
Somatic, Genetic and Epigenetic Changes in Nephrogenic Rests and Their Role in the Transformation to Wilms Tumors, a Systematic Review


 and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy and Eligibility Criteria
2.2. Quality Assessment
2.3. Data Extraction
3. Results
3.1. Search Strategy and Eligibility Criteria
3.2. Quality Assessment
3.3. Characteristics of the Included Studies
3.4. Chromosomal Changes
3.5. Structural Aberrations in Candidate Gene Studies
3.6. Epigenetic Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hennigar, R.A.; O’Shea, P.A.; Grattan-Smith, J.D. Clinicopathologic Features of Nephrogenic Rests and Nephroblastomatosis. Adv. Anat. Pathol. 2001, 8, 276–289. [Google Scholar] [CrossRef]
- Popov, S.D.; Sebire, N.J.; Vujanic, G.M. Wilms’ Tumour—Histology and Differential Diagnosis. In Wilms Tumor; Codon Publications: Singapore, 2016. [Google Scholar]
- Beckwith, J.B.; Kiviat, N.B.; Bonadio, J.F. Nephrogenic Rests, Nephroblastomatosis, and the Pathogenesis of Wilms’ Tumor. Pediatr. Pathol. 1990, 10, 1–36. [Google Scholar] [CrossRef]
- Lee Tsün, H.; Holman, R.L. Bilateral Nephroblastomatosis in a Premature Infant. J. Pathol. Bacteriol. 1961, 82, 249–255. [Google Scholar] [CrossRef]
- Beckwith, J.B. Nephrogenic Rests and the Pathogenesis of Wilms Tumor: Developmental and Clinical Considerations. Am. J. Med. Genet. 1998, 79, 268–273. [Google Scholar] [CrossRef]
- Fukuzawa, R.; Reeve, A.E. Molecular Pathology and Epidemiology of Nephrogenic Rests and Wilms Tumors. J. Pediatr. Hematol. Oncol. 2007, 29, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Caiulo, V.A.; Latini, G.; Cataldi, L.; De Felice, C. Nephrogenic Rests. J. Pediatr. Hematol. Oncol. 2007, 29, 361–363. [Google Scholar] [CrossRef]
- Fukuzawa, R.; Breslow, N.E.; Morison, I.M.; Dwyer, P.; Kusafuka, T.; Kobayashi, Y.; Becroft, D.M.; Beckwith, J.B.; Perlman, E.J.; Reeve, A.E. Epigenetic Differences between Wilms’ Tumours in White and East-Asian Children. Lancet 2004, 363, 446–451. [Google Scholar] [CrossRef]
- Bozlu, G.; Cıtak, E.C. Evaluation of Renal Tumors in Children. Türk. Urol. Derg. Turk. J. Urol. 2018, 44, 268. [Google Scholar] [CrossRef]
- Vujanić, G.M.; Gessler, M.; Ooms, A.H.A.G.; Collini, P.; Coulomb-l’Hermine, A.; D’Hooghe, E.; de Krijger, R.R.; Perotti, D.; Pritchard-Jones, K.; Vokuhl, C.; et al. The UMBRELLA SIOP–RTSG 2016 Wilms Tumour Pathology and Molecular Biology Protocol. Nat. Rev. Urol. 2018, 15, 693–701. [Google Scholar] [CrossRef]
- SIOP-RTSG. Umbrella Protocol 2016 Version 1.8; SIOP-RTSG: Saarbrucken, Germany, 2017. [Google Scholar]
- Dzhuma, K.; Ducou Le Pointe, H.; Coulomb, A.; Tabone, M.; Bergeron, C.; Audry, G.; Irtan, S. Wilms Tumors and Their Precursors: Radiological Diagnosis versus Histology. Pediatr. Blood Cancer 2020, 67, e2841. [Google Scholar] [CrossRef]
- Sandberg, J.K.; Chi, Y.-Y.; Smith, E.A.; Servaes, S.; Hoffer, F.A.; Mullen, E.A.; Perlman, E.J.; Tornwall, B.; Ehrlich, P.F.; Geller, J.I.; et al. Imaging Characteristics of Nephrogenic Rests Versus Small Wilms Tumors: A Report From the Children’s Oncology Group Study AREN03B2. Am. J. Roentgenol. 2020, 214, 987–994. [Google Scholar] [CrossRef]
