
Gene Expression Pattern of ESPL1, PTTG1 and PTTG1IP Can Potentially Predict Response to TKI First-Line Treatment of Patients with Newly Diagnosed CML

 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Molecular Response in Treatment of Chronic Myeloid Leukemia
1.2. ESPL1/Separase, PTTG1/Securin and PTTG1IP/Securin Interacting Protein
2. Materials and Methods
2.1. Patients and Controls
2.2. Sample Preparation and Quantification of BCR::ABL1
2.3. Relative Quantification of ESPL1, PTTG1, PTTG1IP Transcript Levels
2.4. Statistics
3. Results
3.1. Relative Gene Expression of ESPL1, PTTG1 and PTTG1IP
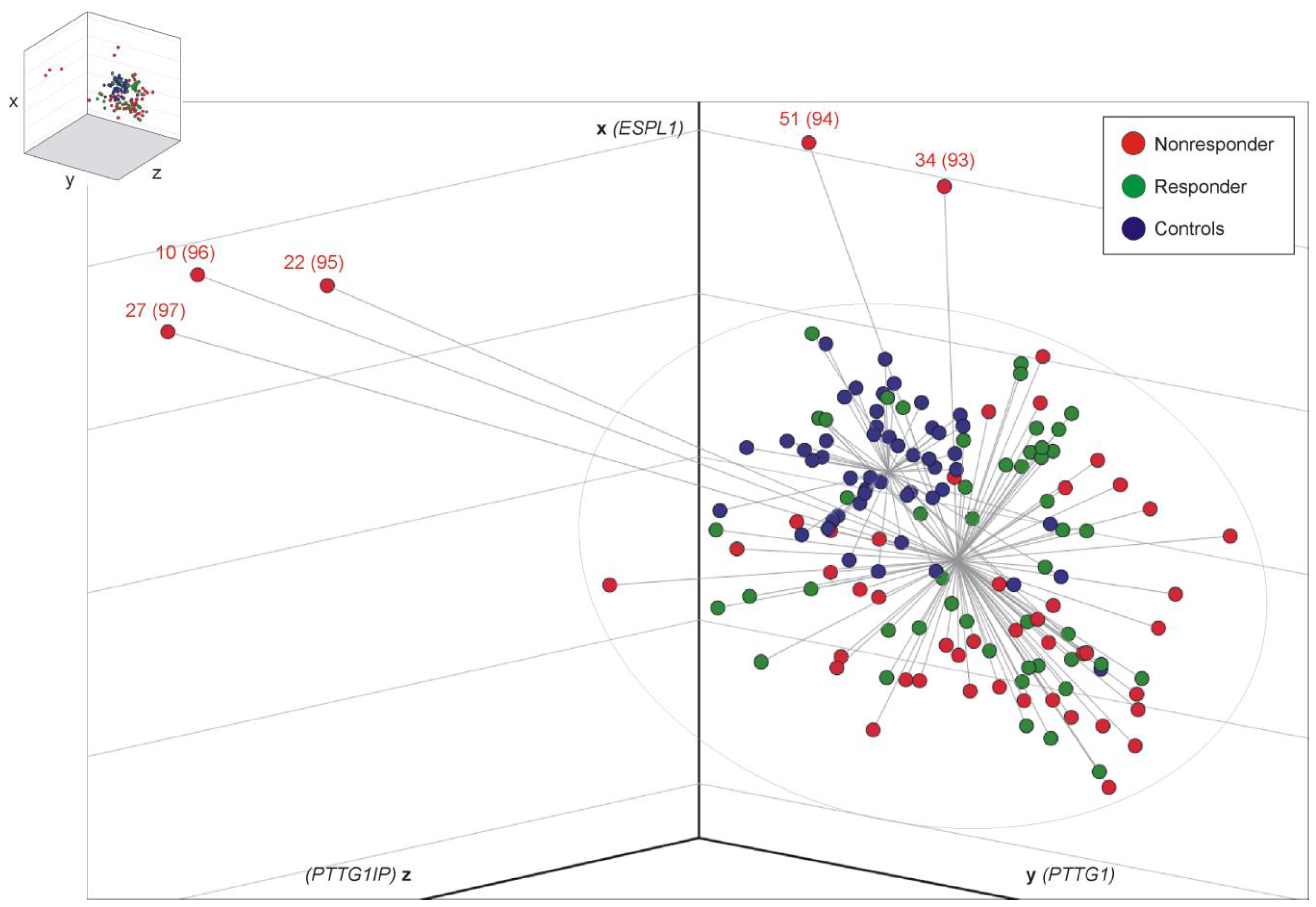
3.2. Distance Analysis for Risk Stratification
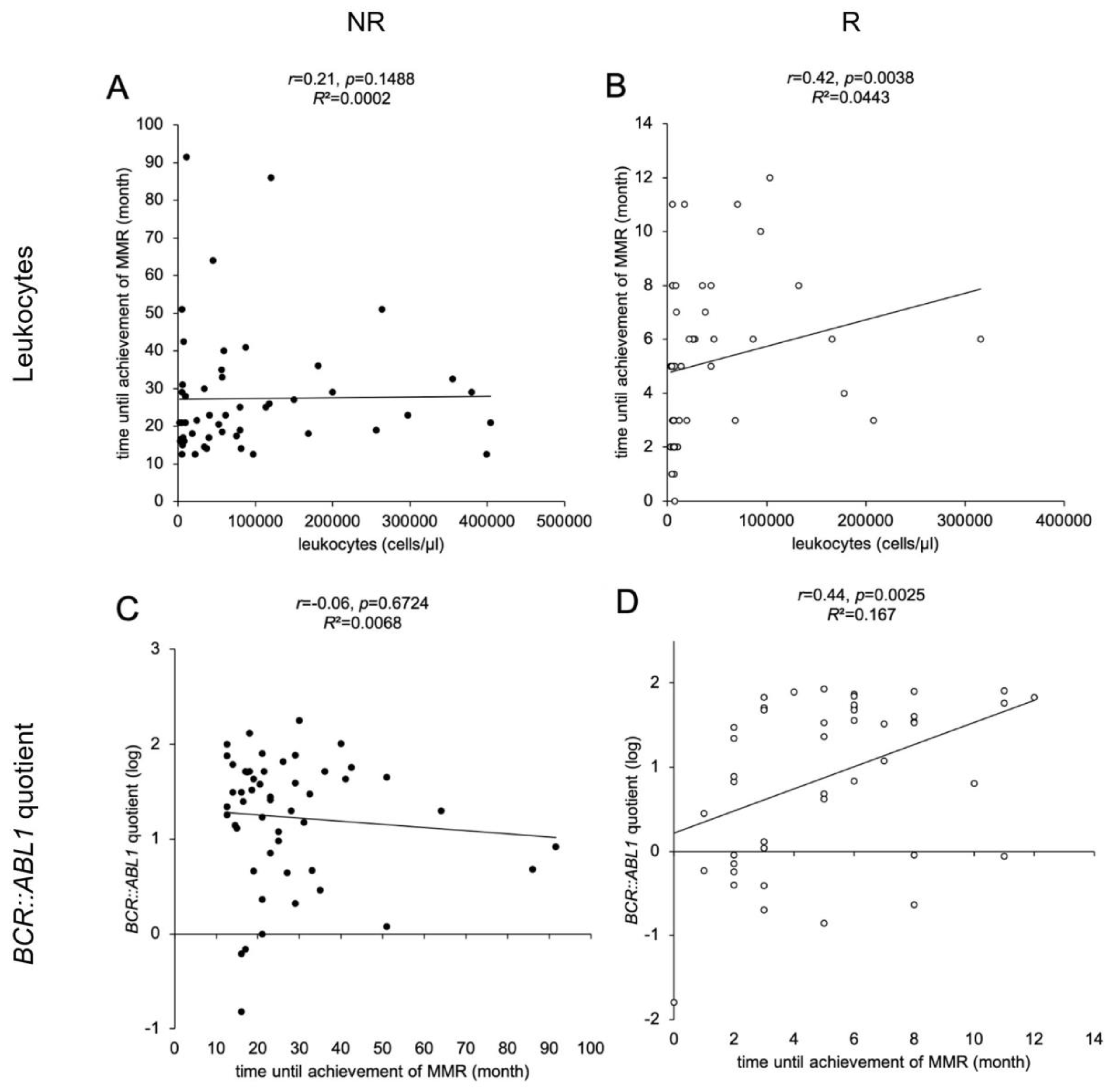
3.3. Leukocyte Count at ID Differs between R and NR Cohort
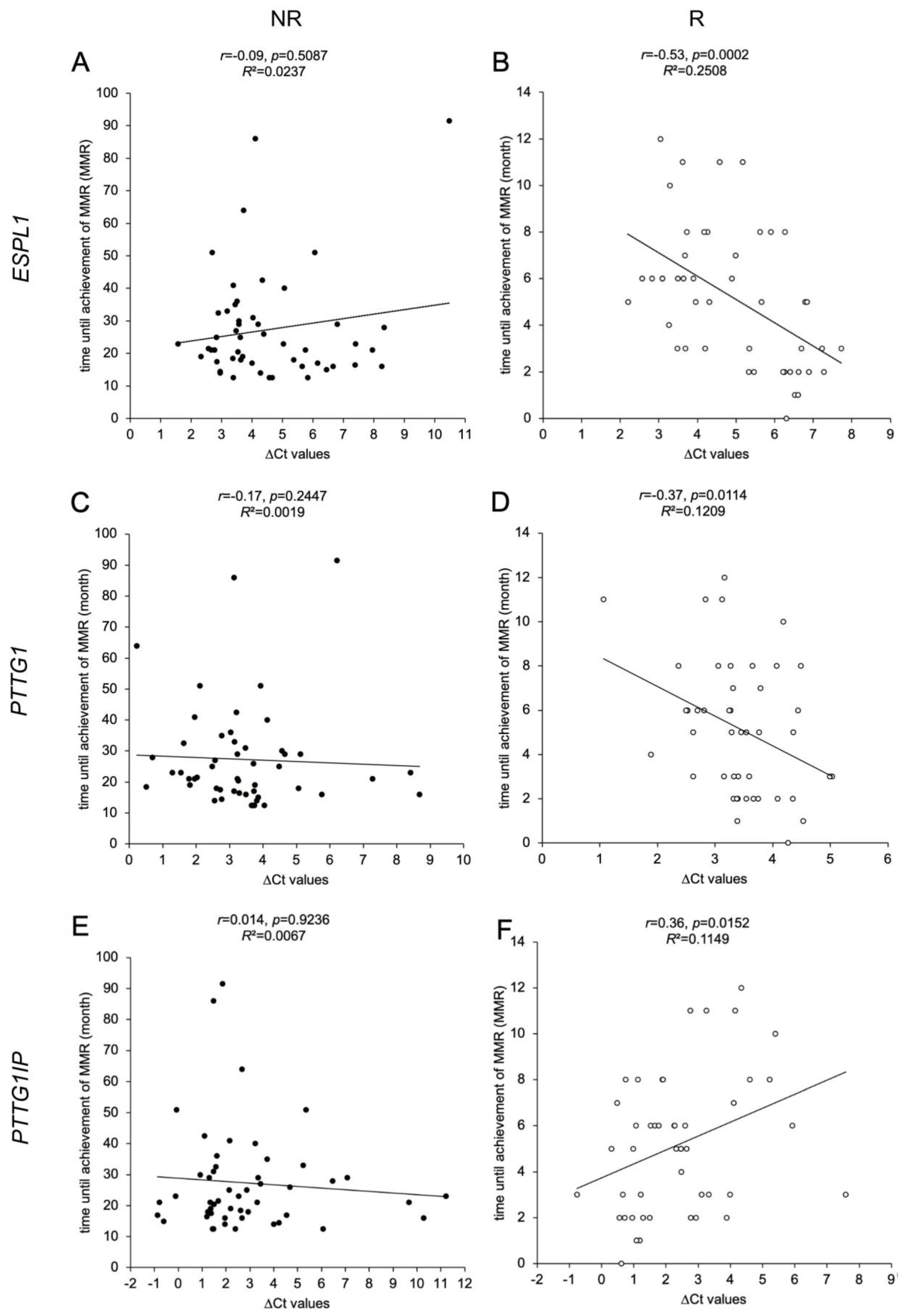
3.4. ESPL1, PTTG1 and PTTG1IP Gene Expression Levels in R Cohort Correlate with Time until Achievement of MMR
3.5. Leukocyte Counts at Time of ID, BCR::ABL1 Quotients and TKI Therapy Correlate with Time until Achievement of MMR
3.6. Predictable NR Display Lower ESPL1 and PTTG1IP Transcript Levels Compared to Corresponding R
3.7. NR and R Cohort Assignment Concurs with Rate of BCR::ABL1 Decline after 3 Months of TKI Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hehlmann, R.; Lauseker, M.; Saussele, S.; Pfirrmann, M.; Krause, S.; Kolb, H.J.; Neubauer, A.; Hossfeld, D.K.; Nerl, C.; Gratwohl, A.; et al. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia 2017, 31, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed]
- Branford, S.; Fletcher, L.; Cross, N.C.; Muller, M.C.; Hochhaus, A.; Kim, D.W.; Radich, J.P.; Saglio, G.; Pane, F.; Kamel-Reid, S.; et al. Desirable performance characteristics for BCR-ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response and comparison of response rates between clinical trials. Blood 2008, 112, 3330–3338. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.; White, H.E.; Muller, M.C.; Saglio, G.; Hochhaus, A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia 2012, 26, 2172–2175. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.; White, H.E.; Colomer, D.; Ehrencrona, H.; Foroni, L.; Gottardi, E.; Lange, T.; Lion, T.; Machova Polakova, K.; Dulucq, S.; et al. Laboratory recommendations for scoring deep molecular responses following treatment for chronic myeloid leukemia. Leukemia 2015, 29, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Castagnetti, F.; Gugliotta, G.; Rosti, G. A review of the European LeukemiaNet recommendations for the management of CML. Ann. Hematol. 2015, 94 (Suppl. S2), S141–S147. [Google Scholar] [CrossRef]
