
A Biterm Topic Model for Sparse Mutation Data

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
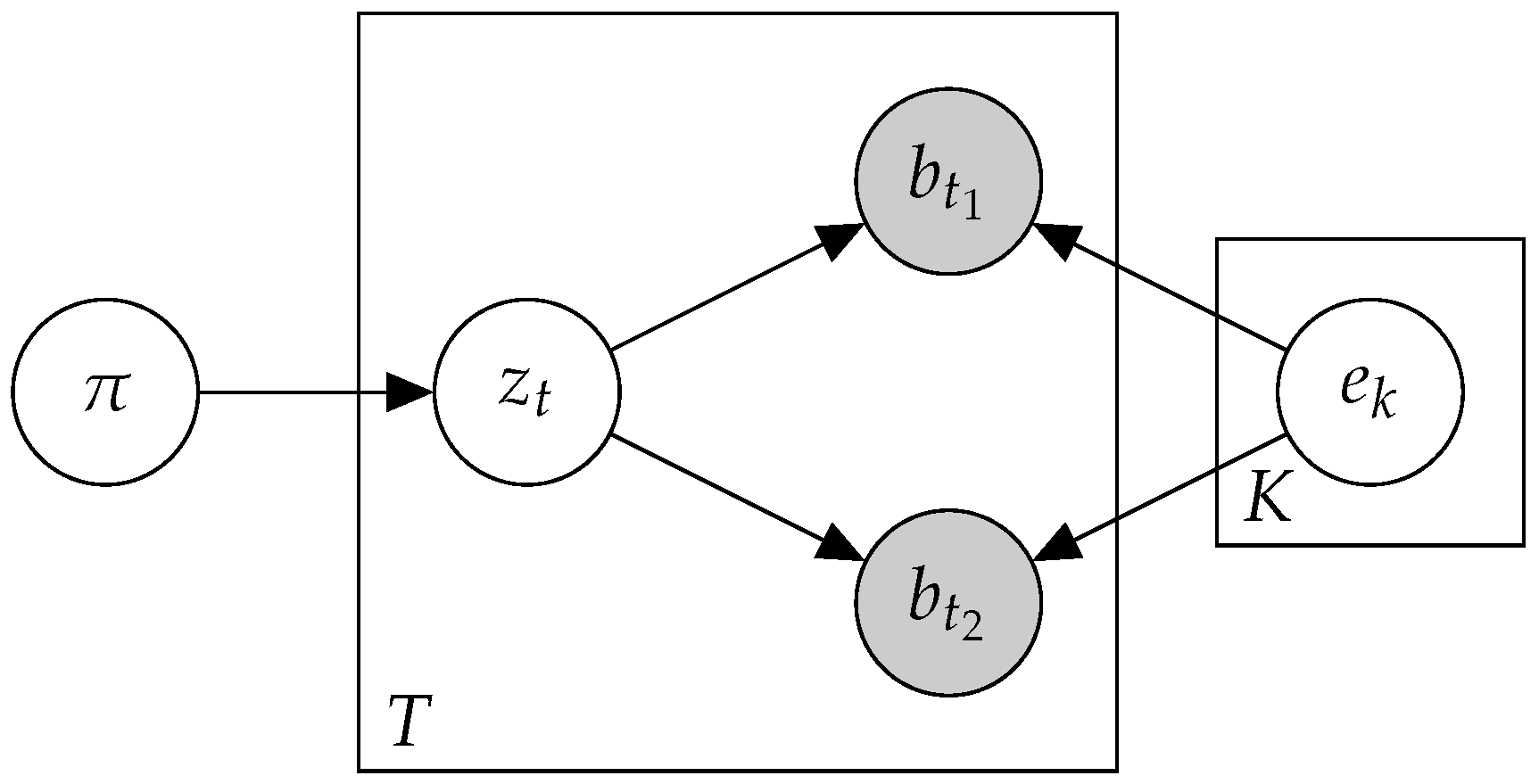
2.1. Preliminaries
2.2. Btm: A Biterm Topic Model
- E-step: Compute for every :
- M-step: Compute for every :
