Deconstructing Intratumoral Heterogeneity through Multiomic and Multiscale Analysis of Serial Sections

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
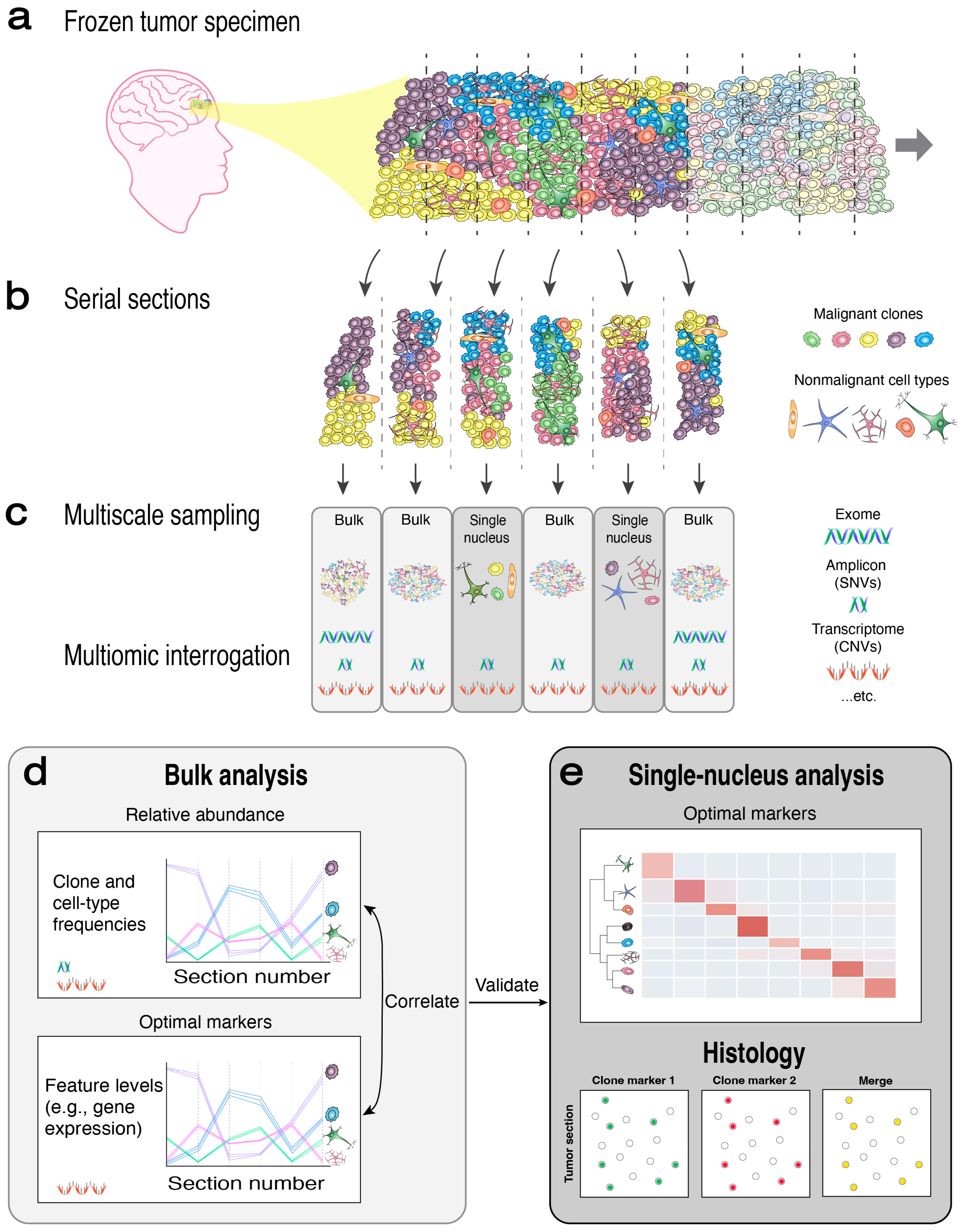
2.1. Overview of MOMA
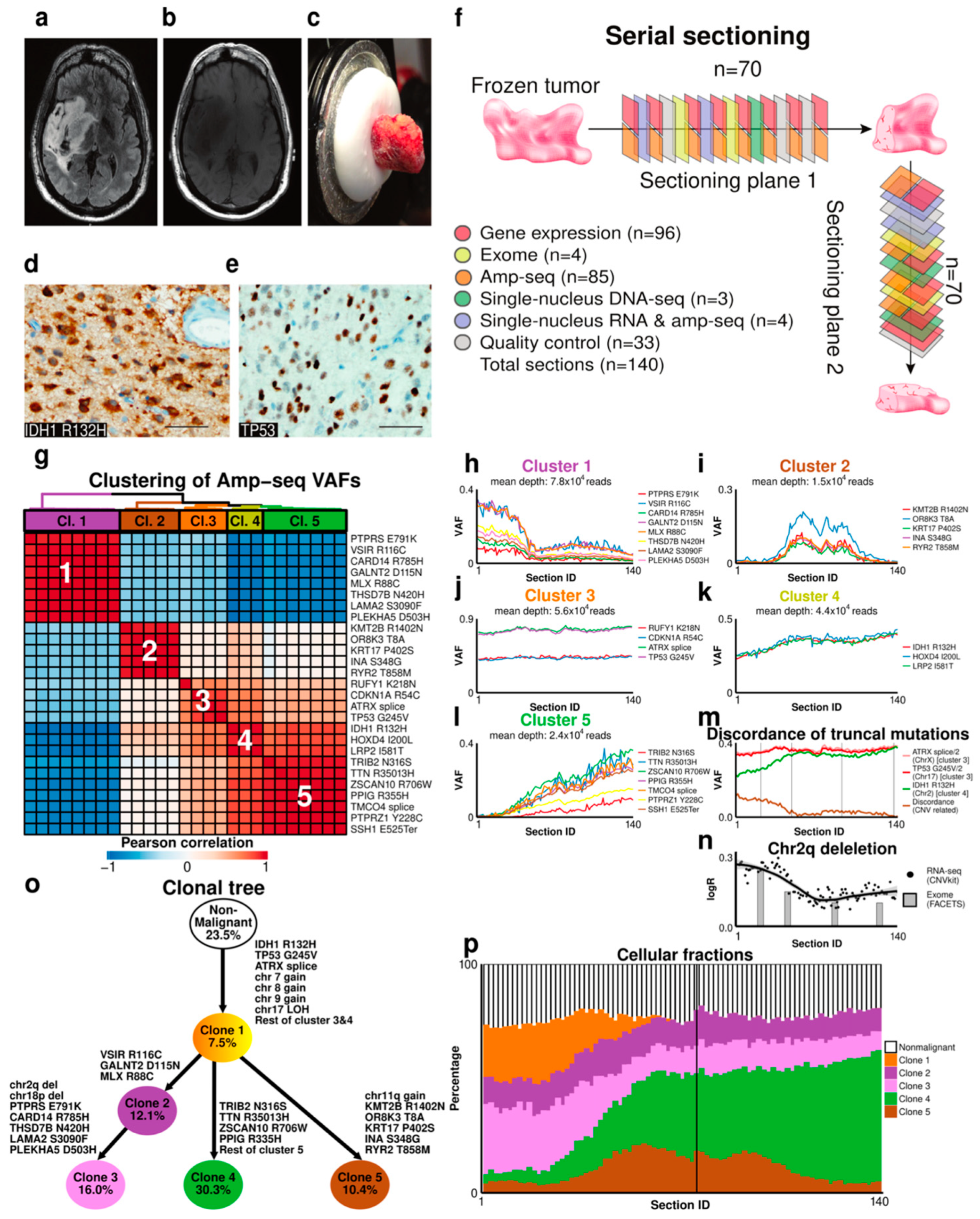
2.2. Case 1: Analysis of Clonal Composition
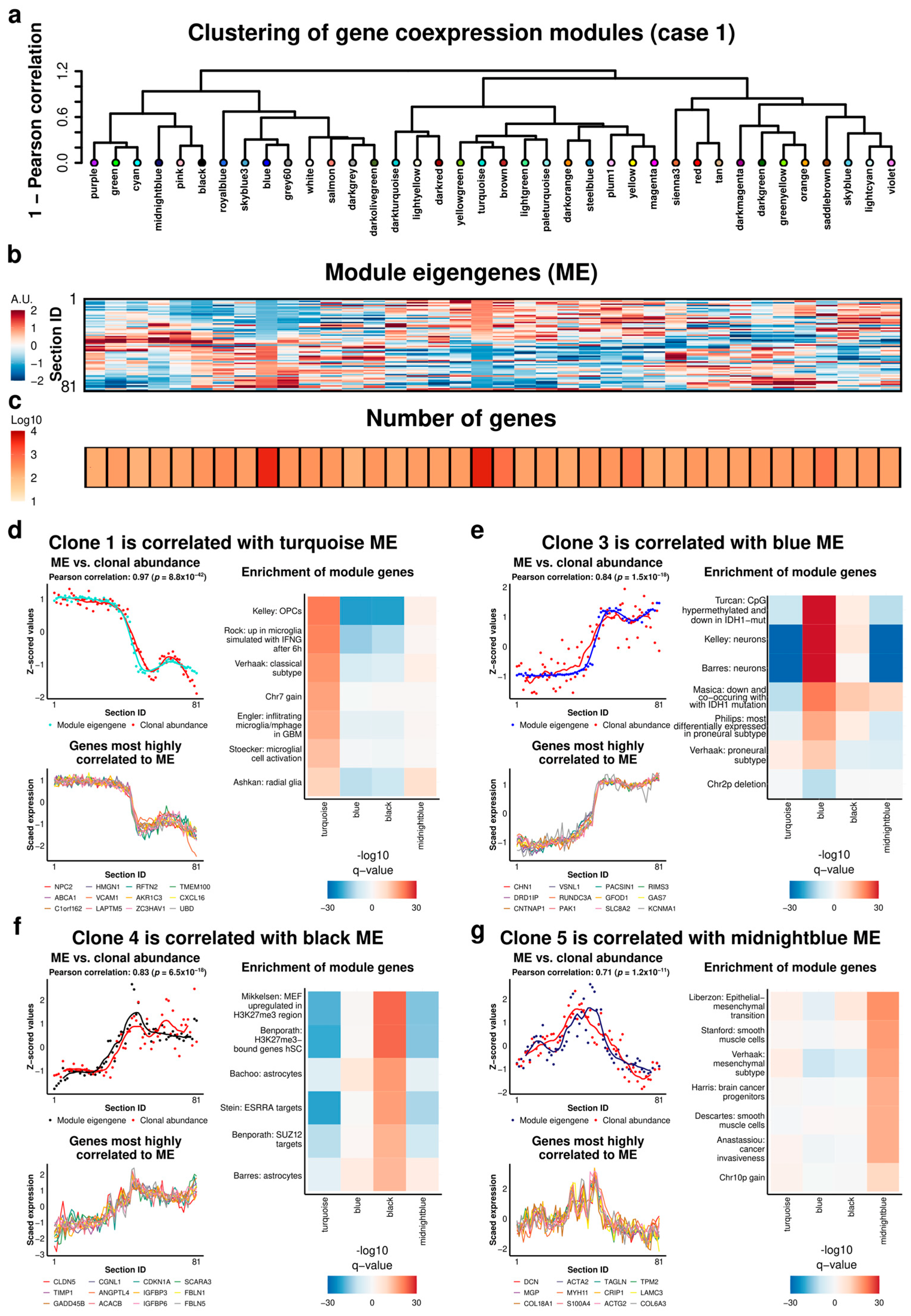
2.3. Case 1: Analysis of Gene Expression
2.4. Case 2: Analysis of Clonal Composition
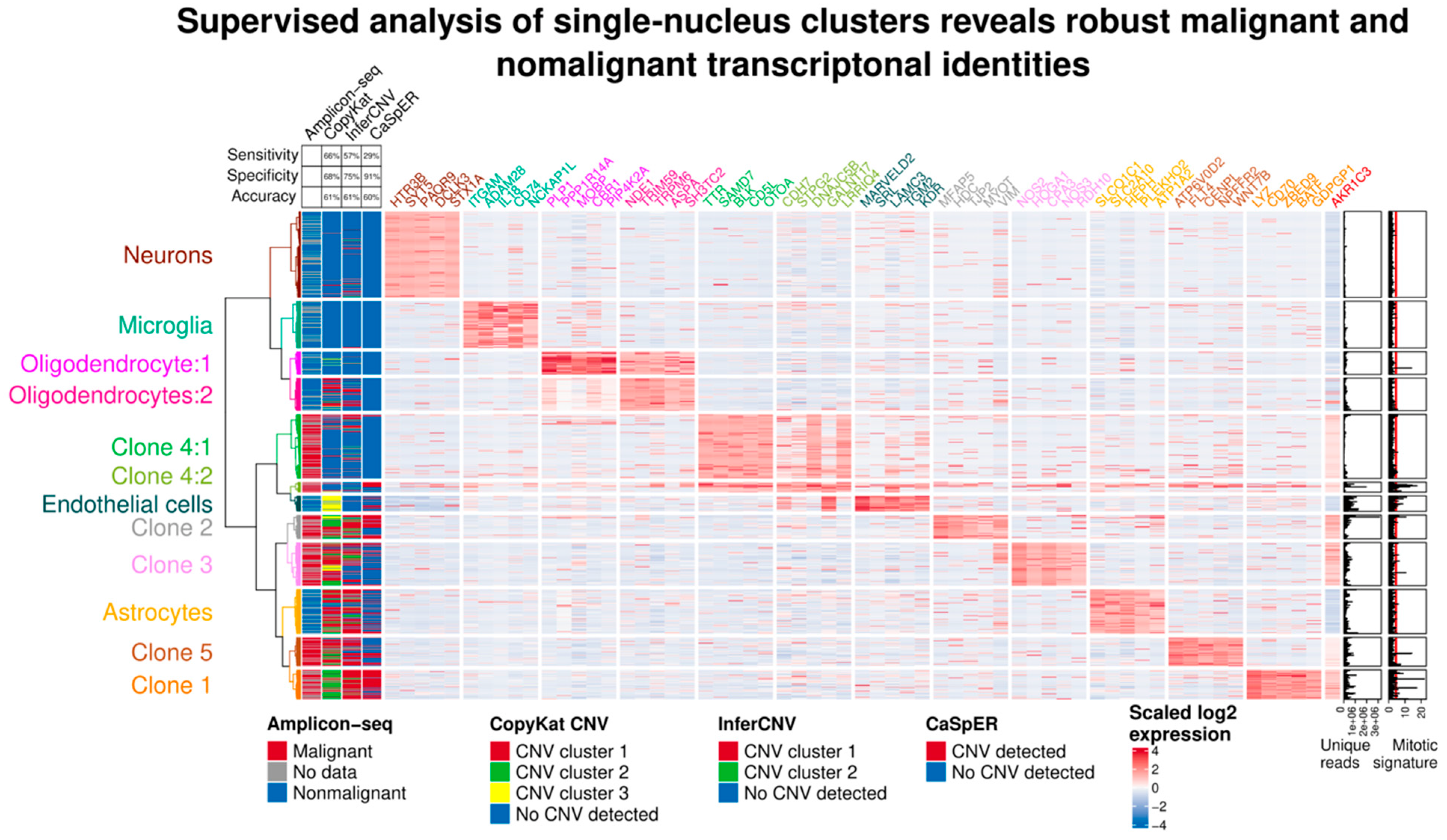
2.5. Case 2: Analysis of Gene Expression
2.6. Integrative Analysis of Gene Expression in Malignant Cells
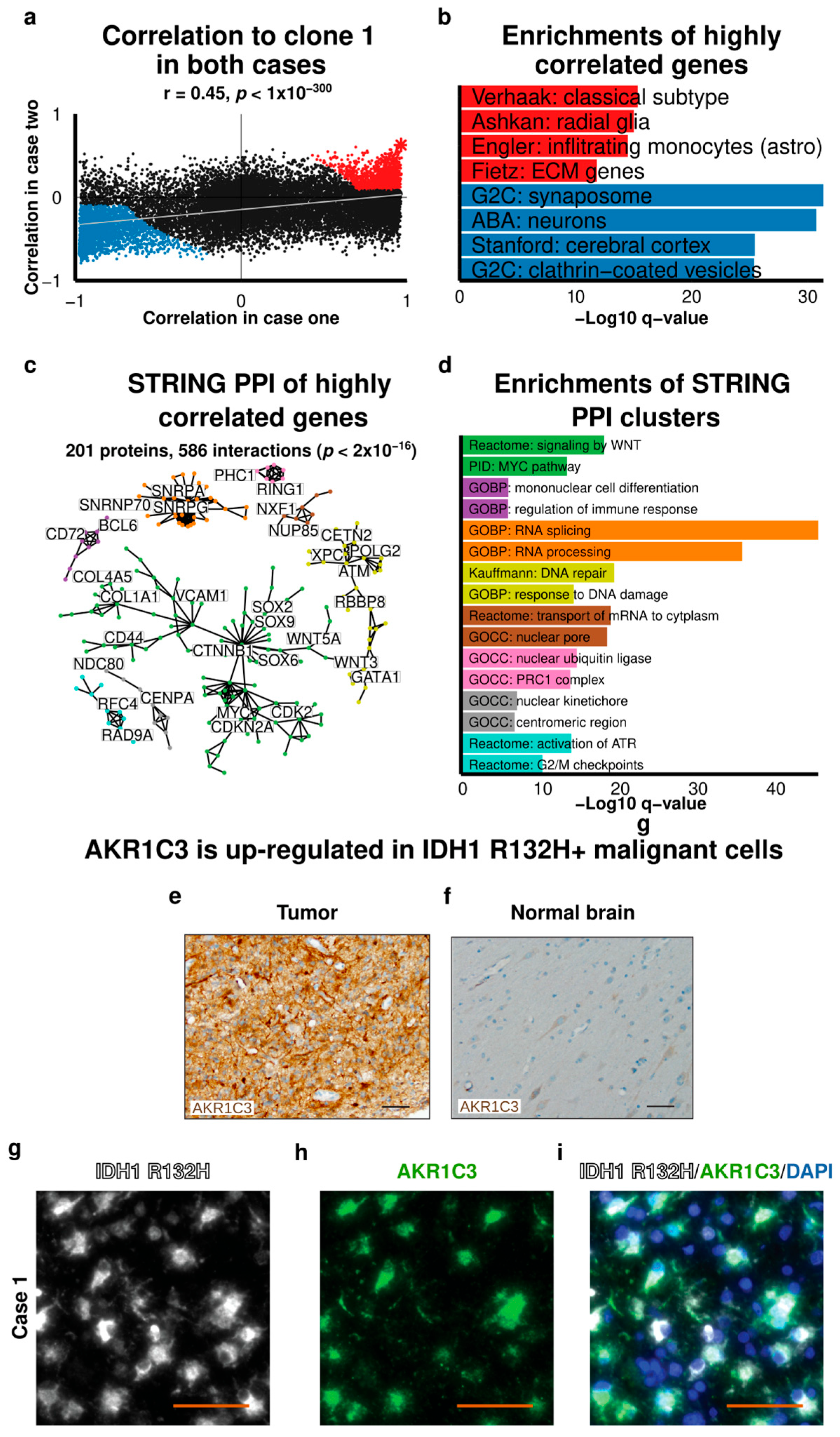
3. Discussion
4. Methods
4.1. Pseudobulk Analysis of scRNA-seq Data
4.2. Sample Acquisition
4.3. Serial Sectioning
4.4. Nucleic Acid Isolation and Quality Control
4.5. Whole-Exome Sequencing (WES) and Data Preprocessing
4.6. Single-Nucleotide Variant (SNV) and Small Insertion/Deletion (Indel) Calling Workflow
4.7. Droplet Digital PCR (ddPCR)
4.8. Amplicon Sequencing (amp-seq) and Data Preprocessing
4.9. Downsampling Analysis of amp-seq Data
4.10. Hierarchical Clustering of Variant Allele Frequencies (VAFs)
4.11. DNA Methylation Data Production and Preprocessing
4.12. Gene Expression Data Production and Preprocessing
4.13. Copy Number Analysis by qPCR
4.14. Copy Number Variation (CNV) Calling (Bulk Data)
4.15. Generation of Clonal Trees with Corresponding Frequencies
4.16. Gene Coexpression Network Analysis
4.17. Module Enrichment Analysis
4.18. Lasso Modeling of Gene Expression
4.19. Differential Gene Coexpression Analysis
4.20. Single-Nucleus DNA-Sequencing and Analysis
4.21. Single-Nucleus RNA-Sequencing and Analysis
4.21.1. Library Prep and Sequencing
4.21.2. Data Preprocessing
4.21.3. snRNA-seq Clustering and Differential Expression Analysis
4.21.4. CNV Calling
4.21.5. UMAP and Trajectory Analysis
4.21.6. Gene Set Enrichment Analysis
4.21.7. Amp-Seq Genotyping
4.22. Inter-Case Analysis
4.23. Histology and Immunostaining
