
Pediatric Extra-Renal Nephroblastoma (Wilms’ Tumor): A Systematic Case-Based Review


Abstract
Simple Summary
Abstract
1. Introduction
2. Case Report
2.1. Clinical Presentation
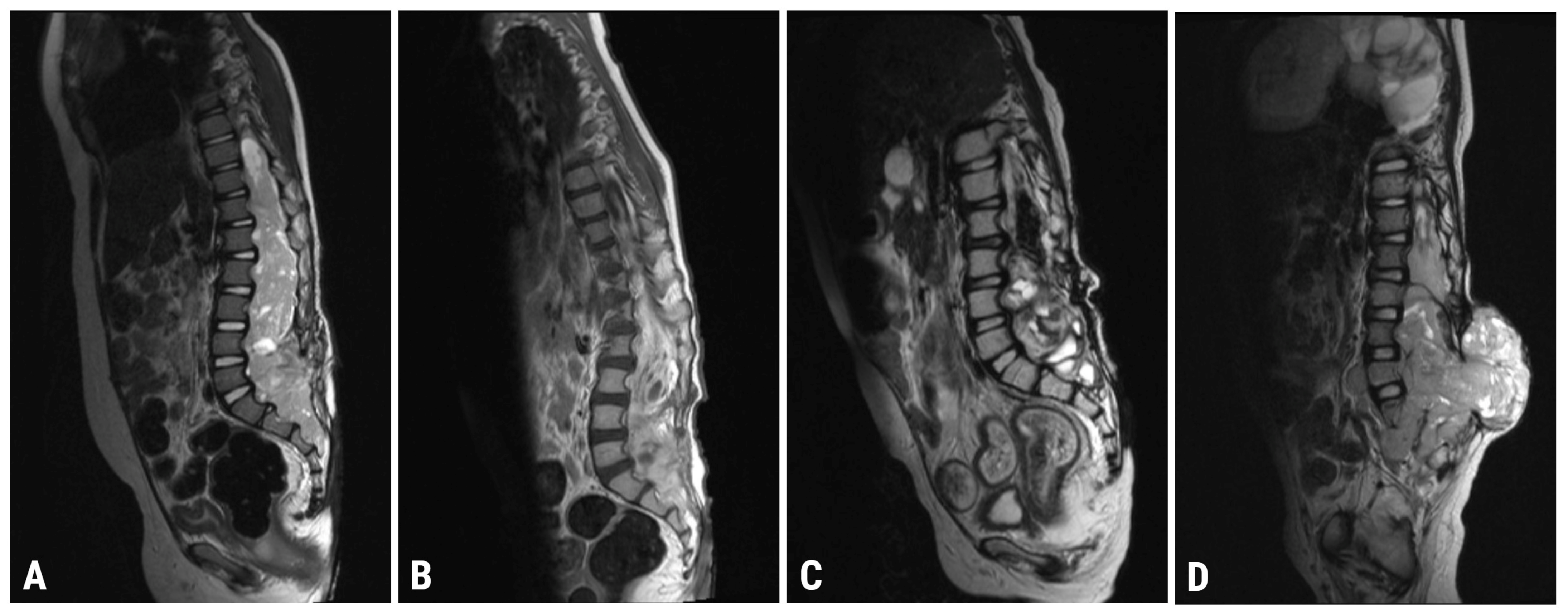
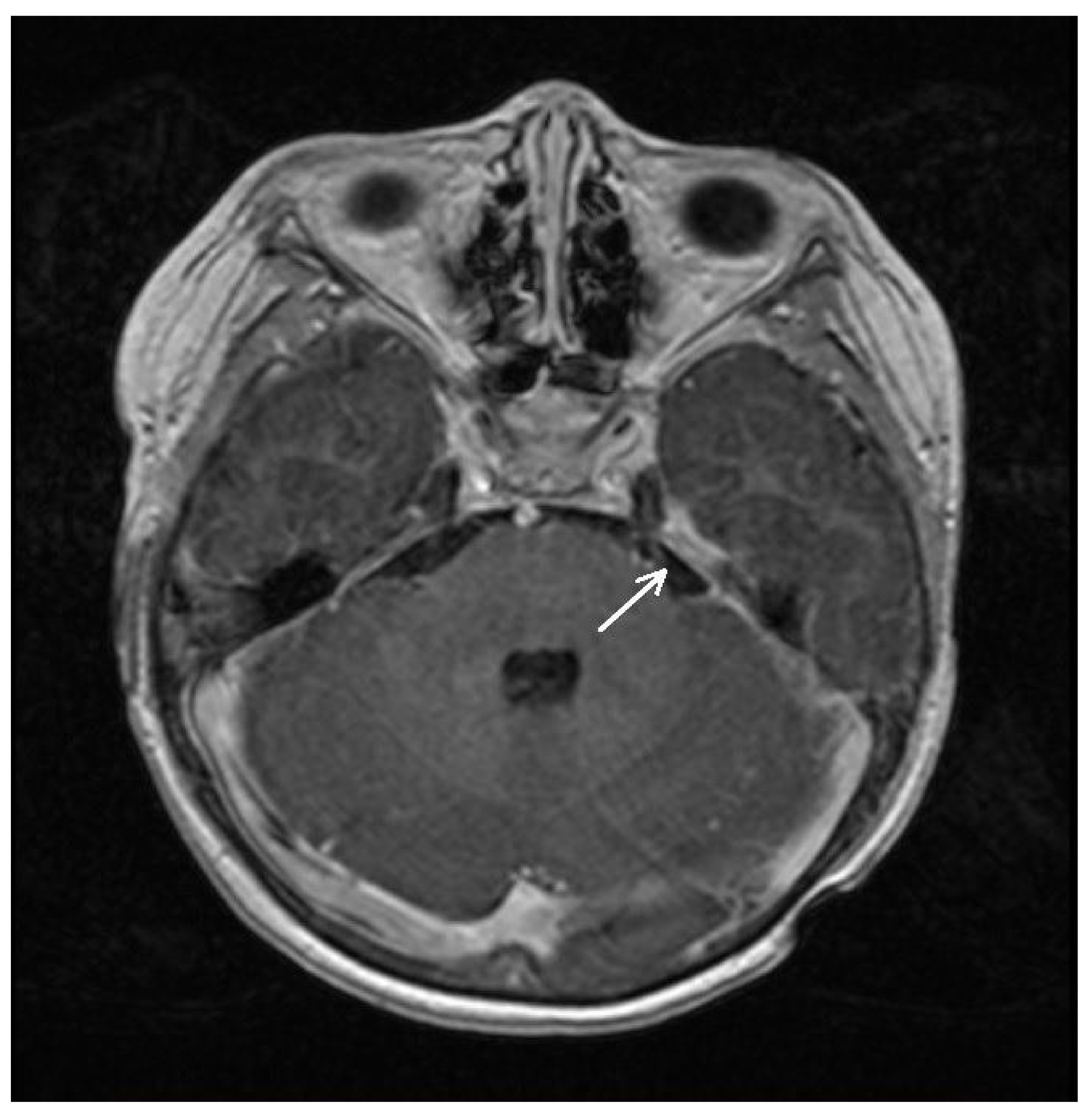
2.2. Imaging
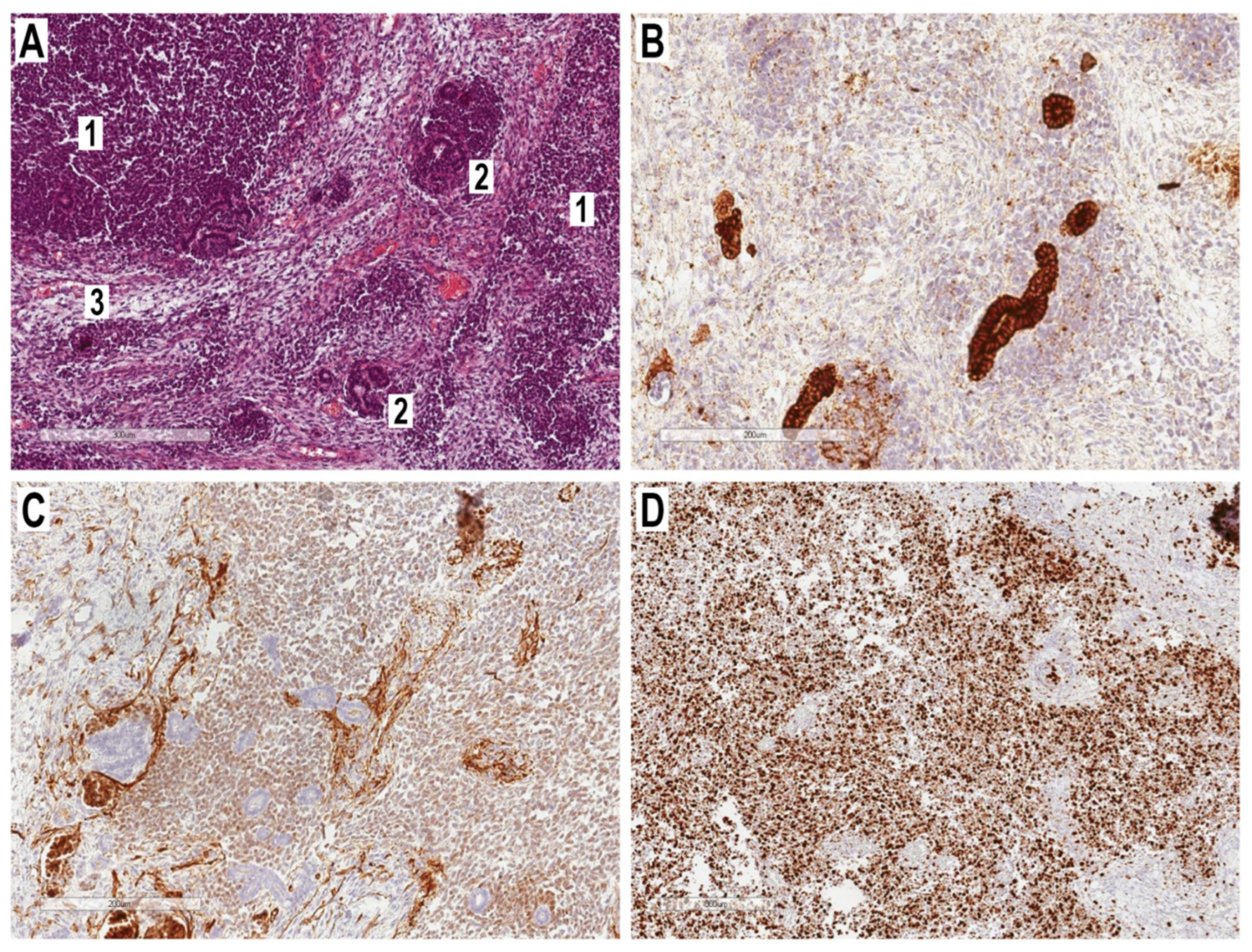
2.3. Histopathological Examination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| January 2020 | Clinical onset (intermittent limp and back/left leg pain) |
| April 2020 | Spinal MRI (mass in the spinal canal at the T12-S3 level) |
| 11 November 2020 | Spinal MRI (confirmation of increased intramedullary mass, with signs of extramedullary growth) |
| 18 December 2020 | Mass biopsy (diagnosis: extra-renal nephroblastoma of the spinal cord) |
| 8 January 2021 | Spinal MRI (T9-S4 mass: 39 × 36 × 205 mm; & C1-C2 mass: 7.4 × 6.2 mm; Figure 1A) Brain MRI (leptomeningeal metastasis of the left temporal lobe: 25 × 5.5 mm) |
| 9 January 2021 | Histopathological examination (ERWT confirmation) |
| 14 January 2021 | Post-operative adjuvant chemotherapy (1st cycle) |
| 2 March 2021 | Spinal MRI (T9-S4 mass: 39 × 33 × 193 mm; & C1-C2 mass: 6 × 3 mm) Brain MRI (unchanged leptomeningeal metastasis of the left temporal lobe: 25 × 5.5 mm in size) |
| 12 April 2021 | Completion of post-operative adjuvant chemotherapy (5th cycle) |
| 4 May 2021 | Spinal MRI (T9-S4 mass: 38 × 32 × 193 mm; & C1-C2 mass: 7 × 3 mm; Figure 1B) Brain MRI (unchanged leptomeningeal metastasis of the left temporal lobe: 25 × 5.5 mm in size) |
| 13 May 2021 | Post-operative adjuvant radiotherapy (1st session) |
| 7 July 2021 | Completion of post-operative adjuvant radiotherapy (34th session) |
| 30 July 2021 | Spinal MRI (T9-S4 mass: 37 × 32 × 190 mm; C1-C2 mass: 7 × 3 mm) Brain MRI (leptomeningeal metastasis of the left temporal lobe: 22 × 4 mm in size) |
| 16 October 2021 | Completion of post-operative adjuvant chemotherapy (9th cycle) |
| 22 October 2021 | Spinal MRI (T9-S4: 36 × 32 × 190 mm; & C1-C2 mass: 7 × 3 mm; Figure 1C) Brain MRI (leptomeningeal metastasis of the left temporal lobe: 20 × 4 mm) |
| 13 December 2021 | PET-CT (high metabolic activity: spinal canal) (weak metabolic activity: neck, left axillary, and right inguinal lymph nodes) |
| 14 December 2021 | Second tumor biopsy |
| 23 December 2021 | Histopathological re-examination (confirmed diagnosis of ERWT) |
| 6 April 2022 | Spinal MRI (T11-S2 mass: 76 × 75 × 195 mm; & C1-C2 mass: 7 × 5 mm; Figure 1D) Brain MRI (leptomeningeal metastasis of the left temporal lobe: 17 × 6 mm in size) |
| 8 April 2022 | Chest CT (multiple bilateral lung metastases, left-sided pneumothorax) |
| 9 April 2022 | Palliative chemotherapy course |
| 17 April 2022 | Death |



2.4. Medical Management and Clinical Course
3. Case-Based Review
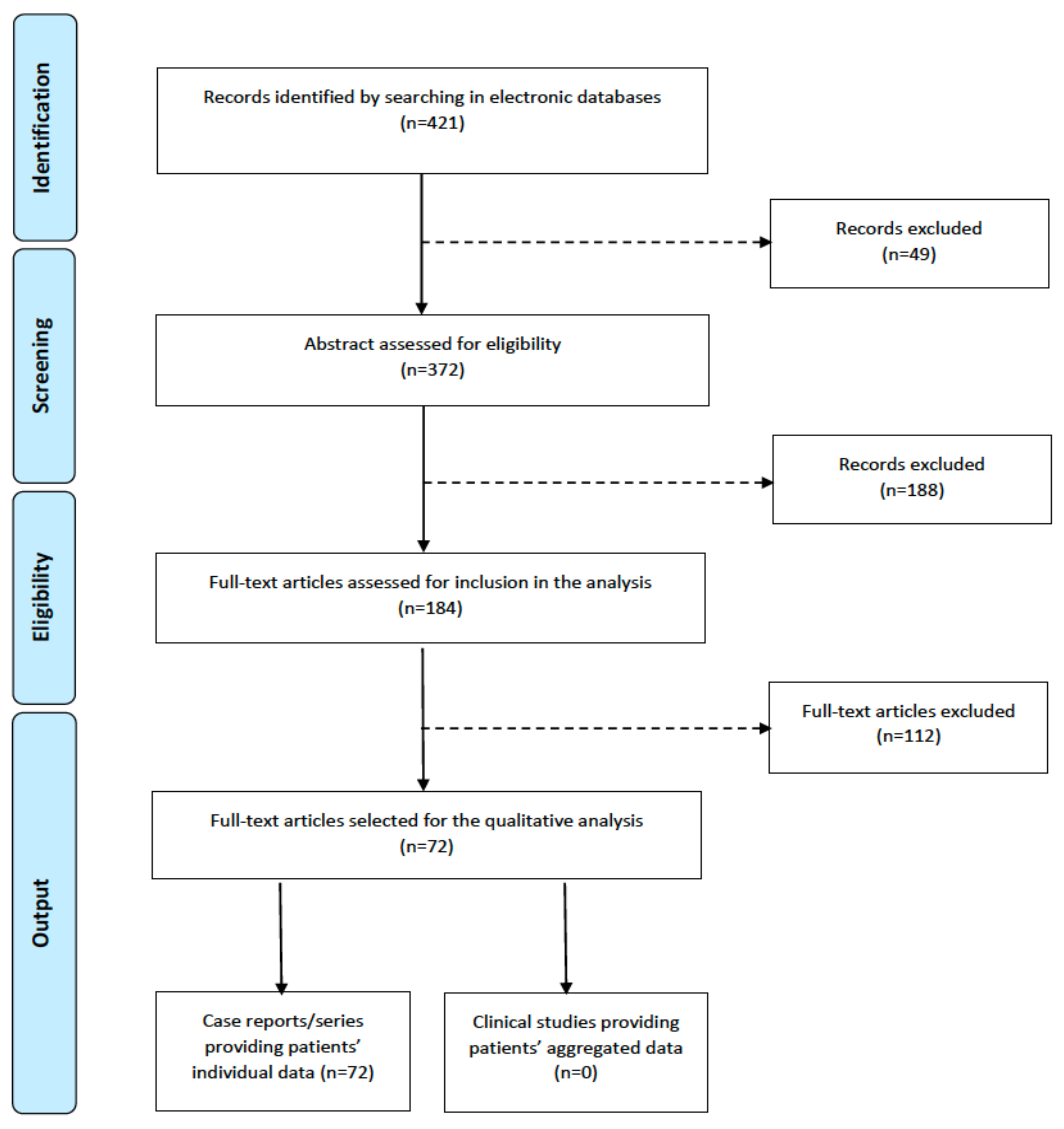
3.1. Systematic Literature Search
3.2. Data Extraction

4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- D’Angio, G.J. The National Wilms Tumor Study: A 40 year perspective. Lifetime Data Anal. 2007, 13, 463–470. [Google Scholar] [CrossRef]
- Shojaeian, R.; Hiradfar, M.; Sharifabad, P.S.; Zabolinejad, N. Extrarenal Wilms’ Tumor: Challenges in Diagnosis, Embryology, Treatment and Prognosis. In Wilms Tumor; van den Heuvel-Eibrink, M.M., Ed.; Codon Publications: Brisbane, Australia, 2016; ISBN 978-0-9944381-1-9. [Google Scholar]
- Moyson, F.; Maurus-Desmarez, R.; Gompel, C. Mediastinal Wilms’ tumor? Acta Chir. Belg. 1961, (Suppl. 2), 118–128. Available online: https://pubmed.ncbi.nlm.nih.gov/14476767/ (accessed on 13 March 2023).
- Taguchi, S.; Shono, T.; Mori, D.; Horie, H. Extrarenal Wilms tumor in children with unfavorable histology: A case report. J. Pediatr. Surg. 2010, 45, e19–e22. [Google Scholar] [CrossRef]
- Cooke, A.; Deshpande, A.V.; La Hei, E.R.; Kellie, S.; Arbuckle, S.; Cummins, G. Ectopic nephrogenic rests in children: The clinicosurgical implications. J. Pediatr. Surg. 2009, 44, e13–e16. [Google Scholar] [CrossRef]
- Bhajekar, A.B.; Joseph, M.; Bhat, H.S. Unattached Nephroblastoma. Br. J. Urol. 1964, 36, 187–190. [Google Scholar] [CrossRef]
- Edelstein, G.; Webb, R.S.; Romsdahl, M.M.; Arboit, J.M. Extrarenal Wilms’ tumor. Am. J. Surg. 1965, 109, 509–512. [Google Scholar] [CrossRef]
- Thompson, M.R.; Emmanuel, I.G.; Campbell, M.S.; Zachary, R.B. Extrarenal Wilms’ tumors. J. Pediatr. Surg. 1973, 8, 37–41. [Google Scholar] [CrossRef]
- Akhtar, M.; Kott, E.; Brooks, B. Extrarenal Wilms’ tumor.Report of a case and review of the literature. Cancer 1977, 40, 3087–3091. [Google Scholar] [CrossRef]