- Vujanić, G.M.; Apps, J.R.; Moroz, V.; Ceroni, F.; Williams, R.D.; Sebire, N.J.; Pritchard-Jones, K. Nephrogenic Rests in Wilms Tumors Treated with Preoperative Chemotherapy: The UK SIOP Wilms Tumor 2001 Trial Experience. Pediatr. Blood Cancer 2017, 64, e26547. [Google Scholar] [CrossRef]
- Liu, E.K.; Suson, K.D. Syndromic Wilms Tumor: A Review of Predisposing Conditions, Surveillance and Treatment. Transl. Androl. Urol. 2020, 9, 2370–2381. [Google Scholar] [CrossRef]
- Kalapurakal, J.A.; Dome, J.S.; Perlman, E.J.; Malogolowkin, M.; Haase, G.M.; Grundy, P.; Coppes, M.J. Management of Wilms’ Tumour: Current Practice and Future Goals. Lancet Oncol. 2004, 5, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussain, T.; Ali, A.; Akhtar, M. Wilms Tumor. Adv. Anat. Pathol. 2014, 21, 166–173. [Google Scholar] [CrossRef]
- Treger, T.D.; Chowdhury, T.; Pritchard-Jones, K.; Behjati, S. The Genetic Changes of Wilms Tumour. Nat. Rev. Nephrol. 2019, 15, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Bossuyt, P.M.; Reitsma, J.B.; Bruns, D.E.; Gatsonis, C.A.; Glasziou, P.P.; Irwig, L.; Lijmer, J.G.; Moher, D.; Rennie, D.; de Vet, H.C.W.; et al. STARD 2015: An Updated List of Essential Items for Reporting Diagnostic Accuracy Studies. Clin. Chem. 2015, 61, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.S.; Barber, M.S.; Kienle, G.S.; Aronson, J.K.; von Schoen-Angerer, T.; Tugwell, P.; Kiene, H.; Helfand, M.; Altman, D.G.; Sox, H.; et al. CARE Guidelines for Case Reports: Explanation and Elaboration Document. J. Clin. Epidemiol. 2017, 89, 218–235. [Google Scholar] [CrossRef]
- Howick, J.; Chalmers, I.G.P.; Glasziou, P.; Greenhalgh, T.; Heneghan, C.; Liberati, A.; Moschetti, I.; Phillips, B.; Thornton, H.; Goddard, O.; et al. The Oxford Levels of Evidence 2; Oxford Centar Evidence-Based Medicine: Oxford, UK, 2011. [Google Scholar]
- Chang, C.A.; Perrier, R.; Kurek, K.C.; Estrada-Veras, J.; Lehman, A.; Yip, S.; Hendson, G.; Diamond, C.; Pinchot, J.W.; Tran, J.M.; et al. Novel Findings and Expansion of Phenotype in a Mosaic RASopathy Caused by Somatic KRAS Variants. Am. J. Med. Genet. Part A 2021, 185, 2829–2845. [Google Scholar] [CrossRef]
- Slack, J.C.; Bründler, M.-A.; Chang, C.A.; Perrier, R.; Lafay-Cousin, L.; Kurek, K.C. Bilateral Nephroblastic Tumors and a Complex Renal Vascular Anomaly in a Patient With a Mosaic RASopathy: Novel Histopathologic Features and Molecular Insights. Pediatr. Dev. Pathol. 2021, 24, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Coorens, T.H.H.; Treger, T.D.; Al-Saadi, R.; Moore, L.; Tran, M.G.B.; Mitchell, T.J.; Tugnait, S.; Thevanesan, C.; Young, M.D.; Oliver, T.R.W.; et al. Embryonal Precursors of Wilms Tumor. Science 2019, 366, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Wegert, J.; Vokuhl, C.; Collord, G.; Del Castillo Velasco-Herrera, M.; Farndon, S.J.; Guzzo, C.; Jorgensen, M.; Anderson, J.; Slater, O.; Duncan, C.; et al. Recurrent Intragenic Rearrangements of EGFR and BRAF in Soft Tissue Tumors of Infants. Nat. Commun. 2018, 9, 2378. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.; Williams, R.D.; Sebire, N.J.; Popov, S.; Vujanic, G.; Chagtai, T.; Alcaide-German, M.; Morris, T.; Butcher, L.M.; Guilhamon, P.; et al. Comparative Methylome Analysis Identifies New Tumour Subtypes and Biomarkers for Transformation of Nephrogenic Rests into Wilms Tumour. Genome Med. 2015, 7, 11. [Google Scholar] [CrossRef]
