EE), we envisaged the use of small 18F-labeled azide-bearing prosthetic groups that could be reliably conjugated to the readily available alkyne derivative of estradiol (EE) under mild click chemistry reaction conditions. All 18F-labeled azides used in this study as prosthetic groups had been previously described in sufficient detail in the literature [23,24,26], and their 18F-syntheses were successfully established in our laboratory: The glycosyl prosthetic group 2,3,4-tri-O-acetyl-6-deoxy-6-[18F]fluoroglucopyranosyl azide (TA-Glyco-N3) was developed in our laboratory by Maschauer et al. [23], and applied to the radiosynthesis of 18F-TA-Glyco-EE and 18F-Glyco-EE by CuAAC in ethanol using 400 nmol of EE. After isolation by semi-preparative HPLC, 18F-TA-Glyco-EE was obtained in an AY of 25% (referred to [18F]fluoride) after a total synthesis time of 77 min, with an Am of up to 57 GBq/µmol. The deacetylation of 18F-TA-Glyco-EE, followed by semi-preparative HPLC isolation, led to 18F-Glyco-EE in an AY of 22% (referred to [18F]fluoride) after a total synthesis time of 90 min.
The 18F-synthesis of the alternative hydrophilic 18F-labeled PEG-N3 was achieved starting from the precursor 2-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)ethyl methanesulfonate [24]. After 18F-fluorination, the 18F-labeled PEG-N3 was conjugated to EE by CuAAC in DMSO. 18F-PEG-EE was purified by semi-preparative HPLC and obtained in an AY of 19% (referred to [18F]fluoride) after a total synthesis time of 81 min, with an Am of up to 18 GBq/µmol.
Aiming at a more lipophilic derivatization of EE for comparison to the above-described hydrophilic derivatization, the well-known isotopic 19F-18F exchange reaction according to Schirrmacher et al. [22] was applied to the radiosynthesis of 18F-SiFA-EE, starting from 19F-SiFA-EE. After cartridge purification and reformulation in HBSS (pH 6.4), 18F-SiFA-EE was obtained in an AY of 44% (referred to [18F]fluoride) after a total synthesis time of 27 min, with an Am of up to 28 GBq/µmol. It should be noted here that the obtained Am of 18F-SiFA-EE was strongly dependent on the starting activity. In addition, it was found that 18F-SiFA-EE was highly amenable to hydrolysis of the Si-18F bond even under slightly acidic conditions.
3.2. In Vitro Characterization
The 18F-labeled EE derivatives were compared in terms of their in vitro properties as shown in Table 1. 18F-SiFA-EE showed high instability in both human serum and plasma (<50% after 10 min), and had therefore to be excluded from further in vitro evaluation. The determination of the octanol–buffer coefficient (logD7.4) confirmed the attempt to provide EE derivatives with increased hydrophilicity. Compared to EE (calculated logKow = 3.7) [27], the 18F-labeled EE derivatives showed lower logD7.4 values, due to the conjugation of hydrophilic prosthetic groups in descending order: 18F-TA-Glyco-EE > 18F-PEG-EE > 18F-Glyco-EE (Table 1). Both compounds, 18F-PEG-EE and 18F-TA-Glyco-EE, showed high plasma protein binding of 71% and 79%, respectively. The in vitro stability of the EE derivatives in human serum and plasma at 37 °C was analyzed by radio-HPLC. 18F-PEG-EE and 18F-Glyco-EE were stable in both plasma and serum over 120 min. As expected, triacetylated 18F-TA-Glyco-EE was rapidly deacetylated in serum and plasma, leading to the formation of 18F-Glyco-EE and partially deacetylated intermediates over 120 min (Figure 3a). In addition, the 90 min incubation of MCF-7 cells with 18F-TA-Glyco-EE and the HPLC analysis of the intracellular radioactivity confirmed that intact 18F-TA-Glyco-EE accumulated in the cells, without any significant deacetylation in the absence of serum (Figure 3b).

Table 1.
In vitro data of EE derivatives.
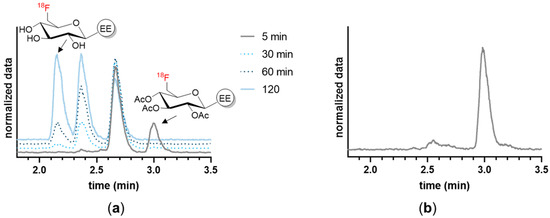
Figure 3.
