
Metabolic Interventions in Tumor Immunity: Focus on Dual Pathway Inhibitors


Abstract
Simple Summary
Abstract
1. Introduction
2. Metabolism of Tumor and Immune Cells
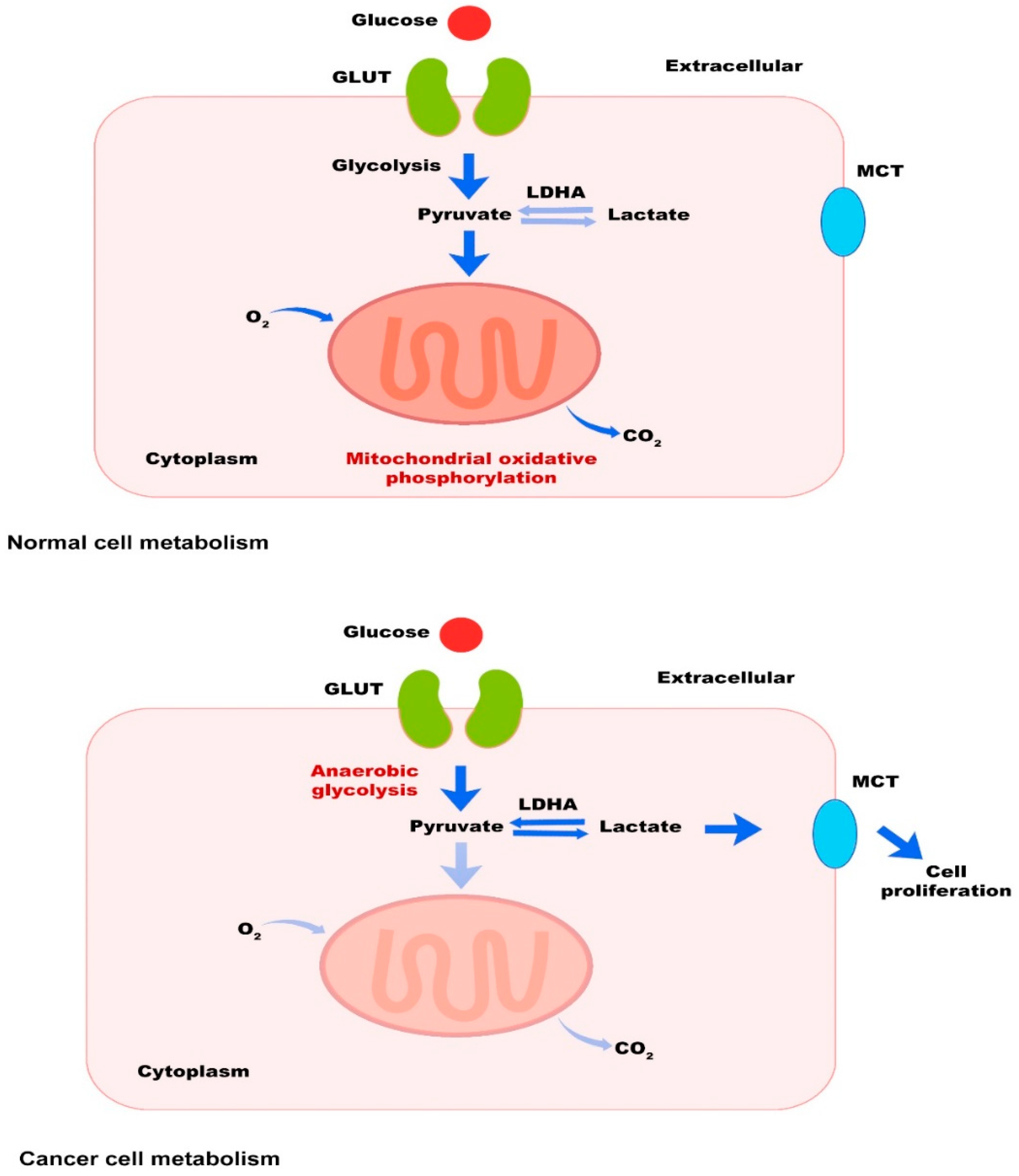
2.1. Tumor Cells
2.2. Immune Cells
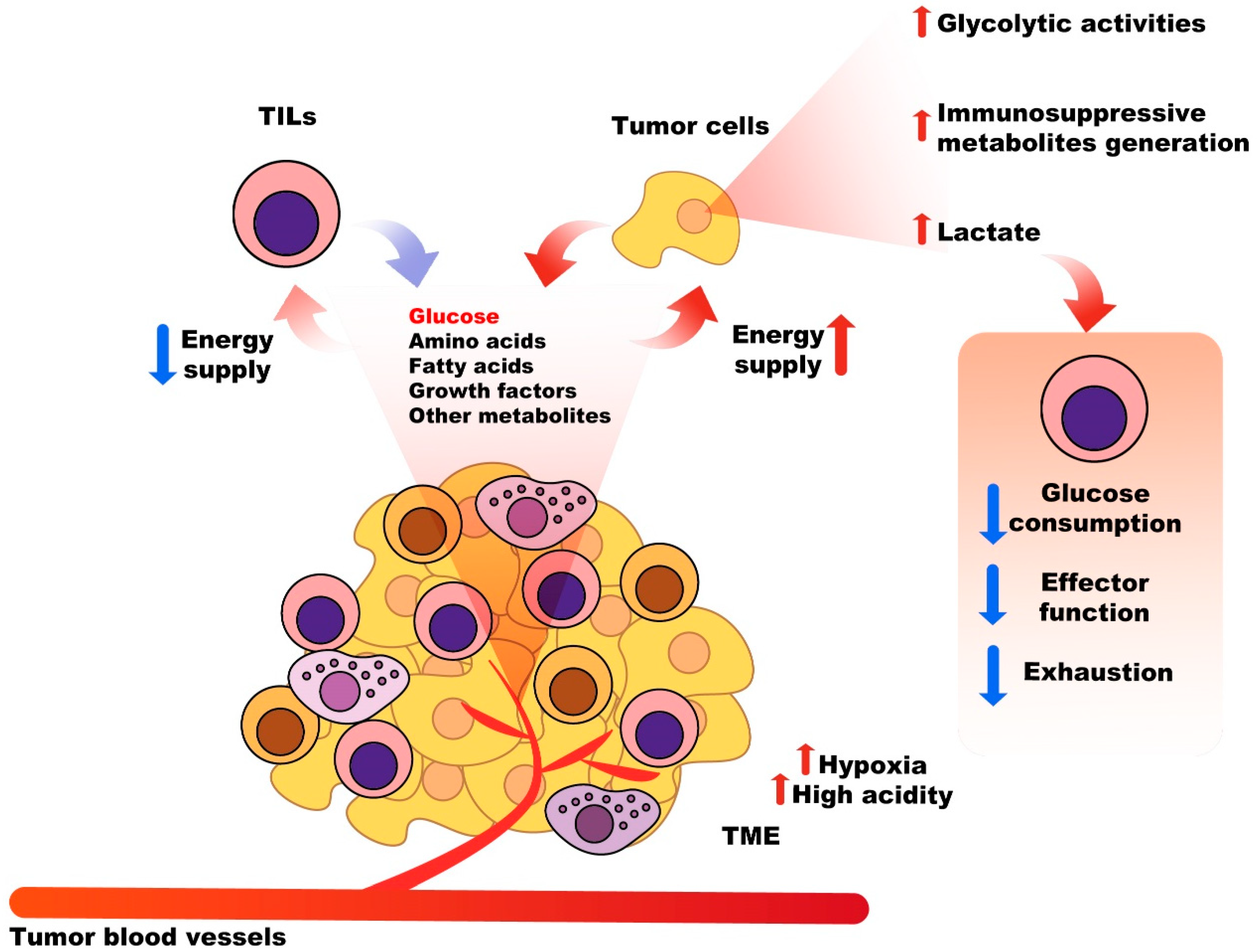
2.3. Nutritional Competition between Tumor Cells and Immune System Cells
3. The Most Important Metabolic Pathways in Cancer and Therapeutic Interventions
3.1. PI3K/AKT/mTOR Pathway
3.2. AMPK Pathway
3.3. Adenosine Pathway
4. Dual Pathway Inhibitors
4.1. Dual PI3K/AKT/mTOR Inhibitors
4.1.1. Dactolisib
4.1.2. Gedatolisib
4.1.3. Voxtalisib
4.1.4. Bimiralisib
4.1.5. Paxalisib
4.1.6. Omipalisib
4.1.7. SF1126
4.1.8. PF-04691502
4.1.9. Samotolisib
4.1.10. PWT33597
4.1.11. Apitolisib
4.2. Other Potential Dual Inhibitors
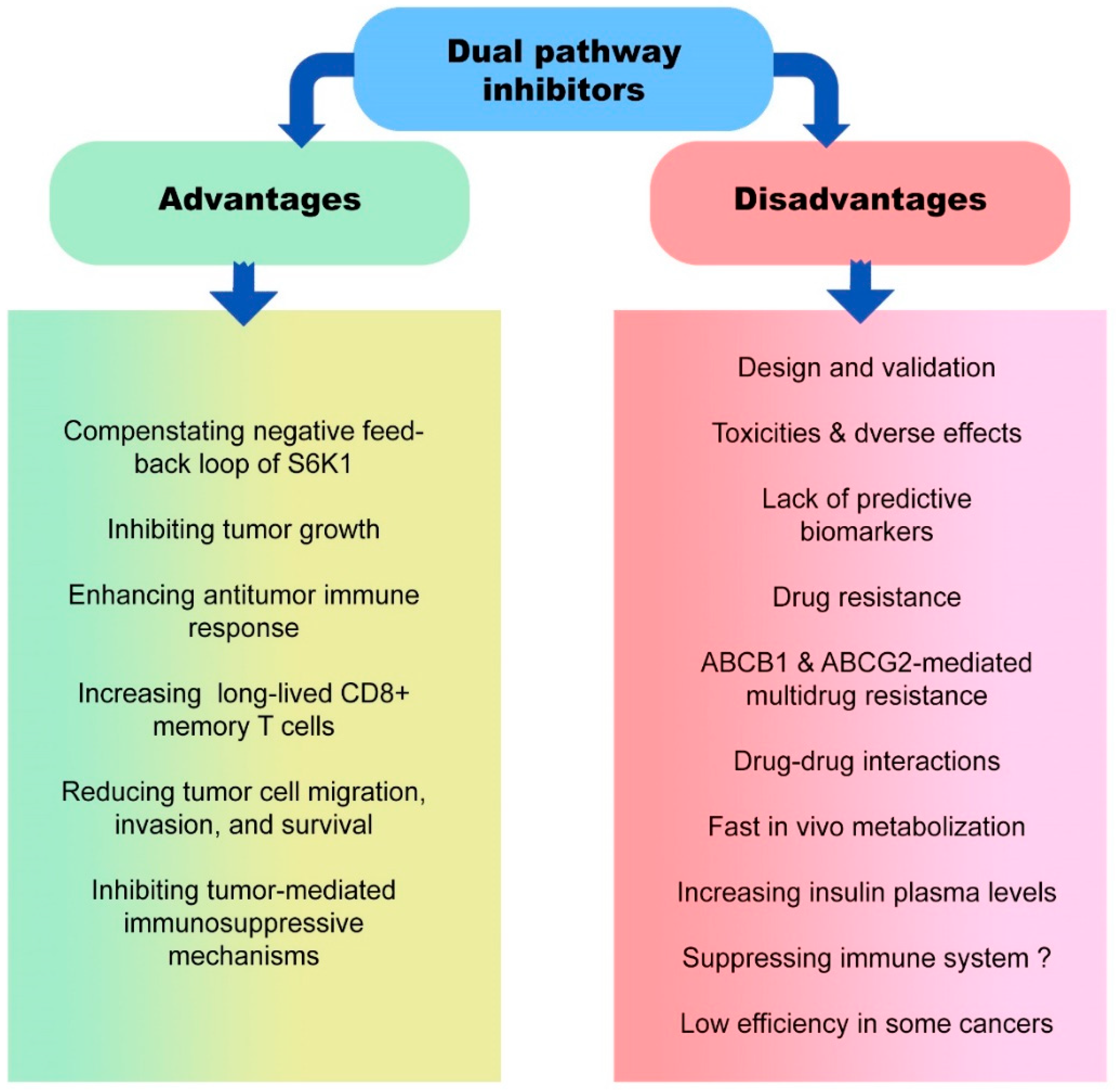
5. Advantages and Disadvantages of Dual Pathway Inhibitors in Cancer Therapy
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Liu, J.; Zhou, Y.; Oltvai, Z.N. Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst. Biol. 2010, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Lapa, B.; Gonçalves, A.C.; Jorge, J.; Alves, R.; Pires, A.S.; Abrantes, A.M.; Coucelo, M.; Abrunhosa, A.; Botelho, M.F.; Nascimento-Costa, J.M. Acute myeloid leukemia sensitivity to metabolic inhibitors: Glycolysis showed to be a better therapeutic target. Med. Oncol. 2020, 37, 72. [Google Scholar] [CrossRef]
- Callao, V.; Montoya, E. Toxohormone-like factor from microorganisms with impaired respiration. Science 1961, 134, 2041–2042. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Domiński, A.; Krawczyk, M.; Konieczny, T.; Kasprów, M.; Foryś, A.; Pastuch-Gawołek, G.; Kurcok, P. Biodegradable pH-responsive micelles loaded with 8-hydroxyquinoline glycoconjugates for Warburg effect based tumor targeting. Eur. J. Pharm. Biopharm. 2020, 154, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.; Lin, C.; Liu, W.; Huo, Y.; Yang, M.; Jiang, S.-H.; Sun, Y.; Hua, R. Endoplasmic Reticulum stress-dependent expression of ERO1L promotes aerobic glycolysis in Pancreatic Cancer. Theranostics 2020, 10, 8400. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.-l.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef]
- Chen, B.; Gao, A.; Tu, B.; Wang, Y.; Yu, X.; Wang, Y.; Xiu, Y.; Wang, B.; Wan, Y.; Huang, Y. Metabolic modulation via mTOR pathway and anti-angiogenesis remodels tumor microenvironment using PD-L1-targeting codelivery. Biomaterials 2020, 255, 120187. [Google Scholar] [CrossRef]
- Terry, S.; Engelsen, A.S.; Buart, S.; Elsayed, W.S.; Venkatesh, G.H.; Chouaib, S. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett. 2020, 492, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chang, L.; Tian, H.; Wang, L.; Zhang, Y.; Yang, T.; Li, G.; Hu, W.; Shah, K.; Chen, G. 1-Pyrroline-5-carboxylate released by prostate Cancer cell inhibit T cell proliferation and function by targeting SHP1/cytochrome c oxidoreductase/ROS Axis. J. Immunother. Cancer 2018, 6, 148. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.; Noguchi, T.; Curtis, J.; Chen, Q.; Gindin, M.; Gubin, M.; Tonc, E. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Amirani, E.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Yousefi, B. Effects of chitosan and oligochitosans on the phosphatidylinositol 3-kinase-AKT pathway in cancer therapy. Int. J. Biol. Macromol. 2020, 164, 456–467. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.S.; Lyon, D.; Kelly, D.L.; Stechmiller, J. Metabolomics: Impact of comorbidities and inflammation on sickness behaviors for individuals with chronic wounds. Adv. Wound Care 2021, 10, 357–369. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef]
- Ali, E.S.; Mitra, K.; Akter, S.; Ramproshad, S.; Mondal, B.; Khan, I.N.; Islam, M.T.; Sharifi-Rad, J.; Calina, D.; Cho, W.C. Recent advances and limitations of mTOR inhibitors in the treatment of cancer. Cancer Cell Int. 2022, 22, 284. [Google Scholar] [CrossRef]
- Viana, S.D.; Reis, F.; Alves, R. Therapeutic use of mTOR inhibitors in renal diseases: Advances, drawbacks, and challenges. Oxidative Med. Cell. Longev. 2018, 2018, 3693625. [Google Scholar] [CrossRef]
- Matthews, D.J.; O’Farrell, M.; James, J.; Giddens, A.C.; Rewcastle, G.W.; Denny, W.A. Preclinical characterization of PWT33597, a dual inhibitor of PI3-kinase alpha and mTOR. Cancer Res. 2011, 71, 4485. [Google Scholar] [CrossRef]