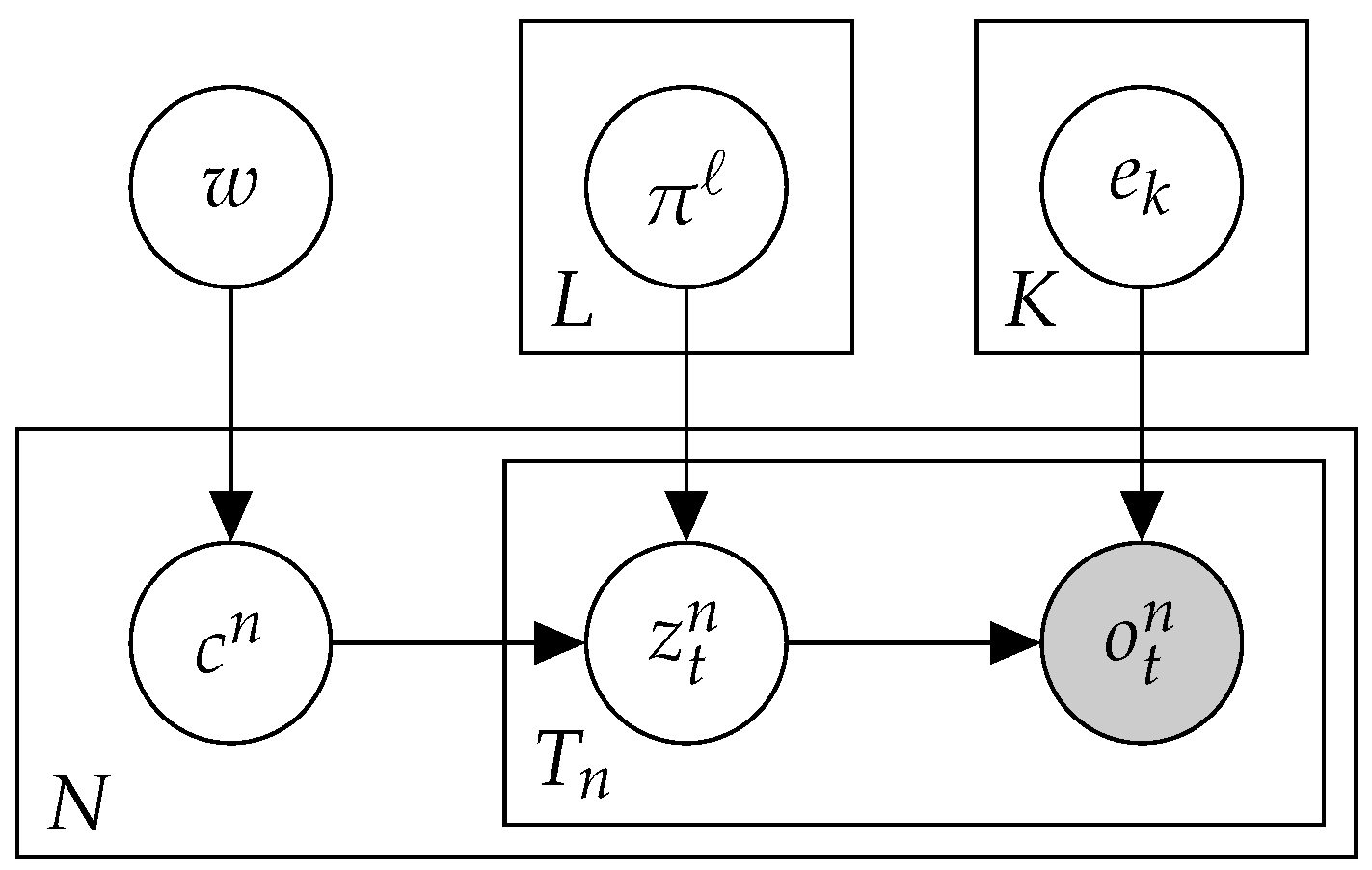
2.3. Mix: A Mixture of MMMs
2.4. Btm2K-Learning the Number of Signatures in a Dataset Using Btm
| Algorithm 1. |
|
2.5. Previous Hyper-Parameter Selection Algorithms
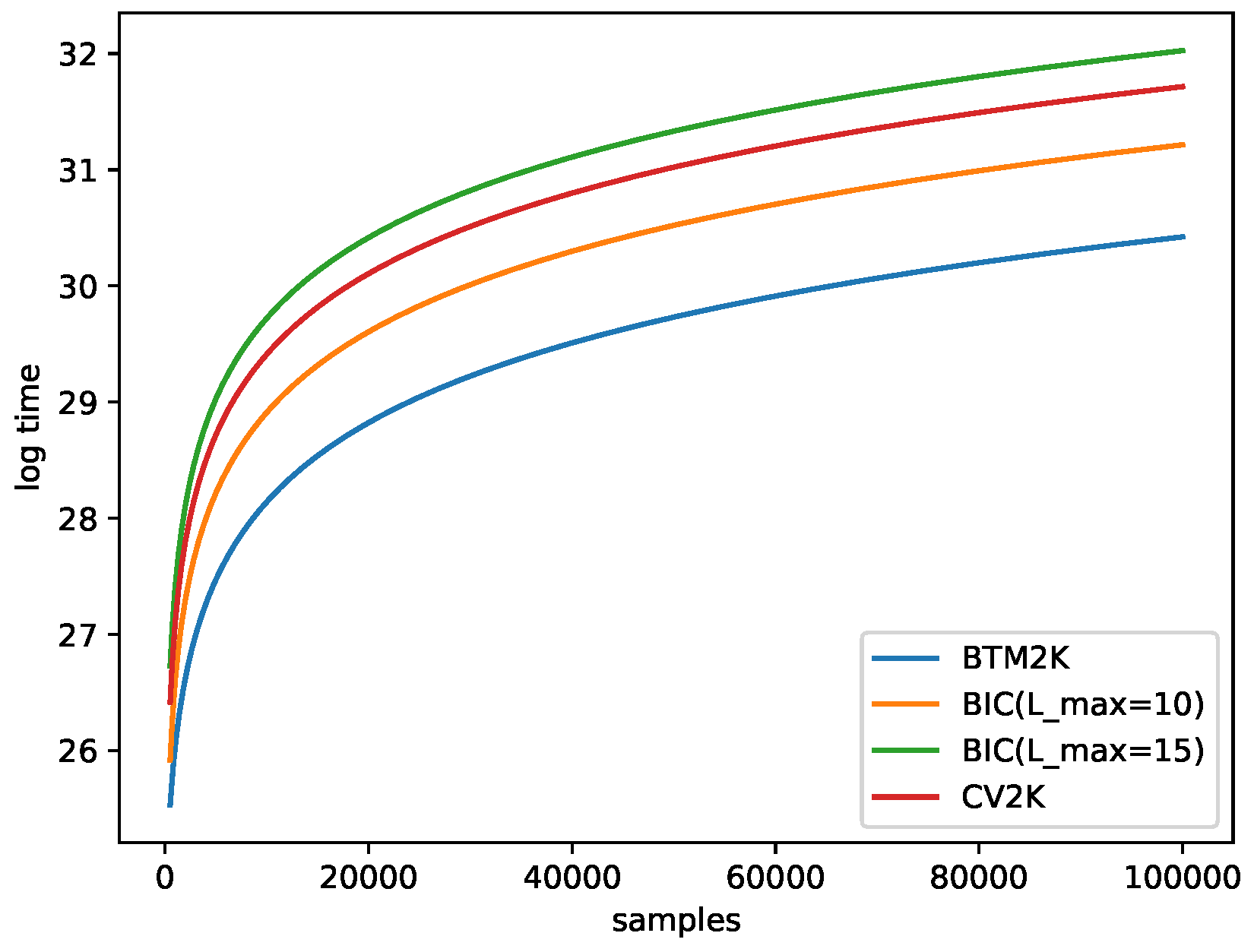
2.6. Running-Time Estimation
- Btm2K: For a given k, we needed to train Btm times (T repetitions of 2 folds). To train Btm, we needed to create biterms with time and training time. In total the cost for k was . Note that we created biterms one time for all k in each run, so in total, the run time was .