HPLC analysis of the stability of 18F-TA-Glyco-EE. (a) The stability of 18F-TA-Glyco-EE in human serum was determined after various incubation periods, indicating time-dependent cleavage of the acetyl groups. (b) The incubation of MCF-7 cells with 18F-TA-Glyco-EE for 90 min at 37 °C confirmed that intact 18F-TA-Glyco-EE was intracellularly detected, without significant deacetylation in the absence of serum.
The 18F-labeled EE derivatives 18F-TA-Glyco-EE, 18F-Glyco-EE, and 18F-PEG-EE, with favorable in vitro properties and sufficient stability, were further evaluated in cellular uptake experiments. The human breast adenocarcinoma cell line MCF-7 was used as the ER+ control, whereas the human breast carcinoma cell line MDA-MB-231 was used as the ER− control [25]. The cells were incubated for a maximum of 120 min with the respective radiotracer in the absence or presence of E2 (72 µM) as a blocking reagent to determine non-specific (ER-independent) tracer uptake (Figure 4). All radioligands exhibited specific ER-dependent uptake in ER+ cells at 60–90 min of incubation that was blocked in the presence of E2 to the level of ER− cells. However, a relatively high amount of non-specific uptake in ER− cells was observed that could be further blocked by the addition of E2. Furthermore, the radiotracers clearly showed a time-dependent total uptake in both ER+ and ER− cells. The most hydrophilic compound, 18F-Glyco-EE, showed a minimal total uptake steadily ascending over time to reach 7%/mg after 90 min, as well as the lowest specific uptake compared to 18F-PEG-EE and 18F-TA-Glyco-EE. 18F-PEG-EE, on the other hand, showed a four times higher total uptake (27%/mg after 60 min), in addition to a higher specific uptake compared to 18F-Glyco-EE. In comparison to 18F-PEG-EE and 18F-Glyco-EE, 18F-TA-Glyco-EE showed a 10-fold higher uptake and thus the highest specific uptake in ER+ cells (270%/mg after 60 min). Based on these results, 18F-TA-Glyco-EE was used as a radioligand for further in vitro and in vivo studies.
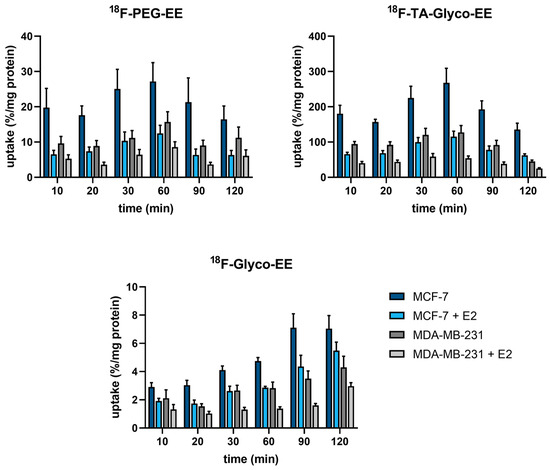
Figure 4.
Cellular uptake (in %/mg) of the different 18F-labeled EE derivatives in ER+ MCF-7 cells and ER− MDA-MB-231 cells in the absence and presence of E2 (72 µM). Each bar represents the mean ± standard error of the mean (SEM) of 2–4 independent experiments, each performed in quadruplicate.
Competitive cellular uptake experiments were performed in ER+ MCF-7 cells at 37 °C for 60 min, using 18F-TA-Glyco-EE as a radioligand and increasing concentrations of E2 or EE derivatives as competitors (Figure 5). The competitive cellular uptake elucidated the findings of the cellular uptake assay. Only at high concentrations of the competitors was the uptake of 18F-TA-Glyco-EE completely diminished, which may be due to various transport mechanisms, including passive transport [28]. In the specific assay set up in this work, IC50 values of 56 µM (E2), 194 µM (TA-Glyco-EE) and 1274 µM (PEG-EE) were determined for the inhibition of cellular uptake of 18F-TA-Glyco-EE in MCF-7 cells (Figure 5).
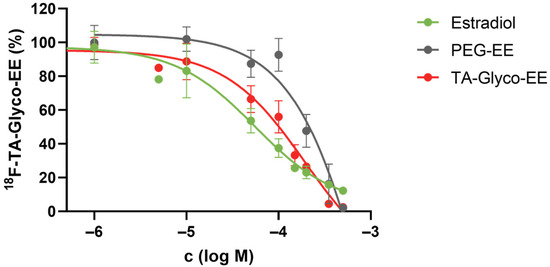
Figure 5.
Competitive cellular uptake experiment of 18F-TA-Glyco-EE in MCF-7 (ER+) breast cancer cells using E2 and EE derivatives. Data are expressed as mean values ± SD from a single experiment performed in triplicate.