- MdZIN, R.; Phillips, M.; Edwards, C.; Murch, A.; Charles, A. Perilobar Nephrogenic Rests and Chromosome 22. Pediatr. Dev. Pathol. 2011, 14, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, R.; Holman, S.K.; Chow, C.W.; Savarirayan, R.; Reeve, A.E.; Robertson, S.P. WTX Mutations Can Occur Both Early and Late in the Pathogenesis of Wilms Tumour. J. Med. Genet. 2010, 47, 791–794. [Google Scholar] [CrossRef]
- Grill, C.; Guelly, C.; Ebner, B.; Leuschner, I.; Hauser-Kronberger, C.; Hoefler, G.; Guertl, B. Loss of PTEN/MMAC1 Activity Is a Rare and Late Event in the Pathogenesis of Nephroblastomas. Hum. Pathol. 2010, 41, 1172–1177. [Google Scholar] [CrossRef]
- Vuononvirta, R.; Sebire, N.J.; Dallosso, A.R.; Reis-Filho, J.S.; Williams, R.D.; Mackay, A.; Fenwick, K.; Grigoriadis, A.; Ashworth, A.; Pritchard-Jones, K.; et al. Perilobar Nephrogenic Rests Are Nonobligate Molecular Genetic Precursor Lesions of Insulin-Like Growth Factor-II-Associated Wilms Tumors. Clin. Cancer Res. 2008, 14, 7635–7644. [Google Scholar] [CrossRef]
- Brown, K.W.; Power, F.; Moore, B.; Charles, A.K.; Malik, K.T.A. Frequency and Timing of Loss of Imprinting at 11p13 and 11p15 in Wilms’ Tumor Development. Mol. Cancer Res. 2008, 6, 1114–1123. [Google Scholar] [CrossRef]
- Chilukamarri, L.; Hancock, A.L.; Malik, S.; Zabkiewicz, J.; Baker, J.A.; Greenhough, A.; Dallosso, A.R.; Huang, T.H.-M.; Royer-Pokora, B.; Brown, K.W.; et al. Hypomethylation and Aberrant Expression of the Glioma Pathogenesis-Related 1 Gene in Wilms Tumors. Neoplasia 2007, 9, 970–978. [Google Scholar] [CrossRef]
- Hancock, A.L.; Brown, K.W.; Moorwood, K.; Moon, H.; Holmgren, C.; Mardikar, S.H.; Dallosso, A.R.; Klenova, E.; Loukinov, D.; Ohlsson, R.; et al. A CTCF-Binding Silencer Regulates the Imprinted Genes AWT1 and WT1-AS and Exhibits Sequential Epigenetic Defects during Wilms’ Tumourigenesis. Hum. Mol. Genet. 2007, 16, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, R.; Heathcott, R.W.; More, H.E.; Reeve, A.E. Sequential WT1 and CTNNB1 Mutations and Alterations of -Catenin Localisation in Intralobar Nephrogenic Rests and Associated Wilms Tumours: Two Case Studies. J. Clin. Pathol. 2006, 60, 1013–1016. [Google Scholar] [CrossRef]
- Ravenel, J.D.; Broman, K.W.; Perlman, E.J.; Niemitz, E.L.; Jayawardena, T.M.; Bell, D.W.; Haber, D.A.; Uejima, H.; Feinberg, A.P. Loss of Imprinting of Insulin-Like Growth Factor-II (IGF2) Gene in Distinguishing Specific Biologic Subtypes of Wilms Tumor. J. Natl. Cancer Inst. 2001, 93, 1698–1703. [Google Scholar] [CrossRef] [PubMed]
- Powlesland, R.M.; Charles, A.K.; Malik, K.T.A.; Reynolds, P.A.; Pires, S.; Boavida, M.; Brown, K.W. Loss of Heterozygosity at 7p in Wilms’ Tumour Development. Br. J. Cancer 2000, 82, 323–329. [Google Scholar] [CrossRef]