- BIC: For a given k, we considered all possible . For a given pair, we trained Mix once for a cost of . In total, for all Ls, we needed . Overall, was needed.
- CV2K: For a given k, we needed to train T times, and each iterations cost time, for a total of time. In total, for all k, we spent .
2.7. Data
- MSK-IMPACT [20,22] Pan-Cancer. We downloaded mutations for a cohort of patients from Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), which was targeted sequencing data from https://www.cbioportal.org/ (accesed on 1 January 2022). The MSK-IMPACT dataset contained 11,369 pan-cancer patients’ sequencing samples across 410 target genes. We restricted our analysis to 18 cancer types with more than 100 samples, which resulted in a dataset of 5931 samples and about 7 mutations per sample.
- Whole genome/exome (WGS/WXS) data. We combined mutations from different sources and cancer types of whole-genome-sequencing and whole-exome-sequencing (WGS/WXS): ovarian cancer (OV), chronic lymphocytic leukemia (CLL), malignant lymphoma (MALY), and colon adenocarcinoma (COAD). We downloaded the OV samples from the Cancer Genome Atlas [29]. For CLL and MALY, we used ICGC release 27, analyzed the sample with the most mutations per patient, and restricted those to samples annotated as “study = PCAWG” [24]. For evaluation purposes, we down-sampled the data to target regions of MSK-IMPACT [20,22]. The data characteristics are summarized in Table 2.