3.3. Biodistribution and Small Animal PET Imaging
The biodistribution of 18F-TA-Glyco-EE was determined in T47D, an ER+ breast cancer cell line, and MDA-MB-231 (ER−) tumor-bearing nude mice (n = 3), and the results are depicted in Figure 6. High radioactivity was detected in the liver (13%ID/g) and intestine (10%ID/g) at 30 min p.i., decreasing over 90 min to less than 3%ID/g, revealing the fast hepatobiliary clearance of 18F-TA-Glyco-EE. The uptake in blood and all other non-target organs was low for all time points (Figure 6a). The uptake of 18F-TA-Glyco-EE in tumors was 1.0%ID/g in T47D tumors and 0.9%ID/g in MDA-MB-231 tumors at 30 min p.i., slightly decreasing over time. The tumor uptake values at 60 min and 90 min p.i. showed statistically significant differences between ER+ T47D and ER− MDA-MB-231 tumors (Figure 6b), indicating the ER-specific uptake of 18F-TA-Glyco-EE in vivo.
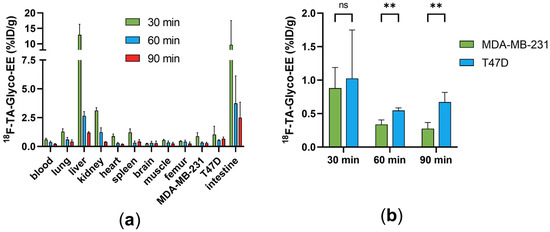
Figure 6.
(a) Biodistribution of 18F-TA-Glyco-EE in T47D (ER+) and MDA-MB-231 (ER−) tumor-bearing nude mice at 30, 60 and 90 min p.i. Data are expressed in %ID/g as mean values ± SD from three animals per group (n = 3–4). (b) Statistical comparison of the mean 18F-TA-Glyco-EE uptake in ER+ and ER− tumors by multiple t-test calculations (ns: not significant (p > 0.05), ** p (60 min) = 0.002, ** p (90 min) = 0.003).
Small animal PET imaging was performed with 18F-TA-Glyco-EE on ER+ tumor-bearing mice over 60 min p.i. (Figure 7). To determine non-specific tumor uptake, fulvestrant (0.5 mg s.c. [29]), as a competitive antagonist, was injected in the same mice and the PET imaging experiment was repeated 48 h later. Pretreatment with fulvestrant resulted in a significantly reduced tumor uptake on PET scans in one of the four animals (Figure 7a). However, in the other three mice, this difference could not be detected (Figure 7b). The low specific uptake of 18F-TA-Glyco-EE could be due to the need to implant an E2-releasing pellet in the back of the mice for proper MCF-7 tumor growth, possibly leading to different levels of available free ER or remaining E2 competing with 18F-TA-Glyco-EE for ER binding in the tumor.
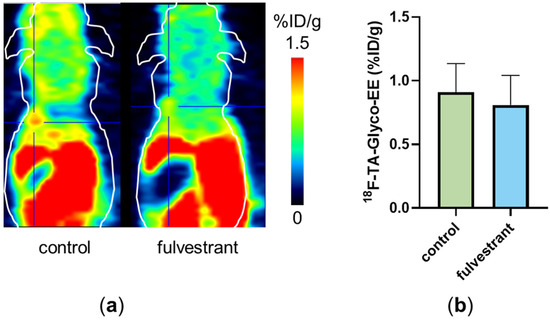
Figure 7.
(a) Small animal PET scan of a single MCF-7 (ER+) tumor-bearing nude mouse injected with 18F-TA-Glyco-EE (control, 60 min p.i.). After the first scan, the same mouse received fulvestrant (0.5 mg s.c.), and 48 h later, the 18F-TA-Glyco-EE-PET was performed again (“fulvestrant”, 60 min p.i.). The tumor is marked by the crosshair. (b) Analysis of tumor uptake of 18F-TA-Glyco-EE at 60 min p.i. in MCF-7 (ER+) tumor-bearing nude mice (“control”) compared to fulvestrant-pretreated mice (“fulvestrant”). Data are expressed as mean values (in %ID/g) ± standard deviation, each n = 4.
Due to the challenging animal model, with ER+ xenografts requiring E2-dependent tumor growth, demonstrating specific tumor binding of 18F-TA-Glyco-EE in vivo appeared difficult. Therefore, we investigated the specificity of 18F-TA-Glyco-EE binding to tumor tissue by the in vitro autoradiography of ER+ (MCF-7 and T47D) and ER− (MDA-MB-231) tumor slices (Figure 8). The incubation of the tumor slices for 60 min at room temperature with either 18F-TA-Glyco-EE or 18F-TA-Glyco-EE together with E2 (72 µM) displayed the high specific binding of 18F-TA-Glyco-EE to the ER+ MCF-7 and T47D tumor sections and only negligible binding in the ER− MDA-MB-231 tumor sections.