- Charles, A.K.; Brown, K.W.; Berry, P.J. Microdissecting the Genetic Events in Nephrogenic Rests and Wilms’ Tumor Development. Am. J. Pathol. 1998, 153, 991–1000. [Google Scholar] [CrossRef]
- Cui, H.; Hedborg, F.; He, L.; Nordenskjöld, A.; Sandstedt, B.; Pfeifer-Ohlsson, S.; Ohlsson, R. Inactivation of H19, an Imprinted and Putative Tumor Repressor Gene, Is a Preneoplastic Event during Wilms’ Tumorigenesis. Cancer Res. 1997, 57, 4469–4473. [Google Scholar]
- Steenman, M.; Redeker, B.; De Meulemeester, M.; Wiesmeijer, K.; Voute, P.A.; Westerveld, A.; Slater, R.; Mannens, M. Comparative Genomic Hybridization Analysis of Wilms Tumors. Cytogenet. Genome Res. 1997, 77, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Austruy, E.; Candon, S.; Henry, I.; Gyapay, G.; Tournade, M.-F.; Mannens, M.; Callen, D.; Junien, C.; Jeanpierre, C. Characterization of Regions of Chromosomes 12 and 16 Involved in Nephroblastoma Tumorigenesis. Genes Chromosom. Cancer 1995, 14, 285–294. [Google Scholar] [CrossRef]
- Hoban, P.; Heighway, J.; White, G.M.; Baker, B.; Gardner, J.; Birch, J.; Morris-Jones, P.; Kelsey, A. Genome-Wide Loss of Maternal Alleles in a Nephrogenic Rest and Wilms’ Tumour from a BWS Patient. Hum. Genet. 1995, 95, 651–656. [Google Scholar] [CrossRef]
- Park, S.; Bernard, A.; Bove, K.E.; Sens, D.A.; Hazen-Martin, D.J.; Garvin, A.J.; Haber, D.A. Inactivation of WT1 in Nephrogenic Rests, Genetic Precursors to Wilms’ Tumour. Nat. Genet. 1993, 5, 363–367. [Google Scholar] [CrossRef]
- Yun, K.; Molenaar, A.J.; Fiedler, A.M.; Mark, A.J.; Eccles, M.R.; Becroft, D.M.; Reeve, A.E. Insulin-like Growth Factor II Messenger Ribonucleic Acid Expression in Wilms Tumor, Nephrogenic Rest, and Kidney. Lab. Invest. 1993, 69, 603–615. [Google Scholar]
- Pritchard-Jones, K.; Fleming, S. Cell Types Expressing the Wilms’ Tumour Gene (WT1) in Wilms’ Tumours: Implications for Tumour Histogenesis. Oncogene 1991, 6, 2211–2220. [Google Scholar] [PubMed]
- Zhang, Y.; Liu, H.; Lv, J.; Xiao, X.; Zhu, J.; Liu, X.; Su, J.; Li, X.; Wu, Q.; Wang, F.; et al. QDMR: A Quantitative Method for Identification of Differentially Methylated Regions by Entropy. Nucleic Acids Res. 2011, 39, e58. [Google Scholar] [CrossRef] [PubMed]
- Cowell, J.K.; Wadey, R.B.; Haber, D.A.; Call, K.M.; Housman, D.E.; Pritchard, J. Structural Rearrangements of the WT1 Gene in Wilms’ Tumour Cells. Oncogene 1991, 6, 595–599. [Google Scholar]
- Rivera, M.N.; Kim, W.J.; Wells, J.; Driscoll, D.R.; Brannigan, B.W.; Han, M.; Kim, J.C.; Feinberg, A.P.; Gerald, W.L.; Vargas, S.O.; et al. An X Chromosome Gene, WTX, Is Commonly Inactivated in Wilms Tumor. Science 2007, 315, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Gadd, S.; Huff, V.; Walz, A.L.; Ooms, A.H.A.G.; Armstrong, A.E.; Gerhard, D.S.; Smith, M.A.; Auvil, J.M.G.; Meerzaman, D.; Chen, Q.-R.; et al. A Children’s Oncology Group and TARGET Initiative Exploring the Genetic Landscape of Wilms Tumor. Nat. Genet. 2017, 49, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Chung, W. Chromosome 11p15.5 Regional Imprinting: Comparative Analysis of KIP2 and H19 in Human Tissues and Wilms’ Tumors. Hum. Mol. Genet. 1996, 5, 1101–1108. [Google Scholar] [CrossRef]