- Simulated data. The simulated data were generated and described in detail in [16] to evaluate SigProfiler (SP) and SignatureAnalyzer (SA). Here, for each of the 12 datasets, we evaluated our method on two sets of realistic synthetic data: SP-realistic, based on SP’s reference signatures and attributes, and SA-realistic, based on SA’s reference signatures and attributes. For each of the (i)–(x) tests, the synthetic datasets were generated based on observed statistics for each signature of each cancer type. Different datasets could differ by the number of signatures, the number of active signatures per samples (sparsity), the number of mutations per sample (whole exome/genome sequencing), whether they reflected a single cancer type or multiple types, and the similarity between signatures. All these factors affected the difficulty of determining the number of components. For each simulated sample, we sampled an MSK-IMPACT patient and down-sampled the simulated sample, so it had the same number of mutations. We removed datasets with missing mutation categories.
2.8. Implementation Details
3. Results
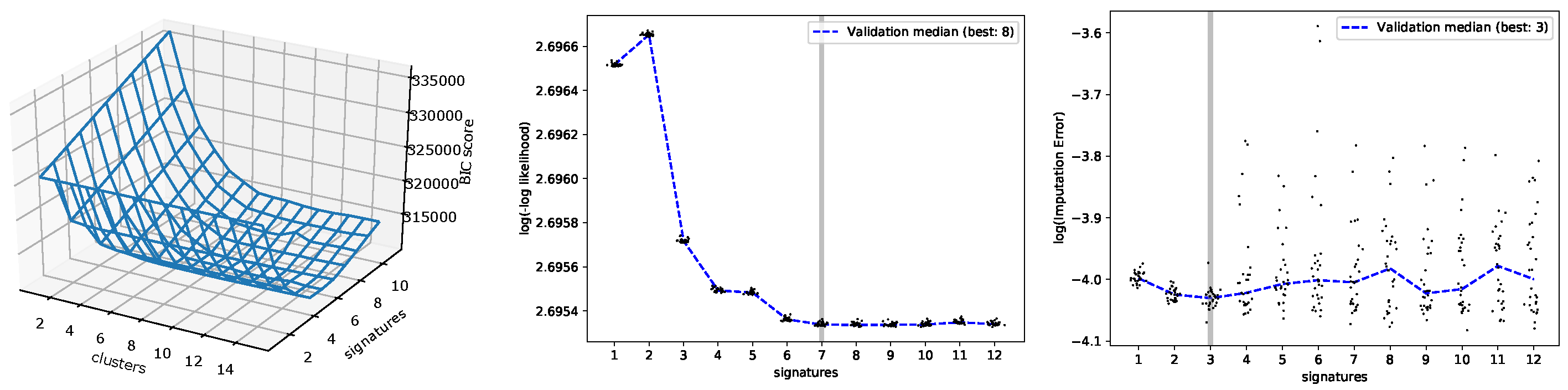
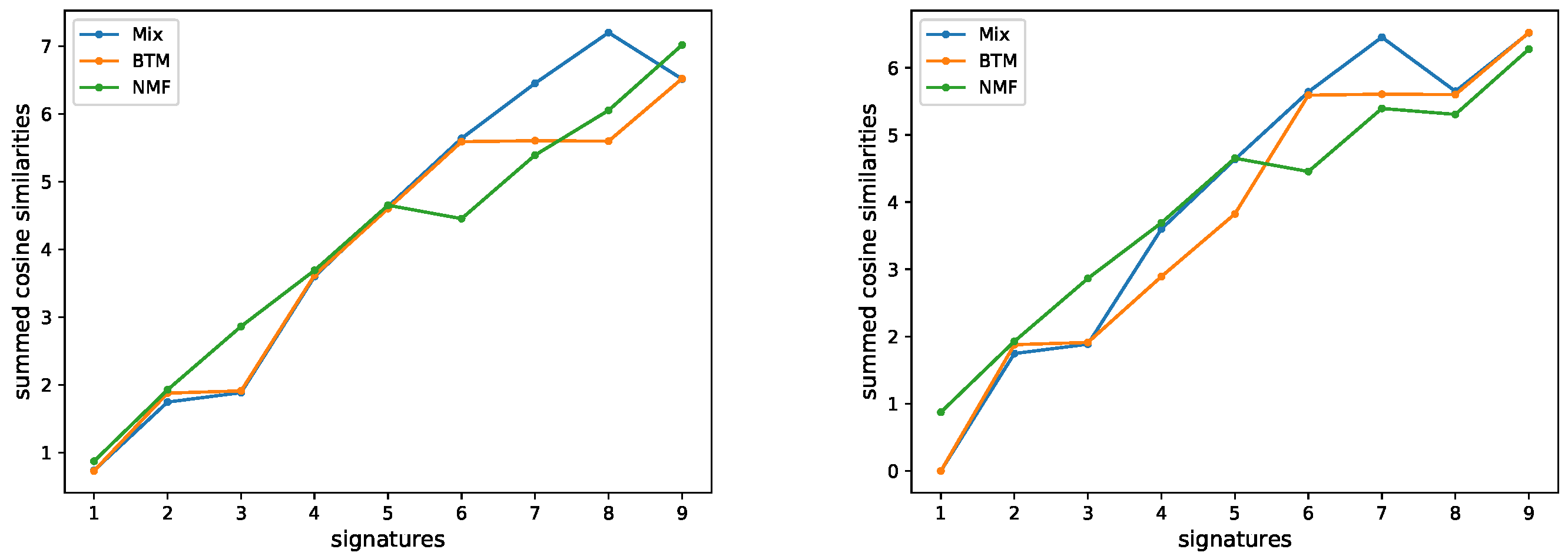
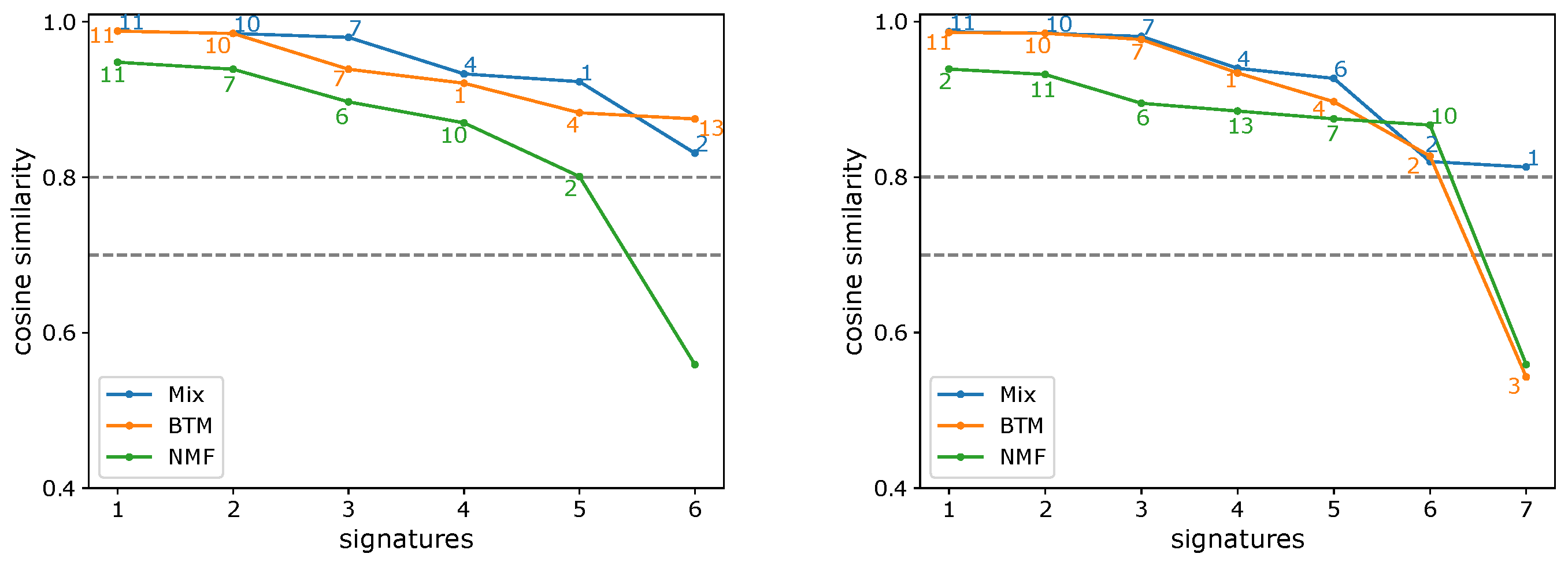
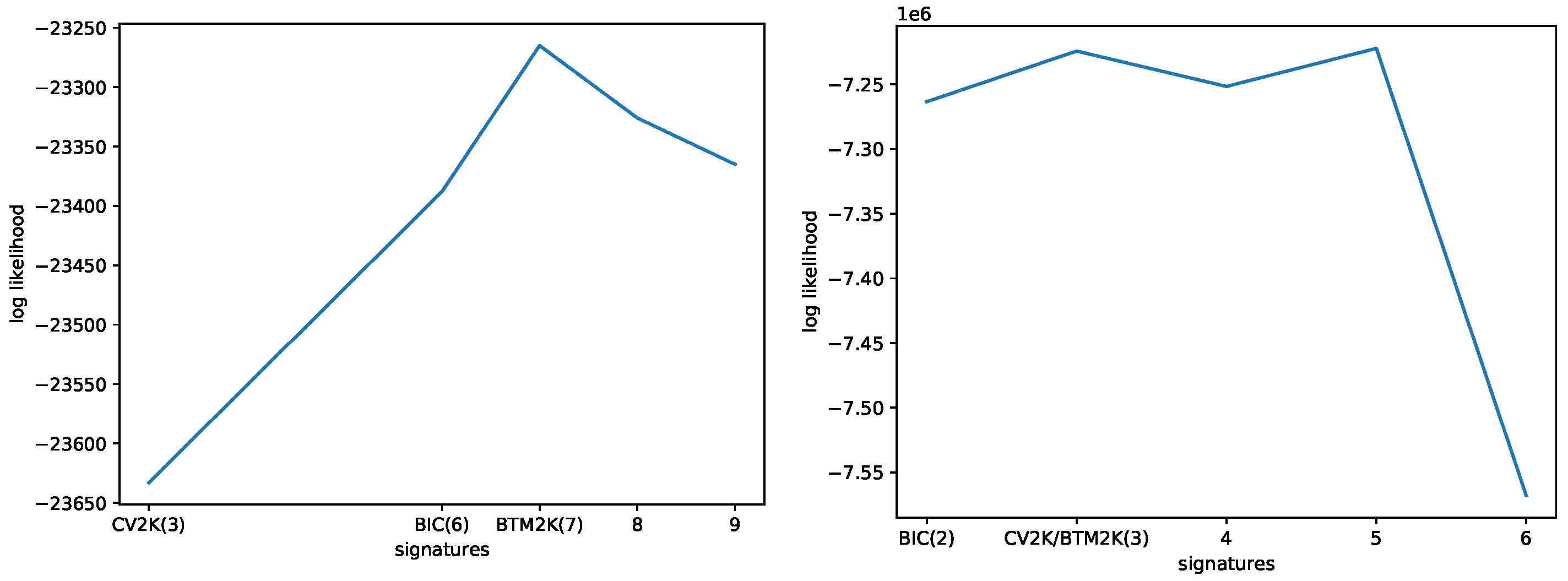
3.1. Evaluating the Number of Signatures from Simulated Data
3.2. Evaluating the Number of Signatures from MSK-IMPACT Data
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Hoeck, A.; Tjoonk, N.H.; van Boxtel, R.; Cuppen, E. Portrait of a cancer: Mutational signature analyses for cancer diagnostics. BMC Cancer 2019, 19, 457. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Covington, K.; Shinbrot, E.; Wheeler, D.A. Mutation signatures reveal biological processes in human cancer. bioRxiv 2016, 036541. [Google Scholar] [CrossRef]