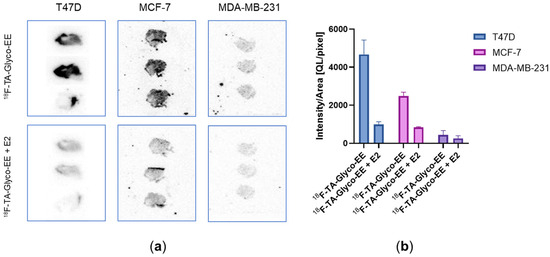
Figure 8.
(a) In vitro autoradiography of T47D, MCF-7 and MDA-MB-231 tumor slices incubated with either 18F-TA-Glyco-EE or 18F-TA-Glyco-EE together with E2 (72 µM) for 60 min. (b) Analysis of 18F-TA-Glyco-EE binding to tumor slices. Data are expressed as mean values of the intensity per area ± SD from 3–4 slices of each group.
4. Discussion
Estradiol derivatives modified in the 17α-position address a hydrophobic binding site of ER that tolerates triazolyl substituents [8,19]. This finding, especially the study by Xu et al. dealing with the 18F-labeled PEG2-triazolyl derivative [18F]FETE [8], combined with the previous report on 17α-triazolyl EE derivatives bearing various substituents in the 4-position of the triazole ring, e.g., phenyl, p-hydroxyphenyl or alkyl alcohols [19], prompted us to select alternative 18F-labeled prosthetic groups with different lipophilicity, including the 18F-glycosyl moiety, for the design of new 18F-labeled EE derivatives as PET ligand candidates.
Applying a straightforward click chemistry-based strategy, four different 18F-labeled EE derivatives were synthesized and evaluated regarding their suitability as radiotracers for ER imaging by PET. All the compounds could be radiolabeled in high yields (AYs between 19 and 44%) in total synthesis times in the range of 27–90 min, which is compatible with the half-life of fluorine-18. Incubation of the radioligands in human serum and plasma showed the high stability of the EE derivatives, except for 18F-SiFA-EE, which showed an unfavorable hydrolysis of the Si-18F bond. Therefore, 18F-SiFA-EE was excluded from further evaluation.
The experimental determination of the logD7.4 values for 18F-PEG-EE, 18F-TA-Glyco-EE and18F-Glyco-EE confirmed higher hydrophilicity compared to E2 and FES (logD7.4 = 2.5 [30]). This finding supports, on the one hand, our aim to develop alternative tracers with suspected improved liver clearance compared to FES. On the other hand, targeting the nuclear ER requires the passive permeability of the tracer to pass the cell membrane, such that highly hydrophilic properties could be assumed unfavorable. This assumption was confirmed by the in vitro results of 18F-Glyco-EE, with the lowest logD7.4 of 1.7, which was less suitable for ER targeting as shown by the low cellular uptake in ER+ MCF-7 cells. This result is in accordance with the fluoroethyl-triazolyl derivative reported by Tejería et al., which had a similar logD7.4 of 1.8 and also showed low cellular uptake in MCF-7 cells [31].
On the contrary, 18F-PEG-EE could be expected to be a promising tracer for ER targeting, due to its structural similarity to the previously reported [18F]FETE, which showed a high tumor uptake of 4%ID/g at 1 h p.i. in MCF-7 tumor-bearing nude mice [8]. In the present study, 18F-PEG-EE, with a PEG3 chain and logD7.4 of 2.2, showed a higher and more specific E2-inhibitable cellular uptake compared to 18F-Glyco-EE.