- Bergman, D.; Halje, M.; Nordin, M.; Engström, W. Insulin-Like Growth Factor 2 in Development and Disease: A Mini-Review. Gerontology 2013, 59, 240–249. [Google Scholar] [CrossRef]
- Charlton, J.; Irtan, S.; Bergeron, C.; Pritchard-Jones, K. Bilateral Wilms Tumour: A Review of Clinical and Molecular Features. Expert Rev. Mol. Med. 2017, 19, e8. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Weirich, A.; Schumacher, V.; Uschkereit, C.; Beier, M.; Leuschner, I.; Graf, N.; Autschbach, F.; Schneider, D.; von Harrach, M. Clinical Relevance of Mutations in the Wilms Tumor Suppressor 1 Gene WT1 and the Cadherin-Associated Protein Β1 Gene CTNNB1 for Patients with Wilms Tumors. Cancer 2008, 113, 1080–1089. [Google Scholar] [CrossRef]
- Duhme, C.; Busch, M.; Heine, E.; de Torres, C.; Mora, J.; Royer-Pokora, B. WT1-Mutant Wilms Tumor Progression Is Associated With Diverting Clonal Mutations of CTNNB1. J. Pediatr. Hematol. Oncol. 2021, 43, e180–e183. [Google Scholar] [CrossRef] [PubMed]
- Hol, J.A.; Kuiper, R.P.; van Dijk, F.; Waanders, E.; van Peer, S.E.; Koudijs, M.J.; Bladergroen, R.; van Reijmersdal, S.V.; Morgado, L.M.; Bliek, J.; et al. Prevalence of (Epi)Genetic Predisposing Factors in a 5-Year Unselected National Wilms Tumor Cohort: A Comprehensive Clinical and Genomic Characterization. J. Clin. Oncol. 2022, 40, 1892–1902. [Google Scholar] [CrossRef] [PubMed]
- van Peer, S.E.; Hol, J.A.; van der Steeg, A.F.W.; van Grotel, M.; Tytgat, G.A.M.; Mavinkurve-Groothuis, A.M.C.; Janssens, G.O.R.; Littooij, A.S.; de Krijger, R.R.; Jongmans, M.C.J.; et al. Bilateral Renal Tumors in Children: The First 5 Years’ Experience of National Centralization in The Netherlands and a Narrative Review of the Literature. J. Clin. Med. 2021, 10, 5558. [Google Scholar] [CrossRef] [PubMed]




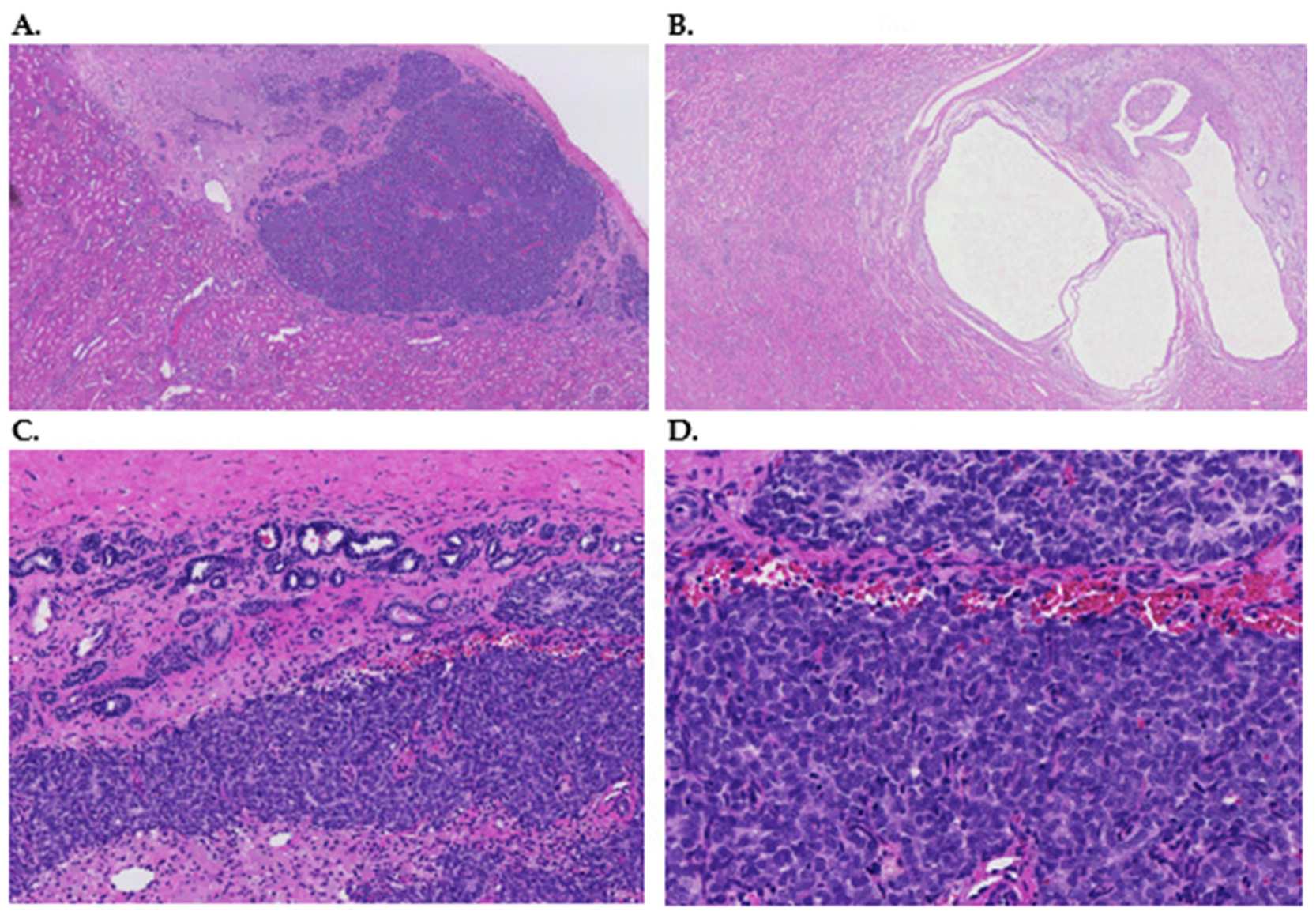{kind=link}
{kind=link}
{kind=link}
| Study | Oxford Level | NR (n) | NR Type | WT (n) | WT Type | WT-NR Pairs | Bilateral WT | Sex | Age at Diagnosis |
|---|---|---|---|---|---|---|---|---|---|
| Chang et al., 2021 * [23] | 4 | Multiple 1 | PLNR NBL | 1 - | epithelial type - | 1 - | Yes - | M F | 21 months 36 months |
| Slack et al., 2021 [24] | 4 | Multiple | PLNR | 1 | epithelial type | 1 | Yes | M | 21 months |
| Coorens et al., 2019 [25] | 4 | 1 | ND | 23 | ND | 1 | No | ND | ND |
| Wegert et al., 2018 [26] | 2 | 12 | ND | 208 | ND | ND | ND | ND | ND |
| Charlton et al., 2015 [27] | 2 | 22 | 17 PLNR, 5 ILNR | 36 | ND | 20 | ND | ND | ND |
| MdZin et al., 2011 [28] | 4 | 9 1 | 9 PLNR 1 PLNR | 2 1 | blastemal type mixed type | 1 1 | Yes No | M M | 42 months 48 months |
| Fukuzawa et al., 2010 [29] | 4 | 4 | 4 ILNR | 4 | 3 stromal type, 1 blastemal type | 4 | ND | 3F, 1M | ND |
| Grill et al., 2010 [30] | 2 | 26 | 18 PLNR, 8 ILNR | 22 | 14 mixed type, 2 stromal type, 4 blastemal type and 1 regressive type, 1 not known | 22 | ND | ND | ND |
| Vuononvirta et al., 2008 [31] | 2 | 50 | 50 PLNR | 25 | ND | 25 | 17/50 NR | ND | ND |
| Brown et al., 2008 ** [32] | 4 | Multiple | PLNR | 51 | ND | 2 | 2; 2 WT-NR pairs | ND | ND |