- Herschbein, L.; Liesveld, J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2018, 32, 235–248. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, K.-N.; Li, R.; Shao, R.; Chen, C. Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and mTOR in endometrial cancer. Curr. Med. Chem. 2014, 21, 3070–3080. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Bhende, P.M.; Sin, S.-H.; Roy, D.; Dittmer, D.P.; Damania, B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood J. Am. Soc. Hematol. 2010, 115, 4455–4463. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Brattain, M.G.; Zhong, H. Dual inhibitors of PI3K/mTOR or mTOR-selective inhibitors: Which way shall we go? Curr. Med. Chem. 2011, 18, 5528–5544. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, Q.; Halim, A.; Song, G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017, 401, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, P. Fatty acid metabolism and cancer development. Sci. Bull. 2016, 61, 1473–1479. [Google Scholar] [CrossRef]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Vande Voorde, J.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, H.; Liu, J.; Deng, Y.; Zhang, N. Comprehensive understanding of anchorage-independent survival and its implication in cancer metastasis. Cell Death Dis. 2021, 12, 629. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Metabolic reprogramming sustains cancer cell survival following extracellular matrix detachment. Redox Biol. 2020, 36, 101643. [Google Scholar] [CrossRef] [PubMed]
- Ghesquière, B.; Wong, B.W.; Kuchnio, A.; Carmeliet, P. Metabolism of stromal and immune cells in health and disease. Nature 2014, 511, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Thwe, P.M.; Amiel, E. The role of nitric oxide in metabolic regulation of dendritic cell immune function. Cancer Lett. 2018, 412, 236–242. [Google Scholar] [CrossRef]
- Williford, J.-M.; Ishihara, J.; Ishihara, A.; Mansurov, A.; Hosseinchi, P.; Marchell, T.M.; Potin, L.; Swartz, M.A.; Hubbell, J.A. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 2019, 5, eaay1357. [Google Scholar] [CrossRef]
- Wang, Y.; Hwang, J.-Y.; Park, H.-b.; Yadav, D.; Oda, T.; Jin, J.-O. Porphyran isolated from Pyropia yezoensis inhibits lipopolysaccharide-induced activation of dendritic cells in mice. Carbohydr. Polym. 2020, 229, 115457. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.-H.; Hong, C.-W.; Kim, E.Y.; Lee, J.M. Current understanding on the metabolism of neutrophils. Immune Netw. 2020, 20, e46. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.; Pearce, E. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef]
- Kobayashi, T.; Lam, P.Y.; Jiang, H.; Bednarska, K.; Gloury, R.; Murigneux, V.; Tay, J.; Jacquelot, N.; Li, R.; Tuong, Z.K. Increased lipid metabolism impairs NK cell function and mediates adaptation to the lymphoma environment. Blood 2020, 136, 3004–3017. [Google Scholar] [CrossRef] [PubMed]
- Domka, K.; Goral, A.; Firczuk, M. cROSsing the line: Between beneficial and harmful effects of reactive oxygen species in B-cell malignancies. Front. Immunol. 2020, 11, 1538. [Google Scholar] [CrossRef]
- Wang, X.-Y.; Wei, Y.; Hu, B.; Liao, Y.; Wang, X.; Wan, W.-H.; Huang, C.-X.; Mahabati, M.; Liu, Z.-Y.; Qu, J.-R. c-Myc-driven glycolysis polarizes functional regulatory B cells that trigger pathogenic inflammatory responses. Signal Transduct. Target. Ther. 2022, 7, 105. [Google Scholar] [CrossRef]
- Kolb, D.; Kolishetti, N.; Surnar, B.; Sarkar, S.; Guin, S.; Shah, A.S.; Dhar, S. Metabolic modulation of the tumor microenvironment leads to multiple checkpoint inhibition and immune cell infiltration. ACS Nano 2020, 14, 11055–11066. [Google Scholar] [CrossRef]
- Palmer, C.S.; Ostrowski, M.; Balderson, B.; Christian, N.; Crowe, S.M. Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 2015, 6, 1. [Google Scholar] [CrossRef]
- Togo, M.; Yokobori, T.; Shimizu, K.; Handa, T.; Kaira, K.; Sano, T.; Tsukagoshi, M.; Higuchi, T.; Yokoo, S.; Shirabe, K. Diagnostic value of 18F-FDG-PET to predict the tumour immune status defined by tumoural PD-L1 and CD8+ tumour-infiltrating lymphocytes in oral squamous cell carcinoma. Br. J. Cancer 2020, 122, 1686–1694. [Google Scholar] [CrossRef]
- Qiu, J.; Villa, M.; Sanin, D.E.; Buck, M.D.; O’Sullivan, D.; Ching, R.; Matsushita, M.; Grzes, K.M.; Winkler, F.; Chang, C.-H. Acetate promotes T cell effector function during glucose restriction. Cell Rep. 2019, 27, 2063–2074.e5. [Google Scholar] [CrossRef]
- Sukumar, M.; Roychoudhuri, R.; Restifo, N.P. Nutrient competition: A new axis of tumor immunosuppression. Cell 2015, 162, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Harmon, C.; O’Farrelly, C.; Robinson, M.W. The immune consequences of lactate in the tumor microenvironment. In Tumor Microenvironment; Springer: Berlin/Heidelberg, Germany, 2020; pp. 113–124. [Google Scholar]