- Guilhot, J.; Preudhomme, C.; Mahon, F.X.; Guilhot, F. Analyzing molecular response in chronic myeloid leukemia clinical trials: Pitfalls and golden rules. Cancer 2015, 121, 490–497. [Google Scholar] [CrossRef]
- Wolf, D.; Sopper, S. Molecular response prediction in CML: Novel ideas? Oncotarget 2017, 8, 80105–80106. [Google Scholar] [CrossRef]
- Branford, S.; Yeung, D.T.; Parker, W.T.; Roberts, N.D.; Purins, L.; Braley, J.A.; Altamura, H.K.; Yeoman, A.L.; Georgievski, J.; Jamison, B.A.; et al. Prognosis for patients with CML and >10% BCR-ABL1 after 3 months of imatinib depends on the rate of BCR-ABL1 decline. Blood 2014, 124, 511–518. [Google Scholar] [CrossRef]
- Breccia, M.; Molica, M.; Colafigli, G.; Massaro, F.; Alimena, G. Early molecular response in chronic myeloid leukemia and halving time: Latest evidences. Leuk. Res. 2016, 48, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Glauche, I.; Kuhn, M.; Baldow, C.; Schulze, P.; Rothe, T.; Liebscher, H.; Roy, A.; Wang, X.; Roeder, I. Quantitative prediction of long-term molecular response in TKI-treated CML—Lessons from an imatinib versus dasatinib comparison. Sci. Rep. 2018, 8, 12330. [Google Scholar] [CrossRef] [PubMed]
- Spiess, B.; Kleiner, H.; Flach, J.; Fabarius, A.; Saussele, S.; Hofmann, W.K.; Seifarth, W. Separase activity distribution can be a marker of major molecular response and proliferation of CD34(+) cells in TKI-treated chronic myeloid leukemia patients. Ann. Hematol. 2020, 99, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Chestukhin, A.; Pfeffer, C.; Milligan, S.; DeCaprio, J.A.; Pellman, D. Processing, localization, and requirement of human separase for normal anaphase progression. Proc. Natl. Acad. Sci. USA 2003, 100, 4574–4579. [Google Scholar] [CrossRef]
- Schockel, L.; Mockel, M.; Mayer, B.; Boos, D.; Stemmann, O. Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat. Cell. Biol. 2011, 13, 966–972. [Google Scholar] [CrossRef]
- Leeke, B.; Marsman, J.; O’Sullivan, J.M.; Horsfield, J.A. Cohesin mutations in myeloid malignancies: Underlying mechanisms. Exp. Hematol. Oncol. 2014, 3, 13. [Google Scholar] [CrossRef]
- McAleenan, A.; Clemente-Blanco, A.; Cordon-Preciado, V.; Sen, N.; Esteras, M.; Jarmuz, A.; Aragon, L. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature 2013, 493, 250–254. [Google Scholar] [CrossRef]