4.24. Data Analysis and Figure Production
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral heterogeneity of the epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Christofides, A.; Strauss, L.; Yeo, A.; Cao, C.; Charest, A.; Boussiotis, V.A. The complex role of tumor-infiltrating macrophages. Nat. Immunol. 2022, 23, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in glioblastoma research: Focus on the tumor microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor heterogeneity: The rosetta stone of therapy resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz JR., L.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz JR., L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connely, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Loo, P.V.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal patterns of selection in cancer and somatic tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; Islam, S.M.A.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Pribluda, A.; de la Cruz, C.C.; Jackson, E.L. Intratumoral heterogeneity: From diversity comes resistance. Clin. Cancer Res. 2015, 21, 2916–2923. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Mooney, J.H.; Ilyas, A.; Chagoya, G.; Estevez-Ordonez, D.; Ibrahim, A.; Nakano, I. Molecular and cellular intratumoral heterogeneity in primary glioblastoma: Clinical and translational implications. J. Neurosurg. 2019, 133, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Seol, H.; Lee, H.J.; Choi, Y.; Lee, H.E.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Park, S.W. Intratumoral heterogeneity of HER2 gene amplification in breast cancer: Its clinicopathological significance. Mod. Pathol. 2012, 25, 938–948. [Google Scholar] [CrossRef]
- Losi, L.; Baisse, B.; Bouzourene, H.; Benhattar, J. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis 2005, 26, 916–922. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Gerlovina, I.; van der Laan, M.J.; Hubbard, A. Big data, small sample. Int. J. Biostat. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 2020, 9, giaa151. [Google Scholar] [CrossRef] [PubMed]