| Chilukamarri et al., 2007 [33] | 4 | 2 | ND | 24 | 5 stromal type, 1 epithelial type, 4 blastemal type, 8 mixed type, 6 not known | 2 | 5; 2 WT-NR pairs | 8M, 16F (WT-NR pairs 2F) | WT 36 ^ months |
| Hancock et al., 2007 [34] | 2 | Multiple | ND | 2 | mixed type | 2 | 2 WT-NR pairs | ND | ND |
| Fukuzawa et al., 2006 [35] | 4 | 3 | 3 ILNR | 2 | stromal type | 2 | ND | ND | ND |
| Ravenel et al., 2001 [36] | 4 | 2 | 2 PLNR | 60 | ND | 1 | ND | ND | ND |
| Powlesland et al., 1999 [37] | 2 | 2 | PLNR | 7 | 3 mixed type, 1 stromal type, 3 blastemal type | 2 | 2, 1 WT-NR pair | 4 M, 3F (WT-NR pair M) | WT 43 months ^ |
| Charles et al., 1998 [38] | 2 | 42 | 22 PLNR, 17 ILNR, 3 both types | 139 | ND | ND | 9; 2 WT-NR pairs | ND | NR 49 months ^ PLNR 54 ^, ILNR 44 ^ |
| Cui et al., 1997 [39] | 2 | 8 | 7 PLNR, 1 ILNR | 14 | ND | 7 | ND | ND | ND |
| Steenman et al., 1997 [40] | 2 | 7 | 7 NBL | 46 | ND | 6 | 2, no WT-NR pairs | ND | ND |
| Austruy et al., 1995 [41] | 2 | 2 | NBL | 28 | ND | 2 | 3, no WT-NR pairs | ND | ND |
| Hoban et al., 1995 [42] | 4 | Multiple | PLNR, ILNR | 1 | ND | 1 | ND | F | 11 months |
| Park et al., 1993 [43] | 4 | Multiple | 1 ILNR, multiple PLNR | 19 | blastemal type | 2 | ND | 2F (WT-NR pair) | 11 months and 48–84/144 months |
| Yun et al., 1993 [44] | 2 | 15 | 13 ILNR, 2 PLNR | 31 | 6 blastemal type, 5 stromal dominant, 20 mixed type | 15 | ND | 14M, 17F (WT-NR pairs 8M, 7F) | WT 43 months ^ |
| Pritchard-Jones et al., 1991 [45] | 2 | Multiple | NBL and not known | 32 | 18 mixed type, 4 epithelial type and 10 not known | ND | ND | ND | ND |
| Study | Gene/Chromosomal Region | Method | Results | Conclusion |
|---|---|---|---|---|
| Chang et al., 2021 [23] | KRAS, FBXW7 | NGS/WGS | Association between nephroblastomatosis and KRAS. | Possible association between KRAS and bilateral WT and between mosaic KRAS and NBL. |
| Slack et al., 2021 [24] | KRAS, FBXW7 | NGS | Mosaic KRAS has similar frequencies in WT and adjacent NR. Bilateral WT, but not two adjacent NR, contained the FBXW7 mutation. | Similar KRAS allele frequencies in WT and NR. FBXW7 mutation seems to be a late event in WT tumorigenesis |
| Coorens et al., 2019 [25] | Genome wide | WGS, WES Methylation analysis | Clonal nephrogenesis in 14/23 (61%) WTs (4/4 bilateral). WT and NR from same patient arose at different times from the same ancestral clone. H19 hypermethylation in 7/12 NK with clonal nephrogenesis but not in NK without clonal nephrogenesis. | There is an association between VAF of embryonal clonal expansions, H19 hypermethylation and development of WT. |
| Wegert et al., 2018 [26] | EGFR, BRAF | WGS | No mutations in both NR (n = 12) and WT (n = 208) | No EGFR, BRAF mutations in NR and WT |
| Charlton et al., 2015 [27] | Genome wide | Comprehensive methylome analysis | NR vs. NK: 629 DMR, 55% showed hypermethylation. NR vs. WT: 2 subgroups WT, one group showed the same epigenetics as NR and one group presented increased methylation variability. | Methylation profiles vary significantly between NK, NRs and WTs and alterations in the methylome lead to NR formation and transformation to WTs. |