- Cucco, F.; Palumbo, E.; Camerini, S.; D’Alessio, B.; Quarantotti, V.; Casella, M.L.; Rizzo, I.M.; Cukrov, D.; Delia, D.; Russo, A.; et al. Separase prevents genomic instability by controlling replication fork speed. Nucleic Acids Res. 2018, 46, 267–278. [Google Scholar] [CrossRef]
- Hellmuth, S.; Gutierrez-Caballero, C.; Llano, E.; Pendas, A.M.; Stemmann, O. Local activation of mammalian separase in interphase promotes double-strand break repair and prevents oncogenic transformation. EMBO J. 2018, 37, e99184. [Google Scholar] [CrossRef]
- Zhang, N.; Ge, G.; Meyer, R.; Sethi, S.; Basu, D.; Pradhan, S.; Zhao, Y.J.; Li, X.N.; Cai, W.W.; El-Naggar, A.K.; et al. Overexpression of Separase induces aneuploidy and mammary tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 13033–13038. [Google Scholar] [CrossRef]
- Haass, W.; Kleiner, H.; Weiss, C.; Haferlach, C.; Schlegelberger, B.; Muller, M.C.; Hehlmann, R.; Hofmann, W.K.; Fabarius, A.; Seifarth, W.; et al. Clonal Evolution and Blast Crisis Correlate with Enhanced Proteolytic Activity of Separase in BCR-ABL b3a2 Fusion Type CML under Imatinib Therapy. PLoS ONE 2015, 10, e0129648. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Ge, G.; Zhang, N.; Huang, E.; Nakamura, L.V.; Minor, M.; Fofanov, V.; Rao, P.H.; Herron, A.; Pati, D. Separase loss of function cooperates with the loss of p53 in the initiation and progression of T- and B-cell lymphoma, leukemia and aneuploidy in mice. PLoS ONE 2011, 6, e22167. [Google Scholar] [CrossRef] [PubMed]
- Pati, D. Oncogenic activity of separase. Cell Cycle 2008, 7, 3481–3482. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Fofanov, V.; Panigrahi, A.; Merchant, F.; Zhang, N.; Pati, D. Overexpression and mislocalization of the chromosomal segregation protein separase in multiple human cancers. Clin. Cancer Res. 2009, 15, 2703–2710. [Google Scholar] [CrossRef]
- Uhlmann, F. Secured cutting: Controlling separase at the metaphase to anaphase transition. EMBO Rep. 2001, 2, 487–492. [Google Scholar] [CrossRef]
- Waizenegger, I.; Gimenez-Abian, J.F.; Wernic, D.; Peters, J.M. Regulation of human separase by securin binding and autocleavage. Curr. Biol. 2002, 12, 1368–1378. [Google Scholar] [CrossRef]
- Holland, A.J.; Taylor, S.S. Cyclin-B1-mediated inhibition of excess separase is required for timely chromosome disjunction. J. Cell Sci. 2006, 119, 3325–3336. [Google Scholar] [CrossRef]
- Hornig, N.C.; Knowles, P.P.; McDonald, N.Q.; Uhlmann, F. The dual mechanism of separase regulation by securin. Curr. Biol. 2002, 12, 973–982. [Google Scholar] [CrossRef]