- Breda, J.; Zavolan, M.; van Nimwegen, E. Bayesian inference of gene expression states from single-cell RNA-seq data. Nat. Biotechnol. 2021, 39, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.J.; Ntranos, V.; Tse, D. Determining sequencing depth in a single-cell RNA-seq experiment. Nat. Commun. 2020, 11, 774. [Google Scholar] [CrossRef]
- Denisenko, E.; Guo, B.B.; Jones, M.; Huo, R.; de Kock, L.; Lassmann, T.; Poppe, D.; Clement, O.; Simmons, R.K.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130. [Google Scholar] [CrossRef]
- Caglayan, E.; Liu, Y.; Konopka, G. Neuronal ambient RNA contamination causes misinterpreted and masked cell types in brain single-nuclei datasets. Neuron 2022, 110, 4043–4056.e5. [Google Scholar] [CrossRef]
- Kelley, K.W.; Nakao-Inoue, H.; Molofsky, A.V.; Oldham, M.C. Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes. Nat. Neurosci. 2018, 21, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Oldham, M.C.; Konopka, G.; Iwamoto, K.; Langfelder, P.; Kato, T.; Horvath, S.; Geschwind, D.H. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008, 11, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.H.; Nowakowski, T.J.; Pollen, A.A.; Javaherian, A.; Kriegstein, A.R.; Oldham, M.C. Radial glia require PDGFD-PDGFRβ signalling in human but not mouse neocortex. Nature 2014, 515, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Raju, C.S.; Spatazza, J.; Stanco, A.; Larimer, P.; Sorrells, S.F.; Kelley, K.W.; Nicholas, C.R.; Paredes, M.F.; Lui, J.H.; Hasenstaub, A.R.; et al. Secretagogin is expressed by developing neocortical gabaergic neurons in humans but not mice and increases neurite arbor size and complexity. Cereb. Cortex 2018, 28, 1946–1958. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Cote, A.