- Madanat, F.; Osborne, B.; Cangir, A.; Sutow, W.W. Extrarenal Wilms tumor. J. Pediatr. 1978, 93, 439–443. [Google Scholar] [CrossRef]
- McCauley, R.G.K. Extrarenal Wilms’ Tumor. Arch. Pediatr. Adolesc. Med. 1979, 133, 1174–1177. [Google Scholar] [CrossRef]
- Johnson, F.; Luttenton, C.; Limbert, D. Extrarenal and urothelial wilms tumor. Urology 1980, 15, 370–373. [Google Scholar] [CrossRef]
- Fried, A.M.; Hatfield, D.R.; Ellis, G.T.; Fitzgerald, K.W. Extrarenal Wilms’ tumor: Sonographic appearance. J. Clin. Ultrasound 1980, 8, 360–362. [Google Scholar] [CrossRef]
- Orlowski, J.P.; Levin, H.S.; Dyment, P.G. Intrascrotal Wilms’ tumor developing in a heterotopic renal anlage of probable mesonephric origin. J. Pediatr. Surg. 1980, 15, 679–682. [Google Scholar] [CrossRef]
- Taylor, W.; Myers, M.; Taylor, W. Extrarenal Wilms’ Tumour in an Infant Exposed to Intrauterine Phenytoin. Lancet 1980, 316, 481–482. [Google Scholar] [CrossRef]
- Bittencourt, A.L.; Britto, J.F.; Fonseca, L.E. Wilms’ Tumor of the uterus: The first report of the literature. Cancer 1981, 47, 2496–2499. [Google Scholar] [CrossRef]
- Adam, Y.C.; Rosen, A.; Oland, J.; Wallach, N.; Reif, R. Extrarenal wilms tumor. J. Surg. Oncol. 1983, 22, 56–58. [Google Scholar] [CrossRef]
- Meng, L.L.; Jagadeesan, K. Extrarenal Wilms’ tumour. Med. J. Malaysia 1983, 38, 134–136. [Google Scholar]
- Luchtrath, H.; Leon, F.; Giesen, H.; Gok, Y. Inguinal nephroblastoma. Virchows Arch. A Pathol. Anat. 1984, 405, 113–118. [Google Scholar] [CrossRef]
- Fernbach, S.K.; Naidich, T.P.; McLone, D.G.; Leestma, J.E. Computed Tomography of Primary Intrathecal Wilms Tumor with Diastematomyelia. J. Comput. Assist. Tomogr. 1984, 8, 523–528. [Google Scholar] [CrossRef]
- Lai, H.-S.; Hung, W.-T.; How, S.-W. Extrarenal Wilms’ tumor—A case report. J. Pediatr. Surg. 1988, 23, 454–456. [Google Scholar] [CrossRef]
- Narasimharao, K.L.; Marwaha, R.K.; Kaushik, S.; Bharati, B.; Katariya, S.; Mitra, S.K.; Walia, B.N.S. Extrarenal Wilms’ tumor. J. Pediatr. Surg. 1989, 24, 212–214. [Google Scholar] [CrossRef]
- Fernandes, E.T.; Kumar, M.; Douglass, E.C.; Wilimas, J.; Parham, D.M.; Rao, B.N. Extrarenal Wilms’ tumor. J. Pediatr. Surg. 1989, 24, 483–485. [Google Scholar] [CrossRef]
- Wakely, P.E.; Sprague, R.I.; Kornstein, M.J. Extrarenal Wilms’ tumor: An analysis of four cases. Hum. Pathol. 1989, 20, 691–695. [Google Scholar] [CrossRef]
- Broecker, B.H.; Caldamone, A.A.; McWilliams, N.B.; Maurer, H.; Salzberg, A. Primary extrarenal Wilms’ tumor in children. J. Pediatr. Surg. 1989, 24, 1283–1288. [Google Scholar] [CrossRef]
- Strand, W.R.; Chou, P.; Pero, J.E.; Kaplan, W.E. Extrarenal Wilms Tumor Occurring in the Inguinal Canal. J. Urol. 1990, 143, 783–785. [Google Scholar] [CrossRef]
- Mirkin, L.D.; Azzarelli, B.; Seo, I.S. Extrarenal Wilms’ Tumor with Cerebellar Metastasis in a Four-Year-Old Girl with Spina Bifida. Am. J. Clin. Pathol. 1990, 93, 805–809. [Google Scholar] [CrossRef]
- Sarode, V.R.; Savitri, K.; Banerjee, C.K.; Narasimharao, K.L.; Khajuria, A. Primary extrarenal Wilms’ tumour: Identification of a putative precursor lesion. Histopathology 1992, 21, 76–78. [Google Scholar] [CrossRef]
- Andrews, P.E.; Kelalis, P.P.; Haase, G.M. Extrarenal Wilms’ tumor: Results of the National Wilms’ Tumor Study. J. Pediatr. Surg. 1992, 27, 1181–1184. [Google Scholar] [CrossRef]
- Suzuki, K.; Miyake, H.; Tashiro, M.; Mori, H.; Fukushige, T.; Tanimura, R.; Yokoyama, S. Extrarenal Wilms’ tumour. Pediatr. Radiol. 1993, 23, 149–150. [Google Scholar] [CrossRef]
- Rasheed, K.; O’Meara, A.; Kelleher, J.; Breatnach, F.; Fitzgerald, R. Extrarenal Wilms’ Tumor. Eur. J. Pediatr. Surg. 1993, 3, 121–123. [Google Scholar] [CrossRef]
- Mount, S.L.; Dickerman, J.D.; Taatjes, D.J. Extrarenal Wilms’ Tumor: An Ultrastructural and Immunoelectron Microscopic Case Report. Ultrastruct. Pathol. 1996, 20, 155–165. [Google Scholar] [CrossRef]
- Arkovitz, M.S.; Ginsburg, H.B.; Eidelman, J.; Greco, M.A.; Rauson, A. Primary extrarenal Wilms’ tumor in the inguinal canal: Case report and review of the literature. J. Pediatr. Surg. 1996, 31, 957–959. [Google Scholar] [CrossRef]
- Kapur, V.K.; Sakalkale, R.P.; Samuel, K.V.; Meisheri, I.V.; Bhagwat, A.D.; Ramprasad, A.; Waingankar, V.S. Association of extrarenal Wilms’ tumor with a horseshoe kidney. J. Pediatr. Surg. 1998, 33, 935–937. [Google Scholar] [CrossRef]
- Benatar, B.; Wright, C.; Freinkel, A.L.; Cooper, K. Primary Extrarenal Wilms’ Tumor of the Uterus Presenting as a Cervical Polyp. Int. J. Gynecol. Pathol. 1998, 17, 277–280. [Google Scholar] [CrossRef]