- Vlotides, G.; Eigler, T.; Melmed, S. Pituitary tumor-transforming gene: Physiology and implications for tumorigenesis. Endocr. Rev. 2007, 28, 165–186. [Google Scholar] [CrossRef]
- Agircan, F.G.; Schiebel, E. Sensors at centrosomes reveal determinants of local separase activity. PLoS Genet. 2014, 10, e1004672. [Google Scholar] [CrossRef]
- Imruetaicharoenchoke, W.; Fletcher, A.; Lu, W.; Watkins, R.J.; Modasia, B.; Poole, V.L.; Nieto, H.R.; Thompson, R.J.; Boelaert, K.; Read, M.L.; et al. Functional consequences of the first reported mutations of the proto-oncogene PTTG1IP/PBF. Endocr. Relat. Cancer 2017, 24, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Read, M.L.; Seed, R.I.; Fong, J.C.; Modasia, B.; Ryan, G.A.; Watkins, R.J.; Gagliano, T.; Smith, V.E.; Stratford, A.L.; Kwan, P.K.; et al. The PTTG1-binding factor (PBF/PTTG1IP) regulates p53 activity in thyroid cells. Endocrinology 2014, 155, 1222–1234. [Google Scholar] [CrossRef]
- Cross, N.C.; Feng, L.; Bungey, J.; Goldman, J.M. Minimal residual disease after bone marrow transplant for chronic myeloid leukaemia detected by the polymerase chain reaction. Leuk. Lymphoma 1993, 11 (Suppl. S1), 39–43. [Google Scholar] [CrossRef] [PubMed]
- Emig, M.; Saussele, S.; Wittor, H.; Weisser, A.; Reiter, A.; Willer, A.; Berger, U.; Hehlmann, R.; Cross, N.C.; Hochhaus, A. Accurate and rapid analysis of residual disease in patients with CML using specific fluorescent hybridization probes for real time quantitative RT-PCR. Leukemia 1999, 13, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Spiess, B.; Rinaldetti, S.; Naumann, N.; Galuschek, N.; Kossak-Roth, U.; Wuchter, P.; Tarnopolscaia, I.; Rose, D.; Voskanyan, A.; Fabarius, A.; et al. Diagnostic performance of the molecular BCR-ABL1 monitoring system may impact on inclusion of CML patients in stopping trials. PLoS ONE 2019, 14, e0214305. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Yu, H.; Wang, L.; Chen, D.; Li, J.; Guo, Y. Conditional transcriptional relationships may serve as cancer prognostic markers. BMC Med. Genom. 2021, 14, 101. [Google Scholar] [CrossRef]
- Smahelova, J.; Kastankova, I.; Polakova, K.M.; Klamova, H.; Zemanova, K.; Tachezy, R.; Hamsikova, E.; Smahel, M. Expression of genes encoding centrosomal proteins and the humoral response against these proteins in chronic myeloid leukemia. Oncol. Rep. 2017, 37, 547–554. [Google Scholar] [CrossRef]
- Haass, W.; Stehle, M.; Nittka, S.; Giehl, M.; Schrotz-King, P.; Fabarius, A.; Hofmann, W.K.; Seifarth, W. The proteolytic activity of separase in BCR-ABL-positive cells is increased by imatinib. PLoS ONE 2012, 7, e42863. [Google Scholar] [CrossRef]