| MdZin et al., 2011 [28] | Chromosome 22 | FISH and microsatellite analysis | Dormant, involuted and sclerosing NR displayed monosomy 22 in 30%, hyperplastic and adenomatous NR in 50%, and 60–80% in nuclei of WT. | More common loss of chromosome 22 in the development of PLNR (from dormant to hyperplastic) to WT. |
| Fukuzawa et al., 2010 [29] | WTX, CTNNB1 | Sequencing analysis, MLPA/microsatellite analysis | CTNNB1 mutation: n = 0 in NR, n = 4 in WT. WTX mutation: n = 1 in NR, n = 4 in WT. | WTX can occur as an early event, or in later stages of development, CTNNB1 is a late event. |
| Grill et al., 2010 [30] | PTEN | LOH-analysis, sequencing analysis | None of the WT and none of the ILNR/PLNR showed LOH. | PTEN does not play a role in tumorigenesis. Downregulation does not cause WNT-pathway activation. |
| Vuononvirta et al., 2008 [31] | Genome wide | aCGH LOH-analysis Methylation analysis | PLNR 3 groups: no copy number changes (44%); single, whole chromosome changes (16%); multiple gains or losses (40%). In 76% NR changes correspond to WT. 11p15 LOH in 10/39 (26%), in NR and tumor (n = 9). H19 hypermethylation in 37/40 (93%) PLNR. | PLNR are non-obligate precursors of WT. |
| Brown et al., 2008 [32] | LOI 11p13/15, LOH 16q, 7p | LOH-analysis Methylation analysis | LOI 11p13 and 11p15; LOH 7p/16q not in NR, but in WT. H19 DMR analysis increased in NR and WT (n = 2). Reduced methylation of WT1 ARR in NR and WT. | LOI at 11p13 and 11p15 are early events in NR, and occur prior to LOH at 7p or 16q. H19 DMR methylation was increased in NR and WT. |
| Chilukamarri et al., 2007 [33] | GLIPR1/RTVP-1 | Methylation analysis | Hypomethylation WT (21/24) and NR (n = 2) | Hypomethylation of GLIPR1/RTVP1 may play a role in WT tumorigenesis. |
| Hancock et al., 2007 [34] | WT1 | Methylation and expression analysis | WT showed hypomethylation of WT1 ARR on both alleles. WT1 methylation differs between FK, NR and WT. | Imprinting defects at 11p13 contribute to WT tumorigenesis. |
| Fukuzawa et al., 2006 [35] | CTNNB1, WT1 11p13 | Sequencing analysis LOH-analysis | No CTNNB1 mutations in NR, n = 2 in WT. WT1 mutation: n = 3 in NR and n = 2 in associated WT. 11p13 LOH in ILNR and tumor (n = 1) | Mutations in the CTNNB1 occur in the later stages of WT tumorigenesis. Mutations of WT1 are early events in ILNR. |
| Ravenel et al., 2001 [36] | IGF-2 | Expression analysis | LOI of IGF-2 in 2 PLNR and associated WT. | LOI of IGF-2 seems to be an early event in the development of WT. |
| Powlesland et al., 1999 [37] | 7p | LOH-analysis | LOH of 7p in 7/77 WT, one associated NR has no LOH of 7p. | LOH of 7p seems to be a late event in WT tumorigenesis. |
| Charles et al., 1998 [38] | WT1 11p15, 11p13, 16q | Sequencing analysis LOH-analysis | Two pairs of ILNR and WT showed WT1 mutation. LOH at 11p15 in 3/25 (12%, all ILNR). LOH at 11p13 in 3/26 (12%, 2 in ILNR, in case with PLNR only in tumor). Loss of 16q in 4/23, only in tumors. | LOH at 11p13 and 11p15 is seen in ILNR and WT. PLNR showed no LOH 11p events occur early in WT development. Genetic changes at 16q are a late event. |