However, the radioligand with the most promising in vitro properties in our series was 18F-TA-Glyco-EE, which revealed a moderate lipophilicity (logD7.4 = 2.3), as well as a substantially high and ER-specific cellular uptake in MCF-7 cells. The 54-fold increased cellular uptake of 18F-TA-Glyco-EE compared to the deacetylated 18F-Glyco-EE leads to the conclusion that the absence of polar hydroxyl groups significantly improved the cellular uptake in ER+ MCF-7 cells, presumably due to increased passive permeability. The ability of intact 18F-TA-Glyco-EE to accumulate in ER+ cells was further proven by the HPLC analysis of cell lysates, confirming the relevance of the acetylation of the hydroxyl groups for adequate cellular uptake. In addition, competitive cellular uptake experiments with 18F-TA-Glyco-EE showed higher IC50 values of PEG-EE compared to the IC50 value for self-competition with TA-Glyco-EE, confirming the favorable properties of TA-Glyco-EE for ER-dependent cellular uptake by direct comparison to PEG-EE and Glyco-EE. As expected, we observed deacetylation of 18F-TA-Glyco-EE in human serum in vitro, due to carbohydrate esterase activity. Deacetylation of 18F-TA-Glyco-EE will also occur in the blood after tracer injection. However, we postulate that at short time points after tracer injection, the acetylation of the tracer and the associated good targeting properties of 18F-TA-Glyco-EE may be useful for adequate tumor accumulation. Deacetylation, which occurs in the blood and competes with the uptake of the acetylated form into the tumor, may support the positive effect of rapid liver clearance that has also been observed.
Combined with its fast and high-yielding radiosynthesis (AY = 25% after 77 min), 18F-TA-Glyco-EE was therefore assumed to be a promising 18F-labeled ER ligand for the PET imaging of ER+ breast cancer and was further evaluated in vivo.
The in vivo biodistribution studies and small animal PET imaging of tumor-bearing mice with 18F-TA-Glyco-EE faced some challenges. It is necessary to implant an E2-releasing pellet to ensure the proper growth of human MCF-7 and T47D xenografts in mice [32]. However, a remaining significant blood concentration of E2 present in the animals during the in vivo studies would compete with the radiotracer for binding to the ER.
The biodistribution study was performed in animals with ER+ T47D and ER− MDA-MB-231 xenografts. Due to the known liver metabolism of estradiol derivatives, 18F-TA-Glyco-EE showed a high uptake in the liver (13%ID/g) at 30 min p.i. However, the radioactivity in the liver substantially decreased over time to reach 1.2%ID/g at 90 min p.i., indicating fast hepatic excretion of the radiometabolites formed from 18F-TA-Glyco-EE. We assume that the majority of liver metabolites were formed after deacetylation of 18F-TA-Glyco-EE in the blood, resulting in polar metabolites in the liver that were rapidly excreted. The uptake in T47D or MDA-MB-231 tumors was low (<1%ID/g); however, at 60–90 min p.i., a statistically significant ER-specific tumor uptake of 18F-TA-Glyco-EE was determined when comparing ER+ to ER− tumors. The dynamic small animal PET imaging of ER+ MCF-7 xenografts showed the specific tumor uptake of 18F-TA-Glyco-EE in only one out of four mice after treatment with fulvestrant to degrade the available estradiol receptors by downregulation [33]. The statistical analysis of all animals did not show a significant difference in tumor uptake of 18F-TA-Glyco-EE comparing non-treated to fulvestrant-treated animals. This could be due to remaining high levels of circulating E2 in the mice after E2-dependent tumor growth. It is therefore tempting to speculate that special attention should be paid to testing the E2 level in the animal’s blood prior to the PET scan, to avoid any potential competition with an ER-targeting tracer. In the paper by Xu et al., the sufficient tumor growth of MCF-7 xenografts in vivo without an E2-releasing pellet was reported, resulting in a relatively high tumor uptake of [18F]FETE of 4%ID/g (60 min p.i.) that could be significantly diminished by fulvestrant pretreatment to 1.5%ID/g (60 min p.i.) [8]. In contrast, Aliaga et al. [34] compared the tumor uptake of FES in different mouse models, reporting a low uptake (<0.4%ID/g) of FES in MCF-7 and T47D tumors. The authors confirmed a loss of available ER in MCF-7 tumors, suggesting that even after the removal of the necessary E2 depot pellet, the intracellular receptors could still be occupied by E2 for a longer time, resulting in the low uptake of an ER-targeting PET ligand. Our results were consistent with those reported by Aliaga et al., as we observed rather low uptake values of 18F-TA-Glyco-EE in MCF-7 tumors of around 1%ID/g.
To verify the presence of estradiol receptor expression in our in vivo model, we performed an in vitro autoradiography using ER+ (MCF-7 and T47D) and ER− (MDA-MB-231) tumor slices obtained from the xenografts. The high specific binding of 18F-TA-Glyco-EE was observed in both ER+ tumor slices, whereas no specific binding was detected in ER− tumor slices. Therefore, our autoradiography study strongly confirmed the presence of ER in T47D and MCF-7 xenografts and revealed a heterogeneous ER distribution in the tumor tissue, while no expression of ER in MDA-MB-231 tumor slices was confirmed. This further corroborates the assumption that the low uptake of 18F-TA-Glyco-EE in ER+ tumors in vivo may be due to high ER occupation by E2 and not due to the adverse binding properties of 18F-TA-Glyco-EE.