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Malikic, S.; McPherson, A.W.; Donmez, N.; Sahinalp, C.S. Clonality inference in multiple tumor samples using phylogeny. Bioinformatics 2015, 31, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cre, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Seshan, V.E. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016, 44, e131. [Google Scholar] [CrossRef] [PubMed]
- Feber, A.; Ghuilhamon, P.; Lechner, M.; Fenton, T.; Wilson, G.A.; Thirlwell, C.; Morris, T.J.; Flanagan, A.M.; Teschendorff, A.E.; Kelly, J.D.; et al. Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol. 2014, 15, R30. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Chesnelong, C.; Pankov, A.; Jalbert, L.E.; Hong, C.; Hayes, J.; Smirnov, I.V.; Marshall, R.; Souza, C.F.; Shen, Y.; et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc. Natl. Acad. Sci. USA 2017, 114, 10743–10748. [Google Scholar] [CrossRef]
- Favero, F.; McGranahan, N.; Salm, M.; Birkbak, N.J.; Sanborn, J.Z.; Benz, S.C.; Becq, J.; Peden, J.F.; Kingsbury, Z.; Grocok, R.J.; et al. Glioblastoma adaptation traced through decline of an IDH1 clonal driver and macro-evolution of a double-minute chromosome. Ann. Oncol. 2015, 26, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Pusch, S.; Sahm, F.; Mayer, J.; Mittelbronn, M.; Harmann, C.; von Deimling, A. IDH1 mutation patterns off the beaten track. Neuropathol. Appl. Neurobiol. 2011, 37, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Eastburn, D.J.; Sciambi, A.; Abate, A.R. Identification and genetic analysis of cancer cells with PCR-activated cell sorting. Nucleic Acids Res. 2014, 42, e128. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Meira, A.; O’Sullivan, J.; Rahman, H.; Mead, A.J. TARGET-Seq: A Protocol for High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. STAR Protocols 2020, 1, 100125. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousous, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305.e8. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schlack, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, M.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef]