- Kareva, I. Metabolism and gut microbiota in cancer immunoediting, CD8/Treg ratios, immune cell homeostasis, and cancer (immuno) therapy: Concise review. Stem Cells 2019, 37, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Donahue, T.R.; Tran, L.M.; Hill, R.; Li, Y.; Kovochich, A.; Calvopina, J.H.; Patel, S.G.; Wu, N.; Hindoyan, A.; Farrell, J.J. Integrative Survival-Based Molecular Profiling of Human Pancreatic CancerIntegrative Profile of Human Pancreatic Cancer. Clin. Cancer Res. 2012, 18, 1352–1363. [Google Scholar] [CrossRef] [PubMed]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, immunity, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B. The PI3K/AKT pathway and renal cell carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Masui, K.; Harachi, M.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. mTOR complex 2 is an integrator of cancer metabolism and epigenetics. Cancer Lett. 2020, 478, 1–7. [Google Scholar] [CrossRef]
- Huang, K.; Fingar, D.C. Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 2014, 36, 79–90. [Google Scholar] [CrossRef]
- Csibi, A.; Lee, G.; Yoon, S.-O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.-M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.-M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, T.; Yang, W.; Zhang, L.; Wu, S.; Yan, C.; Li, Q. Astragalus membranaceus injection suppresses production of interleukin-6 by activating autophagy through the AMPK-mTOR pathway in lipopolysaccharide-stimulated macrophages. Oxidative Med. Cell. Longev. 2020, 2020, 1364147. [Google Scholar]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A Diverse Array of Cancer-Associated MTOR Mutations Are Hyperactivating and Can Predict Rapamycin SensitivityCancer-Associated Hyperactivating MTOR Mutations. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef]
- Pilotto, S.; Simbolo, M.; Sperduti, I.; Novello, S.; Vicentini, C.; Peretti, U.; Pedron, S.; Ferrara, R.; Caccese, M.; Milella, M. OA06. 06 druggable alterations involving crucial carcinogenesis pathways drive the prognosis of squamous cell lung carcinoma (SqCLC). J. Thorac. Oncol. 2017, 12, S266–S267. [Google Scholar] [CrossRef]
- Morrison Joly, M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Estrada, M.V.; Young, C.; Williams, M.; Rexer, B.N.; Sarbassov, D.D.; Muller, W.J. Rictor/mTORC2 Drives Progression and Therapeutic Resistance of HER2-Amplified Breast CancersHER2-Mediated Tumorigenesis Requires mTORC2. Cancer Res. 2016, 76, 4752–4764. [Google Scholar] [CrossRef]
- Mafi, S.; Mansoori, B.; Taeb, S.; Sadeghi, H.; Abbasi, R.; Cho, W.C.; Rostamzadeh, D. mTOR-mediated regulation of immune responses in cancer and tumor microenvironment. Front. Immunol. 2022, 12, 5724. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In Metabolism in Cancer; Springer: Berlin/Heidelberg, Germany, 2016; pp. 39–72. [Google Scholar]
- Buller, C.L.; Loberg, R.D.; Fan, M.-H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Guan, K.-L.; Brosius, F.C., III. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008, 295, C836–C843. [Google Scholar] [CrossRef]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.-H. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Rüegg, M.A.; Hall, M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Driscoll, D.R.; Karim, S.A.; Sano, M.; Gay, D.M.; Jacob, W.; Yu, J.; Mizukami, Y.; Gopinathan, A.; Jodrell, D.I.; Evans, T.R.J.; et al. mTORC2 Signaling Drives the Development and Progression of Pancreatic Cancer. Cancer Res. 2016, 76, 6911–6923. [Google Scholar] [CrossRef]
- Bian, Y.; Wang, Z.; Xu, J.; Zhao, W.; Cao, H.; Zhang, Z. Elevated Rictor expression is associated with tumor progression and poor prognosis in patients with gastric cancer. Biochem. Biophys. Res. Commun. 2015, 464, 534–540. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, X.; Li, M.; Chen, P.; Zhang, B.; Guo, H.; Cao, W.; Wei, X.; Cao, X.; Hao, X.; et al. mTOR Complex Component Rictor Interacts with PKCζ and Regulates Cancer Cell Metastasis. Cancer Res. 2010, 70, 9360–9370. [Google Scholar] [CrossRef]
- Li, H.; Lin, J.; Wang, X.; Yao, G.; Wang, L.; Zheng, H.; Yang, C.; Jia, C.; Liu, A.; Bai, X. Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. Breast Cancer Res. Treat. 2012, 134, 1057–1066. [Google Scholar] [CrossRef]
- Gulhati, P.; Cai, Q.; Li, J.; Liu, J.; Rychahou, P.G.; Qiu, S.; Lee, E.Y.; Silva, S.R.; Bowen, K.A.; Gao, T.; et al. Targeted Inhibition of Mammalian Target of Rapamycin Signaling Inhibits Tumorigenesis of Colorectal Cancer. Clin. Cancer Res. 2009, 15, 7207–7216. [Google Scholar] [CrossRef]
- Xie, S.; Chen, M.; Yan, B.; He, X.; Chen, X.; Li, D. Identification of a Role for the PI3K/AKT/mTOR Signaling Pathway in Innate Immune Cells. PLoS ONE 2014, 9, e94496. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Suresh, M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front. Immunol. 2013, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef]
- Guri, Y.; Nordmann, T.M.; Roszik, J. mTOR at the Transmitting and Receiving Ends in Tumor Immunity. Front. Immunol. 2018, 9, 578. [Google Scholar] [CrossRef]