- Fischer, A.; Illingworth, C.J.; Campbell, P.J.; Mustonen, V. EMu: Probabilistic inference of mutational processes and their localization in the cancer genome. Genome Biol. 2013, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Tiao, G.; Kwiatkowski, D.J.; Rosenberg, J.E.; Allen, E.M.V.; D’Andrea, A.D.; et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Rosales, R.A.; Drummond, R.D.; Valieris, R.; Dias-Neto, E.; da Silva, I.T. signeR: An empirical Bayesian approach to mutational signature discovery. Bioinformatics 2016, 33, 8–16. [Google Scholar] [CrossRef]
- Huang, X.; Wojtowicz, D.; Przytycka, T.M. Detecting presence of mutational signatures in cancer with confidence. Bioinformatics 2017, 34, 330–337. [Google Scholar] [CrossRef]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. deconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Funnell, T.; Zhang, A.; Shiah, Y.J.; Grewal, D.; Lesurf, R.; McKinney, S.; Bashashati, A.; Wang, Y.K.; Boutros, P.C.; Shah, S.P. Integrated single-nucleotide and structural variation signatures of DNA-repair deficient human cancers. bioRxiv 2018, 267500. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Tremmel, G.; Miyano, S.; Stephens, M. A Simple Model-Based Approach to Inferring and Visualizing Cancer Mutation Signatures. PLoS Genet. 2015, 11, e1005657. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, D.; Sason, I.; Huang, X.; Kim, Y.A.; Leiserson, M.D.M.; Przytycka, T.M.; Sharan, R. Hidden Markov models lead to higher resolution maps of mutation signature activity in cancer. Genome Med. 2019, 11, 49. [Google Scholar] [CrossRef]