- Babin, E.A.; Davis, J.R.; Hatch, K.D.; Hallum, A.V. Wilms’ Tumor of the Cervix: A Case Report and Review of the Literature. Gynecol. Oncol. 2000, 76, 107–111. [Google Scholar] [CrossRef]
- Govender, D.; Hadley, G.P.; Nadvi, S.S.; Donnellan, R.B. Primary lumbosacral Wilms tumour associated with occult spinal dysraphism. Virchows Arch. 2000, 436, 502–505. [Google Scholar] [CrossRef]
- Arda, I.; Tüzün, M.; Demirhan, B.; Sevmis, S.; Hicsönmez, A. Lumbosacral extrarenal Wilms’ tumour: A case report and literature review. Eur. J. Pediatr. 2001, 160, 617–619. [Google Scholar] [CrossRef]
- Öner, Ü.; Tokar, B.; Açıkalın, M.F.; İlhan, H.; Tel, N. Wilms’ tumor of the ovary: A case report. J. Pediatr. Surg. 2002, 37, 127–129. [Google Scholar] [CrossRef]
- Deshpande, A.V.; Gawali, J.S.; Sanghani, H.H.; Shenoy, A.S.; Patankar, J.Z.; Borwankar, S.S. Extrarenal Wilm’s tumour—A rare entity. Pediatr. Surg. Int. 2002, 18, 543–544. [Google Scholar] [CrossRef]
- Yunus, M.; Hashmi, R.; Hasan, S.H.; Brohi, H.M.Y. Extrarenal Wilms’ tumor. JPMA J. Pak. Med. Assoc. 2003, 53, 436–439. [Google Scholar]
- Apoznański, W.; Sawicz-Birkowska, K.; Pietras, W.; Dorobisz, U.; Szydełko, T. Extrarenal Wilms Tumour. Eur. J. Pediatr. Surg. 2005, 15, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.C.; Sarat Chandra, P.; Goel, S.; Gupta, V.; Sarkar, C. Primary lumbosacral Wilms tumor associated with diastematomyelia and occult spinal dysraphism. A report of a rare case and a short review of literature. Childs Nerv. Syst. 2005, 21, 240–243. [Google Scholar] [CrossRef]
- Sastri, J.; Dedhia, R.; Laskar, S.; Shet, T.; Kurkure, P.; Muckaden, M. Extra-renal Wilms’ tumour—Is it different? Pediatr. Nephrol. 2006, 21, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Houben, C.H.; Tong, J.H.; Chan, A.W.; Chik, K.W.; Lee, K.H.; Sihoe, J.D.Y.; Tam, Y.H.; Yeung, C.K. Familial extrarenal Wilms tumor. J. Pediatr. Surg. 2007, 42, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, C.; Attili, V.S.S.; Dadhich, H.P.; Kumari, A.; Appaji, L.; Giri, G.V.; Mukharjee, G. Extrarenal Wilms’ tumor: A report of two cases and review of literature. J. Indian Assoc. Pediatr. Surg. 2007, 12, 145–147. [Google Scholar] [CrossRef]
- Leblebici, C.; Behzatoğlu, K.; Yıldız, P.; Koçyıldız, Z.; Bozkurt, S. Extrarenal Wilms’ tumor of the uterus with ovarian dermoid cyst. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 144, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.M.; Zhang, K.R.; Shu, H.; Tian, B.L.; Wang, W.L. Presacral Extrarenal Wilms Tumor in a Child. Urology 2009, 74, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Ngan, K.W.; Shaari, S.; Subramaniam, T. Juxtarenal/pararenal Wilms’ tumour in a six-year-old Malay girl. Singapore Med. J. 2009, 50, e329–e331. [Google Scholar]
- Imran, R.; Aziz, S.A.; Banday, M.A.; Ahmad, S.N. Extrarenal Wilms’ tumour with bone marrow involvement: An index case report. Chin.-Ger. J. Clin. Oncol. 2010, 9, 295–297. [Google Scholar] [CrossRef]
- Teerthanath, S. Extrarenal Nephroblastoma. J. Lab. Physicians 2011, 3, 059–060. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Sohn, M.-H.; Lim, S.T.; Kim, D.W.; Jeong, H.-J.; Yim, C.-Y. F-18 FDG PET/CT Imaging of Metastatic Extrarenal Wilms Tumor Arising in the Inguinal Canal. Clin. Nucl. Med. 2011, 36, 475–478. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nishizawa, S.; Ogiso, Y. Paratesticular extrarenal Wilms’ tumor: Letter to the Editor. Int. J. Urol. 2012, 19, 490–491. [Google Scholar] [CrossRef]
- Armanda, V.; Čulić, S.; Pogorelić, Z.; Kuljiš, D.; Budimir, D.; Kuzmić-Prusac, I. Rare localization of extrarenal nephroblastoma in 1-month-old female infant. J. Pediatr. Urol. 2012, 8, e43–e45. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xiao, X.; Gao, J.; Yao, W.; Chen, H.; Zhang, B. Pelvic Wilms tumor in a child with an absent right kidney and spinal malformations. J. Pediatr. Surg. 2012, 47, e11–e14. [Google Scholar] [CrossRef] [PubMed]
- Gordetsky, J.; Katzman, P.; Rashid, H. Juxtarenal Wilms tumor in an adolescent. Urology 2012, 80, 922–924. [Google Scholar] [CrossRef]
- Marwah, N.; Rattan, K.N.; Rana, P.; Goyal, V.; Sen, R. Extrarenal Wilms’ Tumor of the Ovary: A Case Report and Short Review of the Literature. J. Gynecol. Surg. 2012, 28, 306–308. [Google Scholar] [CrossRef]
- Hiradfar, M.; Shojaeian, R.; Zabolinejad, N.; Saeedi, P.; Joodi, M.; Khazaie, Z.; Hajian, S. Extrarenal Wilms’ tumour presenting as an inguinal mass. Arch. Dis. Child. 2012, 97, 1077. [Google Scholar] [CrossRef]