- Crompton, J.G.; Sukumar, M.; Roychoudhuri, R.; Clever, D.; Gros, A.; Eil, R.L.; Tran, E.; Hanada, K.-i.; Yu, Z.; Palmer, D.C.; et al. Akt Inhibition Enhances Expansion of Potent Tumor-Specific Lymphocytes with Memory Cell Characteristics. Cancer Res. 2015, 75, 296–305. [Google Scholar] [CrossRef]
- Zheng, W.; O’Hear, C.E.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Chae, S.C.; Yang, H.J.; An, D.E.; Lee, S.; Yeo, M.G.; Lee, K.J. Cereblon Deletion Ameliorates Lipopolysaccharide-induced Proinflammatory Cytokines through 5’-Adenosine Monophosphate-Activated Protein Kinase/Heme Oxygenase-1 Activation in ARPE-19 Cells. Immune Netw. 2020, 20, e26. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. 2019, 97, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lin, Y.; Xiong, X.; Wang, L.; Guo, Y.; Chen, Y.; Chen, S.; Wang, G.; Lin, P.; Chen, H.; et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical Trial. Clin. Cancer Res. 2020, 26, 4921–4932. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Brown, J.R.; Sag, D.; Zhang, L.; Suttles, J. Adenosine 5′-Monophosphate–Activated Protein Kinase Regulates IL-10–Mediated Anti-Inflammatory Signaling Pathways in Macrophages. J. Immunol. 2015, 194, 584–594. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef]
- Whiteside, T.; Jackson, E. Adenosine and Prostaglandin E2 Production by Human Inducible Regulatory T Cells in Health and Disease. Front. Immunol. 2013, 4, 212. [Google Scholar] [CrossRef]
- Umansky, V.; Sevko, A. Overcoming immunosuppression in the melanoma microenvironment induced by chronic inflammation. Cancer Immunol. Immunother. 2012, 61, 275–282. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Wu, H.-C.; Wu, J.-E.; Huang, K.-Y.; Yang, S.-C.; Chen, S.-X.; Tsao, C.-J.; Hsu, K.-F.; Chen, Y.-L.; Hong, T.-M. The dual PI3K/mTOR inhibitor BEZ235 restricts the growth of lung cancer tumors regardless of EGFR status, as a potent accompanist in combined therapeutic regimens. J. Exp. Clin. Cancer Res. 2019, 38, 282. [Google Scholar] [CrossRef]
- Rosich, L.; Montraveta, A.; Xargay-Torrent, S.; López-Guerra, M.; Roldán, J.; Aymerich, M.; Salaverria, I.; Beà, S.; Campo, E.; Pérez-Galán, P. Dual PI3K/mTOR inhibition is required to effectively impair microenvironment survival signals in mantle cell lymphoma. Oncotarget 2014, 5, 6788. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of Glucocorticoid Resistance in Pediatric T-cell Acute Lymphoblastic Leukemia by Increasing BIM Expression with the PI3K/mTOR Inhibitor BEZ235BEZ235 plus Dexamethasone in ALL. Clin. Cancer Res. 2016, 22, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Pérez-Fidalgo, A.; Krop, I.E.; Burris, H.; Guerrero-Zotano, A.; Britten, C.D.; Becerra, C.; Schellens, J.; Richards, D.A.; Schuler, M. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharmacol. 2018, 82, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, C.; Li, A.; Liu, X.; Xing, Y.; Shen, J.; Huo, Z.; Zhou, S.; Liu, X.; Xie, Y. PKI-587 enhances chemosensitivity of oxaliplatin in hepatocellular carcinoma through suppressing DNA damage repair pathway (NHEJ and HR) and PI3K/AKT/mTOR pathway. Am. J. Transl. Res. 2019, 11, 5134. [Google Scholar]
- Radovich, M.; Solzak, J.P.; Wang, C.J.; Hancock, B.A.; Badve, S.S.; Althouse, S.K.; Bray, S.M.; Storniolo, A.M.; Ballinger, T.J.; Schneider, B.P. Initial phase I safety study of gedatolisib plus cofetuzumab pelidotin for patients with metastatic triple-negative breast cancer. Clin. Cancer Res. 2022, 28, 3235–3241. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, C.; Zhang, Y.; Li, A.; Sun, C.; Li, R.; Xing, Y.; Shi, M.; Wang, Q. PKI-587 enhances radiosensitization of hepatocellular carcinoma by inhibiting the PI3K/AKT/mTOR pathways and DNA damage repair. PLoS ONE 2021, 16, e0258817. [Google Scholar] [CrossRef]
- Gazi, M.; Moharram, S.A.; Marhäll, A.; Kazi, J.U. The dual specificity PI3K/mTOR inhibitor PKI-587 displays efficacy against T-cell acute lymphoblastic leukemia (T-ALL). Cancer Lett. 2017, 392, 9–16. [Google Scholar] [CrossRef]
- Schram, A.M.; Gandhi, L.; Mita, M.M.; Damstrup, L.; Campana, F.; Hidalgo, M.; Grande, E.; Hyman, D.M.; Heist, R.S. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer 2018, 119, 1471–1476. [Google Scholar] [CrossRef]
- Wen, P.Y.; Omuro, A.; Ahluwalia, M.S.; Fathallah-Shaykh, H.M.; Mohile, N.; Lager, J.J.; Laird, A.D.; Tang, J.; Jiang, J.; Egile, C. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015, 17, 1275–1283. [Google Scholar] [CrossRef]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination TherapyNovel Dual PI3K/mTOR Inhibitor for Lymphomas. Clin. Cancer Res. 2018, 24, 120–129. [Google Scholar] [CrossRef]