- Noll, J.E.; Vandyke, K.; Hewett, D.R.; Mrozik, K.M.; Bala, R.J.; Williams, S.A.; Kok, C.H.; Zannettino, A.C. PTTG1 expression is associated with hyperproliferative disease and poor prognosis in multiple myeloma. J. Hematol. Oncol. 2015, 8, 106. [Google Scholar] [CrossRef]
- Xu, J.; Wang, M.; Gao, X.; Hu, B.; Du, Y.; Zhou, J.; Tian, X.; Huang, X. Separase phosphosite mutation leads to genome instability and primordial germ cell depletion during oogenesis. PLoS ONE 2011, 6, e18763. [Google Scholar] [CrossRef] [PubMed]
- Repo, H.; Gurvits, N.; Loyttyniemi, E.; Nykanen, M.; Lintunen, M.; Karra, H.; Kurki, S.; Kuopio, T.; Talvinen, K.; Soderstrom, M.; et al. PTTG1-interacting protein (PTTG1IP/PBF) predicts breast cancer survival. BMC Cancer 2017, 17, 705. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, L.; Skov, V.; Andersen, M.T.; Aggerholm, A.; Clair, P.; Gniot, M.; Soeby, K.; Udby, L.; Dorff, M.H.; Hasselbalch, H.; et al. Variant-specific discrepancy when quantitating BCR-ABL1 e13a2 and e14a2 transcripts using the Europe Against Cancer qPCR assay. Eur. J. Haematol. 2019, 103, 26–34. [Google Scholar] [CrossRef]
- Bernardi, S.; Bonifacio, M.; Iurlo, A.; Zanaglio, C.; Tiribelli, M.; Binotto, G.; Abruzzese, E.; Russo, D. “Variant-specific discrepancy when quantitating BCR-ABL1 e13a2 and e14a2 transcripts using the Europe Against Cancer qPCR assay.” Is dPCR the key? Eur. J. Haematol. 2019, 103, 272–273. [Google Scholar] [CrossRef]
- Foroni, L.; Wilson, G.; Gerrard, G.; Mason, J.; Grimwade, D.; White, H.E.; de Castro, D.G.; Austin, S.; Awan, A.; Burt, E.; et al. Guidelines for the measurement of BCR-ABL1 transcripts in chronic myeloid leukaemia. Br. J. Haematol. 2011, 153, 179–190. [Google Scholar] [CrossRef] [PubMed]
- White, H.E.; Salmon, M.; Albano, F.; Andersen, C.S.A.; Balabanov, S.; Balatzenko, G.; Barbany, G.; Cayuela, J.M.; Cerveira, N.; Cochaux, P.; et al. Standardization of molecular monitoring of CML: Results and recommendations from the European treatment and outcome study. Leukemia 2022, 36, 1834–1842. [Google Scholar] [CrossRef]
- Experts in Chronic Myeloid, L. The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: From the perspective of a large group of CML experts. Blood 2013, 121, 4439–4442. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ANOVA | ESPL1 | PTTG1 | PTTG1IP |
|---|---|---|---|
| global | p < 0.0001 | p = 0.0036 | p = 0.1736 |
| controls vs. NR * | p < 0.0001 | p = 0.0127 | - |