5. Conclusions
In summary, four different 18F-labeled estradiol derivatives with varied lipophilicities were successfully synthesized by a straightforward click chemistry-based strategy using 18F-labeled prosthetic groups and evaluated preclinically regarding their ability to specifically bind to ER. The observed hydrolytic instability of 18F-SiFA-EE excluded this derivative from further evaluation. 18F-PEG-EE, 18F-Glyco-EE and18F-TA-Glyco-EE showed slight differences in their lipophilicities, but significant differences in their cellular uptake in MCF-7 cells, making 18F-TA-Glyco-EE the most promising candidate for in vivo evaluation. Biodistribution, as well as small animal PET studies, revealed a fast clearance from excreting organs such as the liver, most likely due to deacetylation of the tracer in the blood. The lack of a specific uptake of 18F-TA-Glyco-EE in ER+ tumors in vivo could be explained by ER occupation by E2, since the high specific binding of 18F-TA-Glyco-EE was verified by in vitro autoradiography in ER+ MCF-7 and T47D tumors in comparison to ER− MDA-MB-231 tumors. In conclusion, 18F-TA-Glyco-EE is a promising candidate as an 18F-labeled PET tracer for the imaging of ER+ breast cancer, but has to be further evaluated in a more suitable animal model before translation into the clinic.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers16152639/s1, Figure S1: HPLC chromatograms showing co-injection of 18F-Glyco-EE (top) and reference compound Glyco-EE (bottom). Figure S2: HPLC chromatograms showing co-injection of 18F-TA-Glyco-EE (top) and reference compound TA-Glyco-EE (bottom). Figure S3: HPLC chromatograms showing co-injection of 18F-TA-Glyco-EE (top) and reference compound TA-Glyco-EE (bottom). Figure S4: HPLC chromatograms showing co-injection of 18F-SiFA-EE (top) and reference compound SiFA-EE (bottom).
Author Contributions
Conceptualization, S.M.; methodology, A.F.; validation, A.F., S.M. and O.P.; formal analysis, S.M.; investigation, A.F. and S.M.; resources, S.M. and O.P.; data curation, O.P.; writing—original draft preparation, A.F.; writing—review and editing, S.M. and O.P.; visualization, A.F. and S.M.; supervision, S.M. and O.P.; project administration, S.M.; funding acquisition, S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (DFG grant MA 4295/2-1).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of the government of Franconia in Germany (protocol code 55.2 2532-2-279).
Informed Consent Statement
Not applicable.
Data Availability Statement
All data acquired for this study are stored at the data center of the University Hospital Erlangen, Department of Nuclear Medicine.
Acknowledgments
We gratefully thank Manuel Geisthoff and Ulrike Ittstein for excellent technical assistance, Anke Seitz, and Peter Gmeiner (Chair of Pharmaceutical Chemistry, FAU) for excellent collaboration and help in chemical analyses, and Carmen Wängler (Medical Faculty Mannheim of Heidelberg University) for helpful discussion regarding SiFA synthesis.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Ferlay, J.E.M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Available online: https://gco.iarc.who.int/today (accessed on 6 February 2024).