- Wicki, A.; Brown, N.; Xyrafas, A.; Bize, V.; Hawle, H.; Berardi, S.; Cmiljanović, N.; Cmiljanović, V.; Stumm, M.; Dimitrijević, S. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13). Eur. J. Cancer 2018, 96, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Hillmann, P.; Noack, A.; Römermann, K.; Öhler, L.A.; Rageot, D.; Beaufils, F.; Melone, A.; Sele, A.M.; Wymann, M.P. The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood-brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology 2018, 140, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Cloughesy, T.F.; Olivero, A.G.; Morrissey, K.M.; Wilson, T.R.; Lu, X.; Mueller, L.U.; Coimbra, A.F.; Ellingson, B.M.; Gerstner, E. First-in-Human Phase I Study to Evaluate the Brain-Penetrant PI3K/mTOR Inhibitor GDC-0084 in Patients with Progressive or Recurrent High-Grade GliomaGDC-0084 in Progressive or Recurrent High-Grade Glioma. Clin. Cancer Res. 2020, 26, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Heffron, T.P.; Ndubaku, C.O.; Salphati, L.; Alicke, B.; Cheong, J.; Drobnick, J.; Edgar, K.; Gould, S.E.; Lee, L.B.; Lesnick, J.D. Discovery of clinical development candidate GDC-0084, a brain penetrant inhibitor of PI3K and mTOR. ACS Med. Chem. Lett. 2016, 7, 351–356. [Google Scholar] [CrossRef]
- Stumpf, A.; McClory, A.; Yajima, H.; Segraves, N.; Angelaud, R.; Gosselin, F. Development of an efficient, safe, and environmentally friendly process for the manufacture of GDC-0084. Org. Process Res. Dev. 2016, 20, 751–759. [Google Scholar] [CrossRef]
- Ippen, F.M.; Alvarez-Breckenridge, C.A.; Kuter, B.M.; Fink, A.L.; Bihun, I.V.; Lastrapes, M.; Penson, T.; Schmidt, S.P.; Wojtkiewicz, G.R.; Ning, J. The Dual PI3K/mTOR Pathway Inhibitor GDC-0084 Achieves Antitumor Activity in PIK3CA-Mutant Breast Cancer Brain MetastasesDual PI3K/mTOR Inhibition in Breast Cancer Brain Metastases. Clin. Cancer Res. 2019, 25, 3374–3383. [Google Scholar] [CrossRef]
- Ding, L.-t.; Zhao, P.; Yang, M.-l.; Lv, G.-z.; Zhao, T.-l. GDC-0084 inhibits cutaneous squamous cell carcinoma cell growth. Biochem. Biophys. Res. Commun. 2018, 503, 1941–1948. [Google Scholar] [CrossRef]
- Tinkle, C.; Huang, J.; Campagne, O.; Pan, H.; Onar-Thomas, A.; Chiang, J.; Klimo, P.; Boop, R.; Patay, Z.; Shulkin, B. CTNI-27. first-in-pediatrics phase I study of GDC-0084 (PAXALISIB), a CNS-penetrant PI3K/mTOR inhibitor, in newly diagnosed diffuse intrinsic pontine glioma (DIPG) or other diffuse midline glioma (DMG). Neuro Oncol. 2020, 22, ii48. [Google Scholar] [CrossRef]
- Basu, D.; Salgado, C.M.; Bauer, B.; Khakoo, Y.; Patel, J.R.; Hoehl, R.M.; Bertolini, D.M.; Zabec, J.; Brzozowski, M.R.; Reyes-Múgica, M. The dual PI3K/mToR inhibitor omipalisib/GSK2126458 inhibits clonogenic growth in oncogenically-transformed cells from neurocutaneous melanocytosis. Cancer Genom. Proteom. 2018, 15, 239–248. [Google Scholar] [CrossRef]
- Tseng, C.-Y.; Kuo, C.-Y.; Fu, Y.-H.; Hou, H.-A.; Tien, H.-F.; Lin, L.-I. P422: Omipalisib, a dual PI3K/MTOR inhibitor, targets mitochondria and impairs oxidative phosphorylation in acute myeloid leukemia. HemaSphere 2022, 6, 322–323. [Google Scholar] [CrossRef]
- Lin, L.-I.; Chiyang, T.; Fu, Y.-H.; Hou, H.-A.; Chou, W.-C.; Tien, H.-F. Metabolic Profiling Reveals Cellular Reprogramming of Acute Myeloid Leukemia By Omipalisib through Serine Synthesis Pathway. Blood 2021, 138, 3296. [Google Scholar] [CrossRef]
- Qin, A.-C.; Li, Y.; Zhou, L.-N.; Xing, C.-G.; Lu, X.-S. Dual PI3K-BRD4 inhibitor SF1126 inhibits colorectal cancer cell growth in vitro and in vivo. Cell. Physiol. Biochem. 2019, 52, 758–768. [Google Scholar]
- Mahadevan, D.; Chiorean, E.; Harris, W.; Von Hoff, D.; Stejskal-Barnett, A.; Qi, W.; Anthony, S.; Younger, A.; Rensvold, D.; Cordova, F. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur. J. Cancer 2012, 48, 3319–3327. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Singh, A.R.; Durden, D.L. Pan-PI-3 kinase inhibitor SF1126 shows antitumor and antiangiogenic activity in renal cell carcinoma. Cancer Chemother. Pharmacol. 2015, 75, 595–608. [Google Scholar] [CrossRef]
- Li, Q.; Li, Z.; Luo, T.; Shi, H. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol. Biomed. 2022, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Na, X.; Wang, L.; Yang, Z.; Ren, P. Evaluation of a Dual PI3K/mTOR Inhibitor PF-04691502 against Bladder Cancer Cells. Evid. Based Complement. Altern. Med. 2022, 2022, 8110796. [Google Scholar] [CrossRef] [PubMed]
- Chow, Z.; Johnson, J.; Chauhan, A.; Izumi, T.; Cavnar, M.; Weiss, H.; Townsend Jr, C.M.; Anthony, L.; Wasilchenko, C.; Melton, M.L. PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells 2021, 10, 1261. [Google Scholar] [CrossRef]
- Stoll, J.R.; Willner, J.; Oh, Y.; Pulitzer, M.; Moskowitz, A.; Horwitz, S.; Myskowski, P.; Noor, S.J. Primary cutaneous T-cell lymphomas other than mycosis fungoides and Sézary syndrome. Part I: Clinical and histologic features and diagnosis. J. Am. Acad. Dermatol. 2021, 85, 1073–1090. [Google Scholar] [CrossRef]