- Robinson, W.; Sharan, R.; Leiserson, M.D. Modeling clinical and molecular covariates of mutational process activity in cancer. Bioinformatics 2019, 35, i492–i500. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.; Ng, A.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Trucco, L.D.; Mundra, P.A.; Hogan, K.; Garcia-Martinez, P.; Viros, A.; Mandal, A.K.; Macagno, N.; Gaudy-Marqueste, C.; Allan, D.; Baenke, F.; et al. Ultraviolet radiation–induced DNA damage is prognostic for outcome in melanoma. Nat. Med. 2018, 25, 221–224. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.K.; Melloni, G.E.; Cortés-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Memari, Y.; Davies, H.R. Holistic cancer genome profiling for every patient. Swiss Med. Wkly. 2020, 150, w20158. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056.e10. [Google Scholar] [CrossRef]
- Sason, I.; Chen, Y.; Leiserson, M.D.; Sharan, R. A mixture model for signature discovery from sparse mutation data. Genome Med. 2021, 13, 173. [Google Scholar] [CrossRef]
- Kokalitcheva, K. A year after tweets doubled in size, brevity still rules. Axios 2018. [Google Scholar]
- Yan, X.; Guo, J.; Lan, Y.; Cheng, X. A biterm topic model for short texts. In Proceedings of the 22nd International Conference on World Wide Web, Rio de Janeiro, Brazil, 13–17 May 2013; pp. 1445–1456. [Google Scholar]
- Gilad, G.; Sason, I.; Sharan, R. An automated approach for determining the number of components in non-negative matrix factorization with application to mutational signature learning. Mach. Learn Sci. Technol. 2020, 2, 015013. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, T. Guide to NumPy; Trelgol Publishing: New York, NY, USA, 2006. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | ∼Learning Number of Signatures Complexity | ∼Learning Number of Clusters Complexity (BIC) |