- Rojas, Y.; Slater, B.J.; Braverman, R.M.; Eldin, K.W.; Thompson, P.A.; Wesson, D.E.; Nuchtern, J.G. Extrarenal Wilms tumor: A case report and review of the literature. J. Pediatr. Surg. 2013, 48, e33–e35. [Google Scholar] [CrossRef]
- Morandi, A.; Fagnani, A.M.; Runza, L.; Farris, G.; Zanini, A.; Parolini, F.; Bassi, G.; Gentilino, V.; Macchini, F.; Arnoldi, R.; et al. Extrarenal testicular Wilms’ tumor in a 3-year-old child. Pediatr. Surg. Int. 2013, 29, 961–964. [Google Scholar] [CrossRef]
- Goel, V.; Verma, A.K.; Batra, V.; Puri, S.K. “Primary extrarenal Wilms” tumour’: Rare presentation of a common paediatric tumour. Case Rep. 2014, 2014, bcr2013202172. [Google Scholar] [CrossRef]
- Kumar, S.; Sunilkumar, M.N.; Surendran, D. Extrarenal nephroblastoma in a 7 year old child: A rare case report with review of literature. Int. J. Contemp. Pediatr. 2017, 2, 155–158. [Google Scholar] [CrossRef]
- Thakkar, N.C.; Sarin, Y.K. Extra-Renal Wilms’ Tumor: A Rare Diagnosis. APSP J. Case Rep. 2015, 6, 17. [Google Scholar]
- Park, J. Extrarenal retroperitoneal Wilms’ tumor with subsequent pulmonary and peritoneal metastasis in a 4 year-old girl: A case report and review of literature. J. Pediatr. Surg. Case Rep. 2016, 8, 19–21. [Google Scholar] [CrossRef]
- Wabada, S.; Abubakar, A.; Adamu, A.; Kabir, A.; Gana, L. A retroperitoneal extra-renal wilms’ tumour: A case report. Niger. J. Clin. Pract. 2017, 20, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Itoshima, R.; Kobayashi, R.; Sano, H.; Hori, D.; Kishimoto, K.; Suzuki, D.; Miura, M.; Takagi, Y.; Yamamoto, H.; Fujita, S.; et al. Extrarenal Nephroblastoma of the Retroperitoneal Space in Children: A Case Report and Review of the Literature. J. Pediatr. Hematol. Oncol. 2017, 39, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Igbaseimokumo, U.; Cartwright, C.; Lewing, K.; Hutchison, L.; Habeebu, S. The Rare Association of Spina Bifida and Extrarenal Wilms Tumor: A Case Report and Review of the Literature. World Neurosurg. 2017, 104, 1046.e1–1046.e5. [Google Scholar] [CrossRef]
- Tang, H.; Lu, M.; Jiang, S.; Ren, Y. Two rare cases of abdominal tumor in children: Answers. Pediatr. Nephrol. 2018, 33, 1343–1345. [Google Scholar] [CrossRef]
- Sindhu, I.; Saeed, H.; Wali, R.; Mehreen, A. Primary Extra-renal Wilms’Tumor in Urinary Bladder: Rare Presentation of a Common Pediatric Malignancy. J. Coll. Physicians Surg. Pak. 2019, 29, S31–S33. [Google Scholar] [CrossRef]
- Groth, T.W.; Southern, J.; Goetz, J.T.; Farooq, A. A Case of Primary Paratesticular Wilms Tumor in an Undescended Testis. Urology 2019, 129, 197–199. [Google Scholar] [CrossRef]
- Ismy, J.; Ismy, J.; Kamarlis, R.; Mustafa, A. Rare case of primary bladder Wilm’s tumor in a 1-year old boy. Urol. Case Rep. 2019, 25, 100898. [Google Scholar] [CrossRef]
- Liang, H.; He, Y.; Fu, L.; Tian, J.; Sun, N.; Yu, T.; Huang, Y.; Lin, D.; Wang, G. Extrarenal Wilms tumor in children: A retrospective observational case series. J. Pediatr. Urol. 2020, 16, 664.e1–664.e7. [Google Scholar] [CrossRef] [PubMed]
- Parkhi, M.; Peyam, S.; Peters, N.J.; Sodhi, K.S.; Trehan, A.; Bal, A. Primary Wilms tumor of the urinary bladder. Autopsy Case Rep. 2022, 12, e2021390. [Google Scholar] [CrossRef]
- Qu, Y.; Wu, Y.; Qu, D.; Ge, H. Extrarenal Wilms tumor with hypertension and dilated cardiomyopathy in an infant: A report of an unusual case. Pediatr. Blood Cancer 2022, 69, e29900. [Google Scholar] [CrossRef]
- Albiroty, K.A.; Al Sabahi, A.; Al Shabibi, S.; Al’Ajmi, Z.I.; Al Hinai, K.; Al-Mashaikhi, N. Extrarenal Wilms’ Tumour of the Ovary. Sultan Qaboos Univ. Med. J. 2022, 22, 566–569. [Google Scholar] [CrossRef]
- Spreafico, F.; Fernandez, C.V.; Brok, J.; Nakata, K.; Vujanic, G.; Geller, J.I.; Gessler, M.; Maschietto, M.; Behjati, S.; Polanco, A.; et al. Wilms tumour. Nat. Rev. Dis. Primer 2021, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Daw, N.C.; Chi, Y.-Y.; Kalapurakal, J.A.; Kim, Y.; Hoffer, F.A.; Geller, J.I.; Perlman, E.J.; Ehrlich, P.F.; Mullen, E.A.; Warwick, A.B.; et al. Activity of Vincristine and Irinotecan in Diffuse Anaplastic Wilms Tumor and Therapy Outcomes of Stage II to IV Disease: Results of the Children’s Oncology Group AREN0321 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, C.; Furtwängler, R.; van Tinteren, H.; Teixeira, R.A.P.; Acha, T.; Howell, L.; Vujanic, G.; Godzinski, J.; Melchior, P.; Smets, A.M.; et al. Outcome of patients with stage IV high-risk Wilms tumour treated according to the SIOP2001 protocol: A report of the SIOP Renal Tumour Study Group. Eur. J. Cancer 2020, 128, 38–46. [Google Scholar] [CrossRef]