- Hong, D.S.; Moore, K.N.; Bendell, J.C.; Karp, D.D.; Wang, J.S.; Ulahannan, S.V.; Jones, S.; Wu, W.; Donoho, G.P.; Ding, Y. Preclinical Evaluation and Phase Ib Study of Prexasertib, a CHK1 Inhibitor, and Samotolisib (LY3023414), a Dual PI3K/mTOR InhibitorPreclinical/Phase Ib Evaluation of Prexasertib+ Samotolisib. Clin. Cancer Res. 2021, 27, 1864–1874. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Babu, S.; Cultrera, J.L.; Mehlhaff, B.A.; Goodman, O.B.; Morris, D.S.; Schnadig, I.D.; Albany, C.; Shore, N.D.; Sieber, P.R. Phase 1b/2 Study of Enzalutamide with Samotolisib (LY3023414) or Placebo in Patients with Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 2237–2247. [Google Scholar] [CrossRef]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.; Fink, A. A first-in-human phase 1 study of LY3023414, an oral PI3K/mTOR dual inhibitor, in patients with advanced cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef] [PubMed]
- Gunder, L.C.; Moyer, T.H.; Johnson, H.R.; Auyeung, A.S.; Leverson, G.E.; Zhang, W.; Matkowskyj, K.A.; Carchman, E.H. Anal Cancer Prevention Through the Topical Use of Single or Dual PI3K/mTOR Inhibitors. J. Surg. Res. 2023, 282, 137–146. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, M.; Ventura, R.; Tai, A.; Matthews, D.J. The PI3K/mTOR inhibitor PWT33597 regresses 786-0 renal xenografts. Cancer Res. 2012, 72, 3737. [Google Scholar] [CrossRef]
- Jang, D.K.; Lee, Y.G.; Chae, Y.C.; Lee, J.K.; Paik, W.H.; Lee, S.H.; Kim, Y.-T.; Ryu, J.K. GDC-0980 (apitolisib) treatment with gemcitabine and/or cisplatin synergistically reduces cholangiocarcinoma cell growth by suppressing the PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2020, 529, 1242–1248. [Google Scholar] [CrossRef]
- Omeljaniuk, W.J.; Krętowski, R.; Ratajczak-Wrona, W.; Jabłońska, E.; Cechowska-Pasko, M. Novel dual PI3K/mTOR inhibitor, Apitolisib (GDC-0980), inhibits growth and induces apoptosis in human glioblastoma cells. Int. J. Mol. Sci. 2021, 22, 11511. [Google Scholar] [CrossRef]
- Fang, X.; Yuan, M.; Dai, J.; Lin, Q.; Lin, Y.; Wang, W.; Jiang, Y.; Wang, H.; Zhao, F.; Wu, J. Dual inhibition of glycolysis and oxidative phosphorylation by aptamer-based artificial enzyme for synergistic cancer therapy. Nano Res. 2022, 15, 6278–6287. [Google Scholar] [CrossRef]
- Dasgupta, A.; Sierra, L.; Tsang, S.V.; Kurenbekova, L.; Patel, T.; Rajapakse, K.; Shuck, R.L.; Rainusso, N.; Landesman, Y.; Unger, T. Targeting PAK4 inhibits Ras-mediated signaling and multiple oncogenic pathways in high-risk Rhabdomyosarcoma. Cancer Res. 2021, 81, 199–212. [Google Scholar] [CrossRef]
- Xiang, S.; Huang, D.; He, Q.; Li, J.; Tam, K.Y.; Zhang, S.-L.; He, Y. Development of dual inhibitors targeting pyruvate dehydrogenase kinases and human lactate dehydrogenase A: High-throughput virtual screening, synthesis and biological validation. Eur. J. Med. Chem. 2020, 203, 112579. [Google Scholar] [CrossRef]
- Holshouser, S.L. Dual Inhibitors of KDM1A and Spermine Oxidase: A Novel Approach to Antitumor Therapy. Ph.D Thesis, Medical University of South Carolina, Charleston, SC, USA, 2018. [Google Scholar]
- Dai, X.-J.; Liu, Y.; Xue, L.-P.; Xiong, X.-P.; Zhou, Y.; Zheng, Y.-C.; Liu, H.-M. Reversible lysine specific demethylase 1 (LSD1) inhibitors: A promising wrench to impair LSD1. J. Med. Chem. 2021, 64, 2466–2488. [Google Scholar] [CrossRef]
- Wu, X.; Xu, Y.; Liang, Q.; Yang, X.; Huang, J.; Wang, J.; Zhang, H.; Shi, J. Recent Advances in Dual PI3K/mTOR Inhibitors for Tumour Treatment. Front. Pharmacol. 2022, 13, 875372. [Google Scholar]
- Bharti, R.; Dey, G.; Mandal, M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef]
- Rich, J.N.; Bao, S. Chemotherapy and cancer stem cells. Cell Stem Cell 2007, 1, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H. A phase Ib study of the dual PI3K/mTOR inhibitor dactolisib (BEZ235) combined with everolimus in patients with advanced solid malignancies. Target. Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Garcia-Carbonero, R.; Libutti, S.K.; Hendifar, A.E.; Custodio, A.; Guimbaud, R.; Lombard-Bohas, C.; Ricci, S.; Klümpen, H.J.; Capdevila, J. Phase II study of BEZ235 versus everolimus in patients with mammalian target of rapamycin inhibitor-naïve advanced pancreatic neuroendocrine tumors. Oncologist 2018, 23, 766-e790. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Hamadani, M.; Hayslip, J.; Janssens, A.; Wagner-Johnston, N.; Ottmann, O.; Arnason, J.; Tilly, H.; Millenson, M.; Offner, F. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: An open-label, phase 2 trial. Lancet Haematol. 2018, 5, e170–e180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, G.; Liang, H. Dual PI3K/mTOR Inhibitor, XL765, suppresses glioblastoma growth by inducing ER stress-dependent apoptosis. OncoTargets Ther. 2019, 12, 5415. [Google Scholar] [CrossRef]