- Serin Harmanci, A.; Harmanci, A.O.; Zhou, X. CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nat. Commun. 2020, 11, 89. [Google Scholar] [CrossRef]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Dong, J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Herta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Garcia-Diaz, C.; Poysti, A.; Mereu, E.; Clements, M.P.; Brooks, L.J.; Galvez-Cancino, F.; Castillo, S.P.; Tang, W.; Beattie, G.; Courtot, L.; et al. Glioblastoma cell fate is differentially regulated by the microenvironments of the tumor bulk and infiltrative margin. Cell Rep. 2023, 42, 112472. [Google Scholar] [CrossRef]
- De Silva, M.I.; Stringer, B.W.; Bardy, C. Neuronal and tumourigenic boundaries of glioblastoma plasticity. Trends Cancer 2023, 9, 223–236. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Spassky, N.; Merkle, F.T.; Flames, N.; Tramontin, A.D.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J. Neurosci. 2005, 25, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Redmond, S.A.; Figueres-Onate, M.; Obernier, K.; Nascimento, M.A.; Parraguez, J.I.; Lopez-Mascaraque, L.; Fuentealba, L.C.; Alvarez-Buylla, A. Development of Ependymal and Postnatal Neural Stem Cells and Their Origin from a Common Embryonic Progenitor. Cell Rep. 2019, 27, 429–441.e3. [Google Scholar] [CrossRef]
- Hahn, W.C.; Bader, J.S.; Braun, T.P.; Califano, A.; Clemons, P.A.; Druker, B.J.; Ewald, A.J.; Fu, H.; Jagu, S.; Kemp, C.J.; et al. An expanded universe of cancer targets. Cell 2021, 184, 1142–1155. [Google Scholar] [CrossRef]