| controls vs. R * | p = 0.0061 | p = 0.0167 | - |
| NR vs. R * | p = 0.3910 | p = 0.9998 | - |
| Sample No. | Sensitivity | Specificity | Sum | Cut-Off |
|---|---|---|---|---|
| 1 | 1 | 0 | 1 | 0.549594 |
| 2 | 1 | 0.021739 | 1.021739 | 0.633119 |
| 3 | 1 | 0.043478 | 1.043478 | 0.750284 |
| 4 | 1 | 0.065217 | 1.065217 | 0.773095 |
| 5 | 1 | 0.086957 | 1.086957 | 0.951466 |
| 6 | 1 | 0.108696 | 1.108696 | 0.980357 |
| 7 | 0.980392 | 0.108696 | 1.089088 | 1.150964 |
| 8 | 0.960784 | 0.108696 | 1.06948 | 1.156968 |
| 9 | 0.960784 | 0.130435 | 1.091219 | 1.162309 |
| 10 | 0.960784 | 0.152174 | 1.112958 | 1.17719 |
| 11 | 0.941176 | 0.152174 | 1.09335 | 1.244523 |
| 12 | 0.921569 | 0.152174 | 1.073743 | 1.335997 |
| 13 | 0.901961 | 0.152174 | 1.054135 | 1.355251 |
| 14 | 0.882353 | 0.152174 | 1.034527 | 1.441234 |
| 15 | 0.882353 | 0.173913 | 1.056266 | 1.488975 |
| 16 | 0.862745 | 0.173913 | 1.036658 | 1.534161 |
| 17 | 0.843137 | 0.173913 | 1.01705 | 1.601827 |
| 18 | 0.823529 | 0.173913 | 0.997442 | 1.605338 |
| 19 | 0.823529 | 0.195652 | 1.019182 | 1.61878 |
| 20 | 0.823529 | 0.217391 | 1.040921 | 1.637496 |
| 21 | 0.803922 | 0.217391 | 1.021313 | 1.647798 |
| 22 | 0.803922 | 0.23913 | 1.043052 | 1.652854 |
| 23 | 0.784314 | 0.23913 | 1.023444 | 1.708054 |
| 24 | 0.764706 | 0.23913 | 1.003836 | 1.757046 |
| 25 | 0.764706 | 0.26087 | 1.025575 | 1.793212 |
| 26 | 0.745098 | 0.26087 | 1.005968 | 1.793669 |
| 27 | 0.72549 | 0.26087 | 0.98636 | 1.79974 |
| 28 | 0.705882 | 0.26087 | 0.966752 | 1.810044 |
| 29 | 0.705882 | 0.282609 | 0.988491 | 1.840533 |
| 30 | 0.686275 | 0.282609 | 0.968883 | 1.864345 |
| 31 | 0.686275 | 0.304348 | 0.990622 | 1.95028 |
| 32 | 0.666667 | 0.304348 | 0.971014 | 1.964042 |
| 33 | 0.666667 | 0.326087 | 0.992754 | 2.015026 |
| 34 | 0.666667 | 0.347826 | 1.014493 | 2.039745 |
| 35 | 0.647059 | 0.347826 | 0.994885 | 2.042153 |
| 36 | 0.627451 | 0.347826 | 0.975277 | 2.05187 |
| 37 | 0.627451 | 0.369565 | 0.997016 | 2.057197 |
| 38 | 0.607843 | 0.369565 | 0.977408 | 2.069368 |
| 39 | 0.607843 | 0.391304 | 0.999147 | 2.085874 |
| 40 | 0.588235 | 0.391304 | 0.97954 | 2.096926 |
| 41 | 0.568627 | 0.391304 | 0.959932 | 2.122027 |
| 42 | 0.568627 | 0.413043 | 0.981671 | 2.124711 |
| 43 | 0.568627 | 0.434783 | 1.00341 | 2.180675 |
| 44 | 0.568627 | 0.456522 | 1.025149 | 2.189591 |
| 45 | 0.54902 | 0.456522 | 1.005541 | 2.195555 |
| 46 | 0.54902 | 0.478261 | 1.02728 | 2.20937 |
| 47 | 0.54902 | 0.5 | 1.04902 | 2.277969 |
| 48 | 0.54902 | 0.521739 | 1.070759 | 2.283053 |