- Johnson, F.M.; Janku, F.; Gouda, M.A.; Tran, H.T.; Kawedia, J.D.; Schmitz, D.; Streefkerk, H.; Lee, J.J.; Andersen, C.R.; Deng, D. Inhibition of the Phosphatidylinositol-3 Kinase Pathway Using Bimiralisib in Loss-of-Function NOTCH1-Mutant Head and Neck Cancer. Oncologist 2022, 27, 1004-e1926. [Google Scholar] [CrossRef]
- Zhu, D.-s.; Dong, J.-y.; Xu, Y.-y.; Zhang, X.-t.; Fu, S.-b.; Liu, W. Omipalisib inhibits esophageal squamous cell carcinoma growth through inactivation of phosphoinositide 3-Kinase (PI3K)/AKT/Mammalian target of rapamycin (mTOR) and ERK signaling. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e927106. [Google Scholar] [CrossRef]
- Garlich, J.R.; De, P.; Dey, N.; Su, J.D.; Peng, X.; Miller, A.; Murali, R.; Lu, Y.; Mills, G.B.; Kundra, V. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008, 68, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Chow, Z. Synergistic Cytotoxic Effect of a Novel mTOR/PI3K Dual Inhibitor PF-04691502 and Radiation Therapy on Neuroendocrine Tumor Cells. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, e527. [Google Scholar] [CrossRef]
- Kwiatkowski, D.J.; Choueiri, T.K.; Fay, A.P.; Rini, B.I.; Thorner, A.R.; De Velasco, G.; Tyburczy, M.E.; Hamieh, L.; Albiges, L.; Agarwal, N. Mutations in TSC1, TSC2, and MTOR Are Associated with Response to Rapalogs in Patients with Metastatic Renal Cell CarcinomamTOR Pathway Mutations and Response to Rapalogs in RCC. Clin. Cancer Res. 2016, 22, 2445–2452. [Google Scholar] [CrossRef]
- Powles, T.; Lackner, M.R.; Oudard, S.; Escudier, B.; Ralph, C.; Brown, J.E.; Hawkins, R.E.; Castellano, D.; Rini, B.I.; Staehler, M.D. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2016, 34, 1660. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Heske, C.M. Beyond energy metabolism: Exploiting the additional roles of NAMPT for cancer therapy. Front. Oncol. 2020, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Abu Aboud, O.; Chen, C.-H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R.H. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer GrowthInhibition of PAK4 and NAMPT Decreases RCC Growth. Mol. Cancer Ther. 2016, 15, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kirtane, A.R.; Kiyokawa, J.; Nagashima, H.; Lopes, A.; Tirmizi, Z.A.; Lee, C.K.; Traverso, G.; Cahill, D.P.; Wakimoto, H. Local targeting of NAD+ salvage pathway alters the immune tumor microenvironment and enhances checkpoint immunotherapy in glioblastoma. Cancer Res. 2020, 80, 5024–5034. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Skok, Z.i.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J. Med. Chem. 2019, 63, 884–904. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Wu, T.; Yin, W.; Yan, J.; Sun, Y.; Zhao, D. Therapeutic potential of targeting LSD1/KDM1A in cancers. Pharmacol. Res. 2022, 175, 105958. [Google Scholar] [CrossRef]
- Furbish, A.B.; Alford, A.S.; Burger, P.; Peterson, Y.K.; Murray-Stewart, T.; Casero, R.A., Jr.; Woster, P.M. Identification and Characterization of Novel Small-Molecule SMOX Inhibitors. Med. Sci. 2022, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Sun, D.; Zhang, J.; Xue, R.; Janssen, H.L.; Tang, W.; Dong, L. Spermine oxidase is upregulated and promotes tumor growth in hepatocellular carcinoma. Hepatol. Res. 2018, 48, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Arooj, M.; Sakkiah, S.; Cao, G.P.; Lee, K.W. An innovative strategy for dual inhibitor design and its application in dual inhibition of human thymidylate synthase and dihydrofolate reductase enzymes. PLoS ONE 2013, 8, e60470. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Isaacsson Velho, P.H.; Castro, G., Jr.; Chung, C.H. Targeting the PI3K pathway in head and neck squamous cell carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, 123–128. [Google Scholar] [CrossRef]
- Jung, K.; Kang, H.; Mehra, R. Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC). Cancers Head Neck 2018, 3, 3. [Google Scholar] [CrossRef]
- Langdon, S.P.; Kay, C.; Um, I.H.; Dodds, M.; Muir, M.; Sellar, G.; Kan, J.; Gourley, C.; Harrison, D.J. Evaluation of the dual mTOR/PI3K inhibitors Gedatolisib (PF-05212384) and PF-04691502 against ovarian cancer xenograft models. Sci. Rep. 2019, 9, 18742. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.-S. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2–negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef]
- Park, Y.L.; Kim, H.P.; Cho, Y.W.; Min, D.W.; Cheon, S.K.; Lim, Y.J.; Song, S.H.; Kim, S.J.; Han, S.W.; Park, K.J. Activation of WNT/β-catenin signaling results in resistance to a dual PI3K/mTOR inhibitor in colorectal cancer cells harboring PIK3CA mutations. Int. J. Cancer 2019, 144, 389–401. [Google Scholar] [CrossRef]