- Penning, T.M. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol. Cell. Endocrinol. 2019, 489, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Tian, W.; Jiang, Z.; Huang, T.; Ge, C.; Liu, T.; Zhao, F.; Chen, T.; Cui, Y.; Li, H.; et al. A Positive Feedback Loop of AKR1C3-Mediated Activation of NF-κB and STAT3 Facilitates Proliferation and Metastasis in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 1361–1374. [Google Scholar] [CrossRef]
- Liu, C.; Luo, W.; Zhu, Y.; Yang, J.C.; Nadiminty, N.; Gaikwad, N.W.; Evans, C.P.; Gao, A.C. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Bortolozzi, R.; Bresolin, S.; Rampazzo, E.; Paganin, M.; Maule, F.; Mariotto, E.; Boso, D.; Minuzzo, S.; Agnusdei, V.; Viola, G.; et al. AKR1C enzymes sustain therapy resistance in paediatric T-ALL. Br. J. Cancer 2018, 118, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; van den Bent, M.J.; Blumenthal, D.T.; Touat, M.; Peters, K.B.; Clarke, J.; Mendez, J.; Yust-Katz, S.; Welsh, L.; Mason, W.P.; et al. Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma. N. Engl. J. Med. 2023, 389, 589–601. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.-G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Repository. In Picard Toolkit; Broad Institute: Cambridge, MA, USA, 2019. [Google Scholar]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 11, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennel, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Thorndike, R.L. Who belongs in the family? Psychometrika 1953, 18, 267–276. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions. Version 2.1.4. 2022. Available online: https://CRAN.R-project.org/package=cluster (accessed on 26 June 2024).
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. Champ: 450k chip analysis methylation pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.-P.; Triche, T.J.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017, 33, 558–560. [Google Scholar] [CrossRef]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017, 45, e22. [Google Scholar] [CrossRef]