| Cui et al., 1997 [39] | H19, IGF-2 | ISH | IGF-2 expression present in NR, WT and kidney medulla. H19 is not expressed in NR and WT in contrast to renal medulla. Pattern of IGF-2 expression differs in NR and WT. | Association between expression of IGF-2 and H19, but H19 inactivation also without effect on IGF-2 expression status. Loss of H19 expression possibly involved in blastemal overgrowth. |
| Steenman et al., 1997 [40] | Genome wide | CGH | Losses in 1p, 4q, 7p and gains in 7q, 1q and 12q can occur in both tumor and NBL, even as LOH at 1p and 11p13; loss of 11 only in NBL; loss of 9p, 16q and gain of 8, 10q and 18 are only seen in WT. | Two specific 1p regions involved in WT etiology. |
| Austruy et al., 1995 [41] | 16q | LOH analysis | LOH of 16q in 7/28 WT and in one out of two associated NBL. | LOH of 16q can also occur as an early event Wilms tumorigenesis. |
| Hoban et al., 1995 [42] | Genome wide | LOCH analysis | LOCH present on chromosome 11 (11p13/p15), but also on all other chromosomes. | Loss of all maternal loci, including #11, suggests to be an early genetic event. |
| Park et al., 1993 [43] | WT1 | Sequencing analysis | WT1 mutations in both cases in NR and WT (n = 2), both somatic. | WT1 inactivation seems to be an early genetic event. |
| Yun et al., 1993 [44] | IGF-2 | mRNA ISH | IGF-2 hybridization patterns of NR equal to WTs. IGF-2 expression in NR variable. IGF-2 transcripts more frequent in tumors with blastema. | Occasional NR also displayed different IGF-2 expression, suggesting NR could be precursor lesions of WT. |
| Pritchard-Jones et al., 1991 [45] | WT1 | mRNA ISH | NBL have high levels of expression, similar to WT. | WT1 contributes to WT tumorigenesis. |
| Early Events | Late Events |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bánki, T.; Drost, J.; van den Heuvel-Eibrink, M.M.; Mavinkurve-Groothuis, A.M.C.; de Krijger, R.R. Somatic, Genetic and Epigenetic Changes in Nephrogenic Rests and Their Role in the Transformation to Wilms Tumors, a Systematic Review. Cancers 2023, 15, 1363. https://doi.org/10.3390/cancers15051363
Bánki T, Drost J, van den Heuvel-Eibrink MM, Mavinkurve-Groothuis AMC, de Krijger RR. Somatic, Genetic and Epigenetic Changes in Nephrogenic Rests and Their Role in the Transformation to Wilms Tumors, a Systematic Review. Cancers. 2023; 15(5):1363. https://doi.org/10.3390/cancers15051363
Chicago/Turabian StyleBánki, Tessa, Jarno Drost, Marry M. van den Heuvel-Eibrink, Annelies M. C. Mavinkurve-Groothuis, and Ronald R. de Krijger. 2023. "Somatic, Genetic and Epigenetic Changes in Nephrogenic Rests and Their Role in the Transformation to Wilms Tumors, a Systematic Review" Cancers 15, no. 5: 1363. https://doi.org/10.3390/cancers15051363
APA StyleBánki, T., Drost, J., van den Heuvel-Eibrink, M. M., Mavinkurve-Groothuis, A. M. C., & de Krijger, R. R. (2023). Somatic, Genetic and Epigenetic Changes in Nephrogenic Rests and Their Role in the Transformation to Wilms Tumors, a Systematic Review. Cancers, 15(5), 1363. https://doi.org/10.3390/cancers15051363