- Markman, B.; Tao, J.J.; Scaltriti, M. PI3K pathway inhibitors: Better not left alone. Curr. Pharm. Des. 2013, 19, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Tanaka, K.; Ikegami, S.; Villa, G.R.; Yang, H.; Yong, W.H.; Cloughesy, T.F.; Yamagata, K.; Arai, N.; Cavenee, W.K. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc. Natl. Acad. Sci. USA 2015, 112, 9406–9411. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Aguiar, P.; Campistol, J.M.; Diekmann, F. Safety of mTOR inhibitors in adult solid organ transplantation. Expert Opin. Drug Saf. 2016, 15, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Tiwari, A.K.; Shukla, S.; Robey, R.W.; Singh, S.; Kim, I.-W.; Bates, S.E.; Peng, X.; Abraham, I.; Ambudkar, S.V. Sildenafil reverses ABCB1-and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011, 71, 3029–3041. [Google Scholar] [CrossRef]
- Wu, C.-P.; Hung, C.-Y.; Lusvarghi, S.; Huang, Y.-H.; Tseng, P.-J.; Hung, T.-H.; Yu, J.-S.; Ambudkar, S.V. Overexpression of ABCB1 and ABCG2 contributes to reduced efficacy of the PI3K/mTOR inhibitor samotolisib (LY3023414) in cancer cell lines. Biochem. Pharmacol. 2020, 180, 114137. [Google Scholar] [CrossRef]
- Kovarik, J.M.; Beyer, D.; Schmouder, R.L. Everolimus drug interactions: Application of a classification system for clinical decision making. Biopharm. Drug Dispos. 2006, 27, 421–426. [Google Scholar] [CrossRef]
- Moorthy, G. Clinical Pharmacokinetics of the Novel Combination of BEZ235, PI3K/mTOR Inhibitor, and Everolimus, mTOR Inhibitor: Phase I Clinical Studies and Non-Clinical Mechanistic Assessment; University of Cincinnati: Cincinnati, OH, USA, 2015. [Google Scholar]
- Zhao, X.; Chen, X.; Shen, X.; Tang, P.; Chen, C.; Zhu, Q.; Li, M.; Xia, R.; Yang, X.; Feng, C. IL-36β promotes CD8+ T cell activation and antitumor immune responses by activating mTORC1. Front. Immunol. 2019, 10, 1803. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Heo, J.H.; Park, J.Y.; Jeong, J.Y.; Cho, H.J.; Park, K.S.; Kim, S.H.; Moon, Y.W.; Kim, J.S.; An, H.J. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol. Oncol. 2019, 153, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Yang, J.; Saleh, N.; Chen, S.-C.; Ayers, G.D.; Abramson, V.G.; Mayer, I.A.; Richmond, A. Inhibition of the PI3K/mTOR pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer. Int. J. Mol. Sci. 2021, 22, 5207. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target | Mechanism and Outcomes | Adverse Effects | Ref |
|---|---|---|---|---|
| Dactolisib (BEZ235) | PI3K/mTOR |
|
| [101,102,103,104,105] |
| Gedatolisib (PKI-587) | PI3K/mTOR |
|
| [106,107,108,109] |
| Voxtalisib (SAR245409) | Class-I PI3Ks, mTORC1/ mTORC2 |
|
| [110,111] |
| Bimiralisib (PQR309) | Pan-class I PI3K/mTOR |
|
| [112,113,114] |
| Paxalisib (GDC-0084) | PI3K/mTOR |
|
| [115,116,117,118,119,120] |
| Omipalisib (GSK2126458) | PI3K/mTOR |
| - | [121,122,123] |
| SF1126 | PI3K/mTOR BRD4 |
| - | [124,125,126] |
| PF-04691502 | PI3K/mTOR |
| - | [127,128,129,130] |
| Samotolisib (LY3023414) | PI3K/mTOR |
|
| [131,132,133,134] |
| PWT33597 | PI3K/mTORC1 and mTORC2 |
| - | [135] |
| Apitolisib (GDC-0980) | PI3K/mTOR |
|
| [136,137] |
| AptCCN | Glycolysis & oxidative phosphorylation |
| - | [138] |
| KPT-9274 | NAMPT/p21 PAK4 |
| - | [139] |
| 20e and 20k | PDK/LDHA |
| - | [140] |
| 3,5-diamino-1,2,4-triazole analogs | KDM1A/SMOX |
| - | [141,142] |
| Name | Molecular Formula | 2D Structure |
|---|---|---|
| Dactolisib | C30H23N5O |  |
| Gedatolisib | C32H41N9O4 |  |
| Voxtalisib | C13H14N6O |  |
| Bimiralisib | C17H20F3N7O2 |  |
| Paxalisib | C18H22N8O2 |  |
| Omipalisib | C25H17F2N5O3S |  |
| SF1126 | C39H48N8O14 |  |
| PF-04691502 | C22H27N5O4 |  |
| Samotolisib | C22H27N5O4 |  |
| Apitolisib | C23H30N8O3S |  |
| KPT-9274 | C35H29F3N43 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Lan, H.; Yao, S.; Jin, K.; Chen, Y. Metabolic Interventions in Tumor Immunity: Focus on Dual Pathway Inhibitors. Cancers 2023, 15, 2043. https://doi.org/10.3390/cancers15072043
Chen M, Lan H, Yao S, Jin K, Chen Y. Metabolic Interventions in Tumor Immunity: Focus on Dual Pathway Inhibitors. Cancers. 2023; 15(7):2043. https://doi.org/10.3390/cancers15072043
Chicago/Turabian StyleChen, Min, Huanrong Lan, Shiya Yao, Ketao Jin, and Yun Chen. 2023. "Metabolic Interventions in Tumor Immunity: Focus on Dual Pathway Inhibitors" Cancers 15, no. 7: 2043. https://doi.org/10.3390/cancers15072043
APA StyleChen, M., Lan, H., Yao, S., Jin, K., & Chen, Y. (2023). Metabolic Interventions in Tumor Immunity: Focus on Dual Pathway Inhibitors. Cancers, 15(7), 2043. https://doi.org/10.3390/cancers15072043