| 49 | 0.54902 | 0.543478 | 1.092498 | 2.316287 |
| 50 | 0.54902 | 0.565217 | 1.114237 | 2.358355 |
| 51 | 0.54902 | 0.586957 | 1.135976 | 2.363681 |
| 52 | 0.529412 | 0.586957 | 1.116368 | 2.376338 |
| 53 | 0.509804 | 0.586957 | 1.09676 | 2.405917 |
| 54 | 0.509804 | 0.608696 | 1.1185 | 2.429324 |
| 55 | 0.509804 | 0.630435 | 1.140239 | 2.444764 |
| 56 | 0.509804 | 0.652174 | 1.161978 | 2.456408 |
| 57 | 0.509804 | 0.673913 | 1.183717 | 2.465865 |
| 58 | 0.490196 | 0.673913 | 1.164109 | 2.469809 |
| 59 | 0.470588 | 0.673913 | 1.144501 | 2.516639 |
| 60 | 0.45098 | 0.673913 | 1.124893 | 2.564055 |
| 61 | 0.45098 | 0.695652 | 1.146633 | 2.565945 |
| 62 | 0.45098 | 0.717391 | 1.168372 | 2.611456 |
| 63 | 0.431373 | 0.717391 | 1.148764 | 2.678705 |
| 64 | 0.431373 | 0.73913 | 1.170503 | 2.710391 |
| 65 | 0.431373 | 0.76087 | 1.192242 | 2.73141 |
| 66 | 0.411765 | 0.76087 | 1.172634 | 2.771444 |
| 67 | 0.392157 | 0.76087 | 1.153026 | 2.799222 |
| 68 | 0.392157 | 0.782609 | 1.174766 | 2.815116 |
| 69 | 0.392157 | 0.804348 | 1.196505 | 2.85643 |
| 70 | 0.392157 | 0.826087 | 1.218244 | 2.860967 |
| 71 | 0.372549 | 0.826087 | 1.198636 | 2.880366 |
| 72 * | 0.372549 | 0.847826 | 1.220375 | 3.021803 |
| 73 | 0.352941 | 0.847826 | 1.200767 | 3.028484 |
| 74 | 0.333333 | 0.847826 | 1.181159 | 3.061431 |
| 75 | 0.313725 | 0.847826 | 1.161552 | 3.115181 |
| 76 | 0.313725 | 0.869565 | 1.183291 | 3.149723 |
| 77 | 0.313725 | 0.891304 | 1.20503 | 3.156747 |
| 78 | 0.294118 | 0.891304 | 1.185422 | 3.205288 |
| 79 | 0.27451 | 0.891304 | 1.165814 | 3.209935 |
| 80 | 0.27451 | 0.913043 | 1.187553 | 3.334363 |
| 81 | 0.254902 | 0.913043 | 1.167945 | 3.347152 |
| 82 | 0.235294 | 0.913043 | 1.148338 | 3.452303 |
| 83 | 0.235294 | 0.934783 | 1.170077 | 3.481048 |
| 84 | 0.215686 | 0.934783 | 1.150469 | 3.481381 |
| 85 | 0.215686 | 0.956522 | 1.172208 | 3.547112 |
| 86 | 0.215686 | 0.978261 | 1.193947 | 3.579134 |
| 87 | 0.196078 | 0.978261 | 1.174339 | 3.624489 |
| 88 | 0.176471 | 0.978261 | 1.154731 | 3.698418 |
| 89 | 0.156863 | 0.978261 | 1.135124 | 3.789116 |
| 90 | 0.137255 | 0.978261 | 1.115516 | 3.802186 |
| 91 | 0.117647 | 0.978261 | 1.095908 | 4.781442 |
| 92 | 0.098039 | 0.978261 | 1.0763 | 5.099709 |
| 93 ** | 0.098039 | 1 | 1.098039 | 5.917999 |
| 94 | 0.078431 | 1 | 1.078431 | 6.435286 |
| 95 | 0.058824 | 1 | 1.058824 | 8.635014 |
| 96 | 0.039216 | 1 | 1.039216 | 9.944185 |
| 97 | 0.019608 | 1 | 1.019608 | 10.28212 |
| Parameter | Test | p-Value |
|---|---|---|
| Age | t-test | 0.873 |
| Sex | Chi2-test | 0.990 |