- Godzinski, J.; Graf, N.; Audry, G. Current concepts in surgery for Wilms tumor--the risk and function-adapted strategy. Eur. J. Pediatr. Surg. 2014, 24, 457–460. [Google Scholar] [CrossRef]
- Lopyan, N.M.; Ehrlich, P.F. Surgical Management of Wilms Tumor (Nephroblastoma) and Renal Cell Carcinoma in Children and Young Adults. Surg. Oncol. Clin. 2021, 30, 305–323. [Google Scholar] [CrossRef]
- Pritchard-Jones, K.; Bergeron, C.; de Camargo, B.; van den Heuvel-Eibrink, M.M.; Acha, T.; Godzinski, J.; Oldenburger, F.; Boccon-Gibod, L.; Leuschner, I.; Vujanic, G.; et al. Omission of doxorubicin from the treatment of stage II-III, intermediate-risk Wilms’ tumour (SIOP WT 2001): An open-label, non-inferiority, randomised controlled trial. Lancet 2015, 386, 1156–1164. [Google Scholar] [CrossRef]
- van den Heuvel-Eibrink, M.M.; Hol, J.A.; Pritchard-Jones, K.; van Tinteren, H.; Furtwängler, R.; Verschuur, A.C.; Vujanic, G.M.; Leuschner, I.; Brok, J.; Rübe, C.; et al. Position paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat. Rev. Urol. 2017, 14, 743–752. [Google Scholar] [CrossRef]
- D’angio, G.J.; Breslow, N.; Beckwith, J.B.; Evans, A.; Baum, E.; Delorimier, A.; Fernbach, D.; Hrabovsky, E.; Jones, B.; Kelalis, P.; et al. Treatment of Wilms’ tumor. Results of the third national Wilms’ tumor study. Cancer 1989, 64, 349–360. [Google Scholar] [CrossRef]
- Breslow, N.E.; Ou, S.-S.; Beckwith, J.B.; Haase, G.M.; Kalapurakal, J.A.; Ritchey, M.L.; Shamberger, R.C.; Thomas, P.R.M.; D’Angio, G.J.; Green, D.M. Doxorubicin for favorable histology, Stage II–III Wilms tumor. Cancer 2004, 101, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel-Eibrink, M.M.; van Tinteren, H.; Bergeron, C.; Coulomb-L’Hermine, A.; de Camargo, B.; Leuschner, I.; Sandstedt, B.; Acha, T.; Godzinski, J.; Oldenburger, F.; et al. Outcome of localised blastemal-type Wilms tumour patients treated according to intensified treatment in the SIOP WT 2001 protocol, a report of the SIOP Renal Tumour Study Group (SIOP-RTSG). Eur. J. Cancer 2015, 51, 498–506. [Google Scholar] [CrossRef]
- Kalapurakal, J.A.; Li, S.M.; Breslow, N.E.; Beckwith, J.B.; Ritchey, M.L.; Shamberger, R.C.; Haase, G.M.; Thomas, P.R.M.; Grundy, P.; Green, D.M.; et al. Intraoperative spillage of favorable histology wilms tumor cells: Influence of irradiation and chemotherapy regimens on abdominal recurrence. A report from the National Wilms Tumor Study Group. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, A.; Spreafico, F.; de Krijger, R.R.; Drost, J.; Brok, J.; Perotti, D.; van Tinteren, H.; Venkatramani, R.; Godziński, J.; Rübe, C.; et al. Prognostic Factors for Wilms Tumor Recurrence: A Review of the Literature. Cancers 2021, 13, 3142. [Google Scholar] [CrossRef] [PubMed]
- Vujanić, G.M.; D’Hooghe, E.; Graf, N.; Vokuhl, C.; Al-Saadi, R.; Chowdhury, T.; Pritchard-Jones, K.; Furtwängler, R. Prognostic significance of histopathological response to preoperative chemotherapy in unilateral Wilms’ tumor: An analysis of 899 patients treated on the SIOP WT 2001 protocol in the UK-CCLG and GPOH studies. Int. J. Cancer 2021, 149, 1332–1340. [Google Scholar] [CrossRef]
- Rosen, R.D.; Sapra, A. TNM Classification. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Pillai, R.K.; Jayasree, K. Rare cancers: Challenges & issues. Indian J. Med. Res. 2017, 145, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Alemayehu, C.; Mitchell, G.; Nikles, J. Barriers for conducting clinical trials in developing countries—A systematic review. Int. J. Equity Health 2018, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.; Shaikhyzada, K.; Suleimenova, A.; Ibraimov, B.; Nurgaliev, D.; Poddighe, D. Case report: Atypical teratoid/rhabdoid tumor of the lateral ventricle in a male adolescent (case-based review and diagnostic challenges in developing countries). Front. Oncol. 2022, 12, 985862. [Google Scholar] [CrossRef]
- Khoja, A.; Kazim, F.; Ali, N.A. Barriers to Conducting Clinical Trials in Developing Countries. Ochsner J. 2019, 19, 294–295. [Google Scholar] [CrossRef] [PubMed]
- Poddighe, D.; Abdukhakimova, D. Celiac Disease in Asia beyond the Middle East and Indian subcontinent: Epidemiological burden and diagnostic barriers. World J. Gastroenterol. 2021, 27, 2251–2256. [Google Scholar] [CrossRef] [PubMed]
- Dauyey, Z.; Poddighe, D. Diagnostic Barriers in Children with Immunodeficiencies in Central Asia: A Case-Based Discussion. Pediatr. Rep. 2021, 13, 483–489. [Google Scholar] [CrossRef] [PubMed]