- Dedeurwaerder, S.; Defrance, M.; Calonne, E.; Denis, H.; Sotiriou, C.; Fuks, F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011, 3, 771–784. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabero, D.; Beck, S.; Marabita, F.; Lechner, M.; Bartlett, T.; et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Oldham, M.C.; Langfelder, P.; Horvath, S. Network methods for describing sample relationships in genomic datasets: Application to Huntington’s disease. BMC Syst. Biol. 2012, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Barbosa-Morais, N.L.; Dunning, M.J.; Samarajiwa, S.A.; Darot, J.F.J.; Ritchie, M.E.; Lynch, A.G.; Tavare, S. A re-annotation pipeline for Illumina BeadArrays: Improving the interpretation of gene expression data. Nucleic Acids Res. 2010, 38, e17. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. version 0.11.9. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 26 June 2024).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Rosenbloom, K.R.; Dreszer, T.R.; Long, J.C.; Malladi, V.S.; Sloan, C.A.; Raney, B.J.; Cline, M.S.; Karolchik, D.; Barber, G.P.; Clawson, H.; et al. ENCODE whole-genome data in the UCSC Genome Browser: Update 2012. Nucleic Acids Res. 2012, 40, D912–D917. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef]
- Olshen, A.B.; Bengtsoon, H.; Neuvial, P.; Spellman, P.T.; Olshen, R.A.; Seshan, V.E. Parent-specific copy number in paired tumor-normal studies using circular binary segmentation. Bioinformatics 2011, 27, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, E.S.; Olshen, A.B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 2007, 23, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Glur, C. data.tree: General Purpose Hierarchical Data Structure. Version 1.0.0. 2020. Available online: https://CRAN.R-project.org/package=data.tree (accessed on 26 June 2024).
- Iannone, R. DiagrammeR: Graph/Network Visualization. Version 1.0.10. 2022. Available online: https://CRAN.R-project.org/package=DiagrammeR (accessed on 26 June 2024).