| Time until achievement of MMR | U-test | <0.001 |
| Leukocyte count at ID | U-test | 0.018 |
| BCR::ABL1 gene fusion type | Fisher-test | 0.487 |
| BCR::ABL1 quotient | U-test | 0.067 |
| BCR::ABL1 quotient IS * | U-test | 0.173 |
| Proportion of imatinib to administered TKIs | U-test | 0.007 |
| NR | R | p-Values | |
|---|---|---|---|
| Distance * | 8.42 ± 1.99 | 2.47 ± 0.18 | 0.0028 |
| ΔCt ** ESPL1 | 8.48 ± 1.17 | 6.52 ± 0.25 | 0.018 |
| ΔCt ** PTTG1 | 6.26 ± 3.26 | 3.97 ± 0.57 | 0.1936 |
| ΔCt ** PTTG1IP | 7.89 ± 3.82 | 1.32 ± 0.84 | 0.0168 |
| Leukocytes (cells/μL) | 19760 ± 23302 | 7180 ± 2097 | 0.2946 |
| BCR::ABL1 quotient (%) | 14.29 ± 10.16 | 0.88 ± 1.10 | 0.0414 |
| NR (n = 49) | R (n = 45) | |||
|---|---|---|---|---|
| Halving Time (Days) * 3 M (n = 40) | Doubling Time (Days) * 3 M (n = 9) | Halving Time (Days) * 3 M (n = 41) | Doubling Time (Days) * 3 M (n = 4) | |
| Mean | 86 ± 87 | 72 ± 55 | 51 ± 18 | 268 ± 280 |
| Median | 55 | 52 | 45 | 223 |
| Min | 45 | 11 | 45 | 26 |
| Max | 541 | 186 | 133 | 600 |
| Range | 496 | 175 | 88 | 574 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christiani, E.; Naumann, N.; Weiss, C.; Spiess, B.; Kleiner, H.; Fabarius, A.; Hofmann, W.-K.; Saussele, S.; Seifarth, W. Gene Expression Pattern of ESPL1, PTTG1 and PTTG1IP Can Potentially Predict Response to TKI First-Line Treatment of Patients with Newly Diagnosed CML. Cancers 2023, 15, 2652. https://doi.org/10.3390/cancers15092652
Christiani E, Naumann N, Weiss C, Spiess B, Kleiner H, Fabarius A, Hofmann W-K, Saussele S, Seifarth W. Gene Expression Pattern of ESPL1, PTTG1 and PTTG1IP Can Potentially Predict Response to TKI First-Line Treatment of Patients with Newly Diagnosed CML. Cancers. 2023; 15(9):2652. https://doi.org/10.3390/cancers15092652
Chicago/Turabian StyleChristiani, Eva, Nicole Naumann, Christel Weiss, Birgit Spiess, Helga Kleiner, Alice Fabarius, Wolf-Karsten Hofmann, Susanne Saussele, and Wolfgang Seifarth. 2023. "Gene Expression Pattern of ESPL1, PTTG1 and PTTG1IP Can Potentially Predict Response to TKI First-Line Treatment of Patients with Newly Diagnosed CML" Cancers 15, no. 9: 2652. https://doi.org/10.3390/cancers15092652
APA StyleChristiani, E., Naumann, N., Weiss, C., Spiess, B., Kleiner, H., Fabarius, A., Hofmann, W.-K., Saussele, S., & Seifarth, W. (2023). Gene Expression Pattern of ESPL1, PTTG1 and PTTG1IP Can Potentially Predict Response to TKI First-Line Treatment of Patients with Newly Diagnosed CML. Cancers, 15(9), 2652. https://doi.org/10.3390/cancers15092652