| № | Author | Year | Sex | Age (yrs.) | ERWT Origin Site | Diagnostic Time (wks.) | Stage | Treatment | Relapse/Metastasis | Follow-Up (yrs.) | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Bhajkar et al. [6] | 1964 | M | 2 | Retro- peritoneum | 26 | n/a | Surgery + RAD | None | 0.8 | Alive |
| 2 | Edelstein et al. [7] | 1965 | M | 3 | Retro- peritoneum | 13 | n/a | Surgery + chemo + RAD | None | 0.7 | Alive |
| 3 | Thompson et al. [8] | 1973 | F | 4.5 | Inguinal region | 1 | n/a | Surgery + RAD | Local | 2 | Alive |
| 4 | Thompson et al. [8] | 1973 | M | 3 | Inguinal canal | n/a | n/a | Surgery + chemo + RAD | Local + Right lung | 0.5 | Death |
| 5 | Akhtar et al. [9] | 1977 | M | 0.2 | Inguinal canal | 5 | n/a | Surgery | None | 1.5 | Alive |
| 6 | Madanat et al. [10] | 1978 | F | 9 | Chest wall | n/a | III (NWTS) | Surgery * + chemo + RAD | None | 2.7 | Alive |
| 7 | Madanat et al. [10] | 1978 | M | 0.3 | Inguinal canal | 6 | I (NWTS) | Surgery + chemo | None | 1.8 | Alive |
| 8 | McCauley et al. [11] | 1979 | F | 4.5 | Retro- peritoneum | 0.3 | III (NWTS) | Surgery + chemo + RAD | None | 4 | Alive |
| 9 | Johnson et al. [12] | 1980 | F | 1.5 | Retro- peritoneum | 0.1 | I | Surgery + chemo | None | 1 | Alive |
| 10 | Fried et al. [13] | 1980 | M | 3.5 | Retro- peritoneum | 0.1 | n/a | Surgery + chemo | n/a | n/a | Alive |
| 11 | Orlowski et al. [14] | 1980 | M | 3.5 | Scrotum | n/a | n/a | Surgery + chemo + RAD | Left lung | 1.5 | Alive |
| 12 | Taylor et al. [15] | 1980 | M | 0.5 | Scrotum | n/a | n/a | Surgery + chemo + RAD | None | 0.5 | Alive |
| 13 | Bittencourt et al. [16] | 1981 | F | 14 | Uterus | 52 | n/a | Surgery + chemo + RAD | None | 5.7 | Alive |
| 14 | Adam et al. [17] | 1983 | M | 10 | Retro- peritoneum | n/a | n/a | Surgery | None | 0.1 | Alive |
| 15 | Meng et al. [18] | 1983 | M | 3 | Retro- peritoneum | 9 | n/a | Surgery | n/a | n/a | n/a |
| 16 | Lüchtrath et al. [19] | 1984 | F | 1.2 | Inguinal region | 48 | n/a | Surgery + chemo | None | 3 | Alive |
| 17 | Fernbach et al. [20] | 1984 | F | 2 | Spinal cord (L1) | n/a | n/a | Surgery + chemo + RAD | None | 1 | Alive |
| 18 | Lai et al. [21] | 1988 | F | 5 | Inguinal region | n/a | n/a | Surgery + chemo | Local | 1.6 | Alive |
| 19 | Narasimharao et al. [22] | 1989 | M | 2 | Retro- peritoneum | 13 | n/a | Surgery + chemo | None | 1 | Alive |
| 20 | Fernandes et al. [23] | 1989 | M | 6 | Retro- peritoneum | 0.7 | III (NWTS) | Surgery + chemo + RAD | None | 7 | Alive |
| 21 | Fernandes et al. [23] | 1989 | F | 2 | Retro- peritoneum | n/a | II (NWTS) | Surgery + chemo | None | 5 | Alive |
| 22 | Fernandes et al. [23] | 1989 | F | 2 | Retro- peritoneum | n/a | II (NWTS) | Surgery + chemo | None | 1 | Alive |
| 23 | Wakely et al. [24] | 1989 | F | 0.8 | Uterus | n/a | n/a | Surgery + chemo | None | 2 | Alive |
| 24 | Wakely et al. [24] | 1989 | F | 1.8 | Retro- peritoneum | n/a | n/a | Surgery + chemo + RAD | None | 6 | Alive |
| 25 | Wakely et al. [24] | 1989 | F | 4 | Retro- peritoneum | 3 | n/a | Surgery + chemo | None | 5 | Alive |
| 26 | Wakely et al. [24] | 1989 | M | 4 | Retro- peritoneum | n/a | n/a | Surgery + chemo + RAD | None | 6 | Alive |
| 27 | Broecker et al. [25] | 1989 | F | 0.8 | Pelvis | n/a | II (NWTS) | Surgery + chemo | None | 1 | Alive |
| 28 | Broecker et al. [25] | 1989 | F | 1.8 | Retro- peritoneum | n/a | n/a | Surgery + chemo + RAD | None | 7 | Alive |
| 29 | Broecker et al. [25] | 1989 | F | 1.8 | Retro- peritoneum | n/a | II (NWTS) | Surgery + chemo | Lung node | 1.8 | n/a |
| 30 | Strand et al. [26] | 1990 | M | 0.9 | Inguinal canal | 48 | n/a | Surgery + chemo | n/a | n/a | n/a |
| 31 | Mirkin et al. [27] | 1990 | F | 2 | Spinal cord (T12-L4) | n/a | n/a | Surgery + chemo + RAD | Cerebellum | 1.7 | Alive |
| 32 | Sarode et al. [28] | 1992 | M | 2 | Retro- peritoneum | 9 | n/a | Surgery + chemo | n/a | n/a | n/a |
| 33 | Andrews et al. [29] | 1992 | F | n/a | Sacrococcygeal region | n/a | II (NWTS) | Surgery + chemo | None | 1.3 | Alive |
| 34 | Andrews et al. [29] | 1992 | M | n/a | Retro- peritoneum | n/a | II (NWTS) | Surgery + chemo | None | 0.6 | Alive |
| 35 | Andrews et al. [29] | 1992 | F | n/a | Lumbar region | n/a | II (NWTS) | Surgery + chemo | None | 6.2 | Alive |
| 36 | Andrews et al. [29] | 1992 | M | n/a | Retro- peritoneum | n/a | IV (NWTS) | Surgery + chemo + RAD | Lungs | 2 | Death |
| 37 | Andrews et al. [29] | 1992 | F | n/a | Retro- peritoneum | n/a | I (NWTS) | Surgery + chemo | None | 2.8 | Alive |
| 38 | Andrews et al. [29] | 1992 | F | n/a | Pelvis | n/a | II (NWTS) | Surgery + chemo | Lungs | 4 | Alive |
| 39 | Suzuki et al. [30] | 1993 | M | 2 | Retro- peritoneum | n/a | n/a | Surgery | n/a | n/a | n/a |