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Klosa, J.; Simon, N.; Westermark, P.O.; Liebscher, V.; Wittenburg, D. Seagull: Lasso, group lasso and sparse-group lasso regularization for linear regression models via proximal gradient descent. BMC Bioinform. 2020, 21, 407. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 58, 267–288. [Google Scholar] [CrossRef]
- Yuan, M.; Lin, Y. Model selection and estimation in regression with grouped variables. J. R. Stat. Soc. B 2006, 68, 49–67. [Google Scholar] [CrossRef]
- Laurin, C.; Boomsma, D.; Lubke, G. The use of vector bootstrapping to improve variable selection precision in Lasso models. Stat. Appl. Genet. Mol. Biol. 2016, 15, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.J.; Fan, G.; Plath, K.; Zhou, Q.; Horvath, S. Signed weighted gene co-expression network analysis of transcriptional regulation in murine embryonic stem cells. BMC Genom. 2009, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Tesson, B.M.; Breitling, R.; Jansen, R.C. DiffCoEx: A simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinform. 2010, 11, 497. [Google Scholar] [CrossRef]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Melville, J. uwot: The Uniform Manifold Approximation and Projection (UMAP) Method for Dimensionality Reduction. Version 0.1.16. 2021. Available online: https://CRAN.R-project.org/package=uwot (accessed on 26 June 2024).
- Untergasser, A. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N. BARCRAWL and BARTAB: Software tools for the design and implementation of barcoded primers for highly multiplexed DNA sequencing. BMC Bioinform. 2009, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Butts, C.T. network: A Package for Managing Relational Data in R. J. Stat. Softw. 2008, 24, 1–36. [Google Scholar] [CrossRef]
- Bojanowski, M. intergraph: Coercion Routines for Network Data Objects. 2015. Available online: http://mbojan.github.io/intergraph (accessed on 26 June 2024).
- Briatte, F. ggnetwork: Geometries to Plot Networks with “ggplot2”. 2021. Available online: https://CRAN.R-project.org/package=ggnetwork (accessed on 26 June 2024).
- Karlsson, M.; Zhang, C.; Mear, L.; Zhong, W.; Digre, A.; Katona, B.; Sjostedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Use R!); Springer: Berlin/Heidelberg, Germany, 2016; Volume 276. [Google Scholar]
- Dowle, M.; Srinivasan, A. data.table: Extension of ‘data.frame’. 2021. Available online: https://CRAN.R-project.org/package=data.table (accessed on 26 June 2024).
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes. 2022. Available online: https://CRAN.R-project.org/package=RColorBrewer (accessed on 26 June 2024).
- Auguie, B. gridExtra: Miscellaneous Functions for “Grid” Graphics. 2017. Available online: https://CRAN.R-project.org/package=gridExtra (accessed on 26 June 2024).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Ahlmann-Eltze, C.; Patil, I. ggsignif: R Package for Displaying Significance Brackets for “ggplot2”. Version 0.6.4. 2021. Available online: https://osf.io/preprints/psyarxiv/7awm6 (accessed on 26 June 2024).




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schupp, P.G.; Shelton, S.J.; Brody, D.J.; Eliscu, R.; Johnson, B.E.; Mazor, T.; Kelley, K.W.; Potts, M.B.; McDermott, M.W.; Huang, E.J.; et al. Deconstructing Intratumoral Heterogeneity through Multiomic and Multiscale Analysis of Serial Sections. Cancers 2024, 16, 2429. https://doi.org/10.3390/cancers16132429
Schupp PG, Shelton SJ, Brody DJ, Eliscu R, Johnson BE, Mazor T, Kelley KW, Potts MB, McDermott MW, Huang EJ, et al. Deconstructing Intratumoral Heterogeneity through Multiomic and Multiscale Analysis of Serial Sections. Cancers. 2024; 16(13):2429. https://doi.org/10.3390/cancers16132429
Chicago/Turabian StyleSchupp, Patrick G., Samuel J. Shelton, Daniel J. Brody, Rebecca Eliscu, Brett E. Johnson, Tali Mazor, Kevin W. Kelley, Matthew B. Potts, Michael W. McDermott, Eric J. Huang, and et al. 2024. "Deconstructing Intratumoral Heterogeneity through Multiomic and Multiscale Analysis of Serial Sections" Cancers 16, no. 13: 2429. https://doi.org/10.3390/cancers16132429
APA StyleSchupp, P. G., Shelton, S. J., Brody, D. J., Eliscu, R., Johnson, B. E., Mazor, T., Kelley, K. W., Potts, M. B., McDermott, M. W., Huang, E. J., Lim, D. A., Pieper, R. O., Berger, M. S., Costello, J. F., Phillips, J. J., & Oldham, M. C. (2024). Deconstructing Intratumoral Heterogeneity through Multiomic and Multiscale Analysis of Serial Sections. Cancers, 16(13), 2429. https://doi.org/10.3390/cancers16132429