|---|---|---|
| BIC | ||
| Btm2K | ||
| CV2K | ||
| Cancer | #Samples | #Mutations | #Panel Mutations |
|---|---|---|---|
| OV | 411 | 46,299 | 1812 |
| Maly | 100 | 1,220,526 | 1770 |
| CLL | 100 | 270,870 | 278 |
| COAD | 44 | 52,827 | 1789 |
| Combined | 653 | 1,590,520 | 5604 |
| Data Set | BIC | Btm2K | CV2K | # Signatures with >=5% Down-sampled Mutations |
|---|---|---|---|---|
| ii-sa | 3 | 4->4 | 4->2 | 8 |
| ii-sp | 3 | 10->7 | 4->2 | 6 |
| v-sa | 2 | 3->3 | 3->2 | 6 |
| v-sp | 2 | 3->2 | 6->2 | 5 |
| vii.a(pri.)-sp | 1 | 2->2 | 3->1 | 2 |
| vii.b(sec.)-sa | 1 | 1->1 | 5->2 | 3 |
| viii-sp | 1 | 2->2 | 5->1 | 7 |
| ix-sa | 2 | 4->4 | 4->2 | 8 |
| ix-sp | 4 | 6->6 | 4->3 | 6 |
| x-sa | 1 | 3->3 | 5->1 | 8 |
| x-sp | 1 | 6->6 | 5->4 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sason, I.; Chen, Y.; Leiserson, M.D.M.; Sharan, R. A Biterm Topic Model for Sparse Mutation Data. Cancers 2023, 15, 1601. https://doi.org/10.3390/cancers15051601
Sason I, Chen Y, Leiserson MDM, Sharan R. A Biterm Topic Model for Sparse Mutation Data. Cancers. 2023; 15(5):1601. https://doi.org/10.3390/cancers15051601
Chicago/Turabian StyleSason, Itay, Yuexi Chen, Mark D. M. Leiserson, and Roded Sharan. 2023. "A Biterm Topic Model for Sparse Mutation Data" Cancers 15, no. 5: 1601. https://doi.org/10.3390/cancers15051601
APA StyleSason, I., Chen, Y., Leiserson, M. D. M., & Sharan, R. (2023). A Biterm Topic Model for Sparse Mutation Data. Cancers, 15(5), 1601. https://doi.org/10.3390/cancers15051601