- Blamey, R.W.; Hornmark-Stenstam, B.; Ball, G.; Blichert-Toft, M.; Cataliotti, L.; Fourquet, A.; Gee, J.; Holli, K.; Jakesz, R.; Kerin, M.; et al. ONCOPOOL—A European database for 16,944 cases of breast cancer. Eur. J. Cancer 2010, 46, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Chapter Three—Estrogen receptor signaling mechanisms. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 116, pp. 135–170. [Google Scholar]
- van Kruchten, M.; de Vries, E.G.E.; Brown, M.; de Vries, E.F.J.; Glaudemans, A.W.J.M.; Dierckx, R.A.J.O.; Schröder, C.P.; Hospers, G.A.P. PET imaging of oestrogen receptors in patients with breast cancer. Lancet Oncol. 2013, 14, e465–e475. [Google Scholar] [CrossRef] [PubMed]
- Kiesewetter, D.O.; Kilbourn, M.R.; Landvatter, S.W.; Heiman, D.F.; Katzenellenbogen, J.A.; Welch, M.J. Preparation of four fluorine-18-labeled estrogens and their selective uptakes in target tissues of immature rats. J. Nucl. Med. 1984, 25, 1212–1221. [Google Scholar] [PubMed]
- Liu, G.; Wang, W.; Lin, J.; Li, K.; Lv, G.; Zhao, X.; Wang, S.; Luo, S.; Qiu, L. Kit-like 18F-labeling of an estradiol derivative as a potential PET imaging agent for estrogen receptor-positive breast cancer. J. Radioanal. Nucl. Chem. 2017, 312, 599–607. [Google Scholar] [CrossRef]
- Xu, D.; Zhuang, R.; You, L.; Guo, Z.; Wang, X.; Peng, C.; Zhang, D.; Zhang, P.; Wu, H.; Pan, W.; et al. 18F-labeled estradiol derivative for targeting estrogen receptor-expressing breast cancer. Nucl. Med. Biol. 2018, 59, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Seimbille, Y.; Rousseau, J.; Benard, F.; Morin, C.; Ali, H.; Avvakumov, G.; Hammond, G.L.; van Lier, J.E. 18F-labeled difluoroestradiols: Preparation and preclinical evaluation as estrogen receptor-binding radiopharmaceuticals. Steroids 2002, 67, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.K.; Hathaway, H.J.; Ramesh, C.; Arterburn, J.B.; Dai, D.; Sklar, L.A.; Norenberg, J.P.; Prossnitz, E.R. Preclinical Development of a Neutral, Estrogen Receptor–Targeted, Tridentate 99mTc(I)-Estradiol-Pyridin-2-yl Hydrazine Derivative for Imaging of Breast and Endometrial Cancers. J. Nucl. Med. 2008, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, K.; Arun, A.; Singh, S.; Manohar, M.; Chuttani, K.; Konwar, R.; Dwivedi, A.; Soni, R.; Singh, A.K.; Mishra, A.K.; et al. Bivalent Approach for Homodimeric Estradiol Based Ligand: Synthesis and Evaluation for Targeted Theranosis of ER(+) Breast Carcinomas. Bioconjug Chem. 2016, 27, 961–972. [Google Scholar] [CrossRef]
- Xu, D.; Lin, X.; Zeng, X.; Wen, X.; Li, J.; Li, Y.; Huang, J.; Chen, X.; Guo, Z.; Zhang, X. Radioiodinated 4-(p-Iodophenyl) Butanoic Acid-Modified Estradiol Derivative for ER Targeting SPECT Imaging. Anal. Chem. 2021, 93, 13998–14006. [Google Scholar] [CrossRef]
- Xu, D.; Peng, C.; Gao, F.; Guo, Z.; Zhuang, R.; Su, X.; Zhang, X. Radioiodinated estradiol dimer for estrogen receptor targeted breast cancer imaging. Chem. Biol. Drug Des. 2020, 96, 1332–1340. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Drug Trial Snapshot: CERIANNA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trial-snapshot-cerianna (accessed on 6 February 2024).
- Katzenellenbogen, J.A. The quest for improving the management of breast cancer by functional imaging: The discovery and development of 16α-[18F]fluoroestradiol (FES), a PET radiotracer for the estrogen receptor, a historical review. Nucl. Med. Biol. 2021, 92, 24–37. [Google Scholar] [CrossRef]
- O’Brien, S.R.; Edmonds, C.E.; Katz, D.; Mankoff, D.A.; Pantel, A.R. 18F-Fluoroestradiol (FES) PET/CT: Review of current practice and future directions. Clin. Transl. Imaging 2022, 10, 331–341. [Google Scholar] [CrossRef]
- Oliveira, M.; Neto, C.; Ribeiro Morais, G.; Thiemann, T. Steroid receptor ligands for breast cancer targeting: An insight into their potential role as pet imaging agents. Curr. Med. Chem. 2013, 20, 222–245. [Google Scholar] [CrossRef] [PubMed]
- von Schoultz, B.; Carlström, K.; Collste, L.; Eriksson, A.; Henriksson, P.; Pousette, Å.; Stege, R. Estrogen therapy and liver function—Metabolic effects of oral and parenteral administration. Prostate 1989, 14, 389–395. [Google Scholar] [CrossRef]