| 40 | Rasheed et al. [31] | 1993 | M | 3 | Retro- peritoneum | 3 | III (UKCCSG) | Surgery + chemo + RAD | None | 7 | Alive |
| 41 | Rasheed et al. [31] | 1993 | F | 4 | Retro- peritoneum | 1.4 | III (UKCCSG) | Surgery + chemo + RAD | None | 1.7 | Alive |
| 42 | Mount et al. [32] | 1996 | F | 5 | Retro- peritoneum | n/a | n/a | Surgery + chemo | None | 2 | Alive |
| 43 | Arkovitz et al. [33] | 1996 | M | 3.5 | Inguinal canal | n/a | III (NWTS) | Surgery + chemo + RAD | None | 2 | Alive |
| 44 | Kapur et al. [34] | 1998 | M | 1.5 | Retro- peritoneum | n/a | I (TNM) | Surgery + chemo | None | 0.6 | Alive |
| 45 | Kapur et al. [34] | 1998 | M | 2 | Retro- peritoneum | 2 | III (TNM) | Surgery + chemo + RAD | None | 3 | Alive |
| 46 | Benatar et al. [35] | 1998 | F | 11 | Uterus | n/a | n/a | Surgery | Local | 0.6 | n/a |
| 47 | Babin et al. [36] | 2000 | F | 13 | Uterus | 9 | n/a | Surgery + chemo + RAD | Local | 5 | Alive |
| 48 | Govender et al. [37] | 2000 | F | 4 | Spinal cord (T10-Sx) | 13 | n/a | Surgery * + chemo + RAD | n/a | n/a | n/a |
| 49 | Arda et al. [38] | 2001 | F | 5 | Retro- peritoneum | n/a | III | Surgery + chemo + RAD | None | 3 | Alive |
| 50 | Oner et al. [39] | 2002 | F | 3.5 | Ovary | n/a | n/a | Surgery + chemo | None | 0.6 | Alive |
| 51 | Deshpande et al. [40] | 2002 | M | 1 | Lumbar region (L2-L4) | 9 | n/a | Surgery + chemo + RAD | n/a | n/a | Alive |
| 52 | Yunus et al. [41] | 2003 | M | <0.1 | Retro- peritoneum | 0.7 | n/a | Surgery + chemo | None | 1.8 | Alive |
| 53 | Apoznański et al. [42] | 2005 | M | 17 | Retro- peritoneum | n/a | III (SIOP) | Surgery + chemo + RAD | None | 1 | Alive |
| 54 | Sharma et al. [43] | 2005 | F | 1.5 | Spinal cord (L2-5) | n/a | n/a | Surgery + chemo | n/a | n/a | Alive |
| 55 | Sastri et al. [44] | 2006 | M | 2 | Paravertebral region | 26 | n/a | Surgery + chemo + RAD | None | 5 | Alive |
| 56 | Sastri et al. [44] | 2006 | M | 0.8 | Lumbar region | 0.7 | n/a | Surgery + chemo + RAD | None | 4 | Alive |
| 57 | Sastri et al. [44] | 2006 | F | 15 | Retro- peritoneum | 9 | n/a | Surgery + chemo + RAD | None | 5 | Alive |
| 58 | Houben et al. [45] | 2007 | M | 3.7 | Retro- peritoneum | n/a | IV (NWTS) | Surgery + chemo | None | 4 | Alive |
| 59 | Houben et al. [45] | 2007 | M | 2.8 | Retro- peritoneum | n/a | I (NWTS) | Surgery + chemo | None | 1 | Alive |
| 60 | Ramachandra et al. [46] | 2007 | M | 4 | Retro- peritoneum | 8 | III (NWTS) | Surgery + chemo + RAD | None | 1 | Alive |
| 61 | Ramachandra et al. [46] | 2007 | F | 3 | Retro- peritoneum | n/a | II (NWTS) | Surgery + chemo + RAD | None | 1.3 | Alive |
| 62 | Leblebici et al. [47] | 2009 | F | 16 | Uterus | 26 | n/a | Surgery + chemo | n/a | n/a | Death |
| 63 | Jiaet al. [48] | 2009 | F | 3 | Retro- peritoneum | 1.4 | n/a | Surgery | n/a | 0.3 | n/a |
| 64 | Ngan et al. [49] | 2009 | F | 6 | Retro- peritoneum (juxtarenal) | 0.7 | I | Surgery | None | 1 | Alive |
| 65 | Cooke et al. [5] | 2009 | M | 1.2 | Inguinal canal | n/a | n/a | Surgery | None | 3 | Alive |
| 66 | Imran et al. [50] | 2010 | F | 7 | Retro- peritoneum | n/a | n/a | Surgery + chemo + RAD | None | n/a | Alive |
| 67 | Taguchi et al. [4] | 2010 | F | 2.8 | Retro- peritoneum | n/a | n/a | Surgery + chemo | None | 2 | Alive |
| 68 | Teerthanath [51] | 2011 | F | 6 | Retro- peritoneum | 26 | n/a | Surgery + chemo | None | 4 | Alive |
| 69 | Jeong et al. [52] | 2011 | M | 9 | Inguinal canal | 1.4 | n/a | Surgery + chemo | Lungs, mediastinal lymph nodes | n/a | n/a |
| 70 | Yamamoto et al. [53] | 2012 | M | 0.6 | Scrotum | n/a | n/a | Surgery | None | 3 | Alive |
| 71 | Armanda et al. [54] | 2012 | F | 0.1 | Lumbosacral region | 1.4 | I (SIOP) | Surgery + chemo | None | 2 | Alive |
| 72 | Li et al. [55] | 2012 | F | 1.8 | Pelvis | 2 | III (NWTS) | Surgery + chemo + RAD | None | 3 | Alive |
| 73 | Gordetsky et al. [56] | 2012 | M | 17 | Retro- peritoneum (juxtarenal) | 9 | II | Surgery + chemo + RAD | n/a | n/a | n/a |
| 74 | Marwah et al. [57] | 2012 | F | 1.2 | Ovary | n/a | n/a | Surgery + chemo | n/a | n/a | n/a |
| 75 | Hiradfar et al. [58] | 2012 | F | 9 | Inguinal region | n/a | n/a | Surgery | n/a | n/a | n/a |
| 76 | Rojas et al. [59] | 2013 | M | 2 | Retro- peritoneum | n/a | I/II | Surgery + chemo | n/a | n/a | n/a |
| 77 | Morandi et al. [60] | 2013 | M | 3 | Pelvis | n/a | n/a | Surgery + chemo | None | 2 | Alive |
| 78 | Goel et al. [61] | 2014 | n/a | 5 | Retro- peritoneum | 9 | n/a | Surgery + chemo + RAD | None | 2 | Alive |
| 79 | Kumar et al. [62] | 2015 | F | 7 | Retro- peritoneum | 1 | n/a | Surgery | None | 0.8 | Alive |