- Wetzel, E.A.; Hanson, A.M.; Troutfetter, C.L.; Burkett, D.J.; Sem, D.S.; Donaldson, W.A. Synthesis and evaluation of 17α-triazolyl and 9α-cyano derivatives of estradiol. Bioorg. Med. Chem. 2020, 28, 115670. [Google Scholar] [CrossRef] [PubMed]
- Radford, L.L.; Lapi, S.E. Methods for the Production of Radionuclides for Medicine. In Radiopharmaceutical Chemistry; Lewis, J.S., Windhorst, A.D., Zeglis, B.M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 63–83. [Google Scholar]
- Tietze, L.F.; Schmuck, K. SiFA Azide: A New Building Block for PET Imaging Using Click Chemistry. Synlett 2011, 2011, 1697–1700. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Bradtmöller, G.; Schirrmacher, E.; Thews, O.; Tillmanns, J.; Siessmeier, T.; Buchholz, H.G.; Bartenstein, P.; Wängler, B.; Niemeyer, C.M.; et al. 18F-Labeling of Peptides by means of an Organosilicon-Based Fluoride Acceptor. Angewandte Chem. Int. Ed. 2006, 45, 6047–6050. [Google Scholar] [CrossRef]
- Maschauer, S.; Haubner, R.; Kuwert, T.; Prante, O. 18F-Glyco-RGD Peptides for PET Imaging of Integrin Expression: Efficient Radiosynthesis by Click Chemistry and Modulation of Biodistribution by Glycosylation. Mol. Pharm. 2014, 11, 505–515. [Google Scholar] [CrossRef]
- Breyholz, H.-J.; Wagner, S.; Faust, A.; Riemann, B.; Höltke, C.; Hermann, S.; Schober, O.; Schäfers, M.; Kopka, K. Radiofluorinated Pyrimidine-2,4,6-triones as Molecular Probes for Noninvasive MMP-Targeted Imaging. ChemMedChem 2010, 5, 777–789. [Google Scholar] [CrossRef]
- Witt, B.L.; Tollefsbol, T.O. Molecular, Cellular, and Technical Aspects of Breast Cancer Cell Lines as a Foundational Tool in Cancer Research. Life 2023, 13, 2311. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Wängler, B.; Bailey, J.; Bernard-Gauthier, V.; Schirrmacher, E.; Wängler, C. Small Prosthetic Groups in 18F-Radiochemistry: Useful Auxiliaries for the Design of 18F-PET Tracers. Semin. Nucl. Med. 2017, 47, 474–492. [Google Scholar] [CrossRef]
- Hansch, C.L.A.; Hoekman, D. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants; American Chemical Society: Washington, DC, USA, 1995; Volume 2. [Google Scholar]
- Van de Wiele, C.; De Vos, F.; Slegers, G.; Van Belle, S.; Dierckx, R.A. Radiolabeled estradiol derivatives to predict response to hormonal treatment in breast cancer: A review. Eur. J. Nucl. Med. 2000, 27, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; Phoenix, S.; Ouellet, R.; Langlois, R.; van Lier, J.E.; Turcotte, É.E.; Bénard, F.; Lecomte, R. Assessment of the Novel Estrogen Receptor PET Tracer 4-Fluoro-11β-methoxy-16α-[18F]fluoroestradiol (4FMFES) by PET Imaging in a Breast Cancer Murine Model. Mol. Imaging Biol. 2013, 15, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Antunes, I.F.; van Waarde, A.; Dierckx, R.A.J.O.; de Vries, E.G.E.; Hospers, G.A.P.; de Vries, E.F.J. Synthesis and Evaluation of the Estrogen Receptor β–Selective Radioligand 2-18F-Fluoro-6-(6-Hydroxynaphthalen-2-yl)Pyridin-3-ol: Comparison with 16α-18F-Fluoro-17β-Estradiol. J. Nucl. Med. 2017, 58, 554–559. [Google Scholar] [CrossRef][Green Version]
- Tejería, M.E.; Pereira, M.P.; Gambini, J.P.; Duarte, P.; Giglio, J.G.; Rey, A.M. Synthesis of a [18F]F Estradiol Derivative via Click Chemistry Using an Automated Synthesis Module: In Vitro Evaluation as Potential Radiopharmaceutical for Breast Cancer Imaging. Pharmaceuticals 2024, 17, 388. [Google Scholar] [CrossRef] [PubMed]
- Holen, I.; Walker, M.; Nutter, F.; Fowles, A.; Evans, C.A.; Eaton, C.L.; Ottewell, P.D. Oestrogen receptor positive breast cancer metastasis to bone: Inhibition by targeting the bone microenvironment in vivo. Clin. Exp. Metastasis 2016, 33, 211–224. [Google Scholar] [CrossRef]
- Carlson, R.W. The History and Mechanism of Action of Fulvestrant. Clin. Breast Cancer 2005, 6, S5–S8. [Google Scholar] [CrossRef]
- Aliaga, A.; Rousseau, J.A.; Ouellette, R.; Cadorette, J.; van Lier, J.E.; Lecomte, R.; Bénard, F. Breast cancer models to study the expression of estrogen receptors with small animal PET imaging. Nucl. Med. Biol. 2004, 31, 761–770. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).