| 80 | Thakkar et al. [63] | 2015 | F | 5 | Retro- peritoneum | 3 | III (NWTS) | Surgery + chemo + RAD | None | n/a | Alive |
| 81 | Park [64] | 2016 | F | 4 | Retro- peritoneum | n/a | n/a | Surgery + chemo + RAD | Lungs, peritoneum | 4 | Alive |
| 82 | Wabada et al. [65] | 2017 | M | 2 | Retro- peritoneum | 13 | III (SIOP) | Surgery + chemo | None | 0.3 | Alive |
| 83 | Itoshima et al. [66] | 2017 | M | 4 | Retro- peritoneum | n/a | III (NWTS) | Surgery + chemo + RAD | None | 3 | Alive |
| 84 | Igbaseimokumo et al. [67] | 2017 | F | <0.1 | Spinal cord (L5) | 13 | n/a | Surgery + chemo | None | 2.5 | Alive |
| 85 | Tang et al. [68] | 2018 | M | 2 | Retro- peritoneum | n/a | n/a | n/a | n/a | n/a | n/a |
| 86 | Tang et al. [68] | 2018 | F | 2 | Mesentery | n/a | n/a | n/a | n/a | n/a | n/a |
| 87 | Sindhu et al. [69] | 2019 | M | 6 | Bladder | 65 | III (SIOP) | Surgery + chemo + RAD | None | n/a | Alive |
| 88 | Groth et al. [70] | 2019 | M | 0.7 | Inguinal canal | n/a | III (NWTS) | Surgery + chemo + RAD | Local | 1.3 | Alive |
| 89 | Ismy et al. [71] | 2019 | M | 1 | Bladder | 13 | n/a | Surgery | n/a | n/a | n/a |
| 90 | Liang et al. [72] | 2020 | M | 5 | Retro- peritoneum | n/a | III (NWTS) | Surgery + chemo | Local + Lungs + Liver | 1 | Death |
| 91 | Liang et al. [72] | 2020 | F | 3.4 | Retro- peritoneum | n/a | III (NWTS) | Surgery + chemo + RAD | None | 10.8 | Alive |
| 92 | Liang et al. [72] | 2020 | F | 3.4 | Sigmoid colon | 4 | II (NWTS) | Surgery + chemo | None | 3.3 | Alive |
| 93 | Liang et al. [72] | 2020 | M | 9.8 | Retro- peritoneum | n/a | III (NWTS) | Surgery + chemo + RAD | Local + Lungs | 1.8 | Alive |
| 94 | Liang et al. [72] | 2020 | M | 2.8 | Inguinal canal | n/a | II (NWTS) | Surgery + chemo | None | 1.5 | Alive |
| 95 | Parkhi et al. [73] | 2022 | F | 4 | Bladder | 4 | n/a | Surgery + chemo | None | 0.8 | Alive |
| 96 | Qu et al. [74] | 2022 | M | 0.5 | Pelvis | 4 | n/a | Surgery + chemo | None | 0.3 | Alive |
| 97 | Albiroty et al. [75] | 2022 | F | 2 | Ovary | 9 | n/a | Surgery + chemo + RAD | None | 1 | Alive |
| 98 | Our case | 2022 | M | 4 | Spinal cord (T9-S4) | 48 | IV (SIOP) | Surgery * + chemo + RAD | Local + Lungs | 0.3 | Death |
| № | Article | Sex | Age (yrs.) | Site | Spinal Malformation | External Malformation | Surgery | Chemo-Therapy (Main Drugs) | Radio- Therapy (Regimen) | Recurrence/ Metastasis | Follow-Up (yrs.) | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Fernbach et al., 1984 [26] | F | 2 | L1 | Diastemato-myelia | Lipoma with hypertrichosis | Near-total excision | Yes (n/a) | Yes (n/a) | No | 1 | Alive |
| 2 | Mirkin et al., 1990 [34] | F | 2 | T12-L4 | Diastemato-myelia | Lipoma with hypertrichosis | Gross total excision | ARA-C VCR ACT-D DXR | Local + Metastasis (2700 rads) | Yes (Cerebellum) | 1.7 | Alive |
| 3 | Govender et al., 2000 [51] | F | 4 | T10-Sx | Spina bifida | No | Biopsy only | CSP ETO IFO | Palliative (n/a) | n/a | n/a | n/a |
| 4 | Sharma et al., 2005 [57] | F | 1.5 | L2-L5 | Diastemato-myelia | Lipoma with hypertrichosis | Gross total excision | Yes (n/a) | n/a | n/a | n/a | Alive |
| 5 | Igbaseimo-kumo et al., 2017 [67] | F | <0.1 | L5 | Occult dysraphism | Lipoma with hypertrichosis | Gross total excision | VCR ACT-D | No | No | 2.5 | Alive |
| 6 | Our case | M | 4 | T9-S4 | Spina bifida occulta | No | Biopsy only | CPT ETO CYC DXR | Craniospinal (25.5 Gy) + Local (25.5 Gy) | Yes (Local + Lungs) | 0.3 | Death |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karim, A.; Shaikhyzada, K.; Abulkhanova, N.; Altyn, A.; Ibraimov, B.; Nurgaliyev, D.; Poddighe, D. Pediatric Extra-Renal Nephroblastoma (Wilms’ Tumor): A Systematic Case-Based Review. Cancers 2023, 15, 2563. https://doi.org/10.3390/cancers15092563
Karim A, Shaikhyzada K, Abulkhanova N, Altyn A, Ibraimov B, Nurgaliyev D, Poddighe D. Pediatric Extra-Renal Nephroblastoma (Wilms’ Tumor): A Systematic Case-Based Review. Cancers. 2023; 15(9):2563. https://doi.org/10.3390/cancers15092563
Chicago/Turabian StyleKarim, Akzhol, Kundyz Shaikhyzada, Nazgul Abulkhanova, Akzhunis Altyn, Bakytkali Ibraimov, Dair Nurgaliyev, and Dimitri Poddighe. 2023. "Pediatric Extra-Renal Nephroblastoma (Wilms’ Tumor): A Systematic Case-Based Review" Cancers 15, no. 9: 2563. https://doi.org/10.3390/cancers15092563
APA StyleKarim, A., Shaikhyzada, K., Abulkhanova, N., Altyn, A., Ibraimov, B., Nurgaliyev, D., & Poddighe, D. (2023). Pediatric Extra-Renal Nephroblastoma (Wilms’ Tumor): A Systematic Case-Based Review. Cancers, 15(9), 2563. https://doi.org/10.3390/cancers15092563