
Glucose Metabolism Reprogramming in Bladder Cancer: Hexokinase 2 (HK2) as Prognostic Biomarker and Target for Bladder Cancer Therapy

 ,
,  , ,
, ,  ,
,  and
and
Abstract
Simple Summary
Abstract
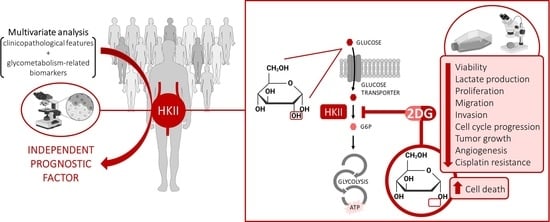
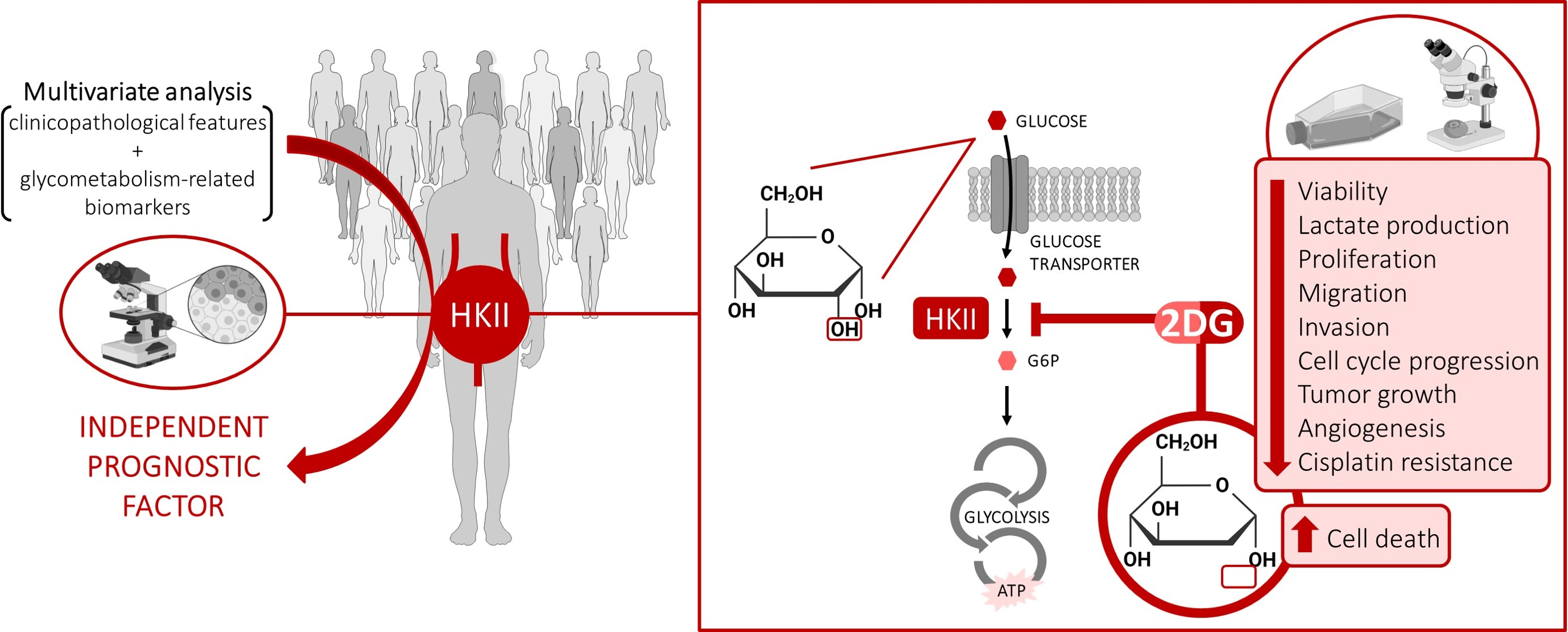
1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Samples
2.2. Immunohistochemistry and Evaluation of Immunohistochemistry Results
2.3. Cell Lines and General Cell Culture Conditions
2.4. Protein Extraction and Western Blotting
2.5. Immunofluorescence
2.6. Cell Viability Assay
2.7. Lactate Production Quantification
2.8. Cell Proliferation Assay
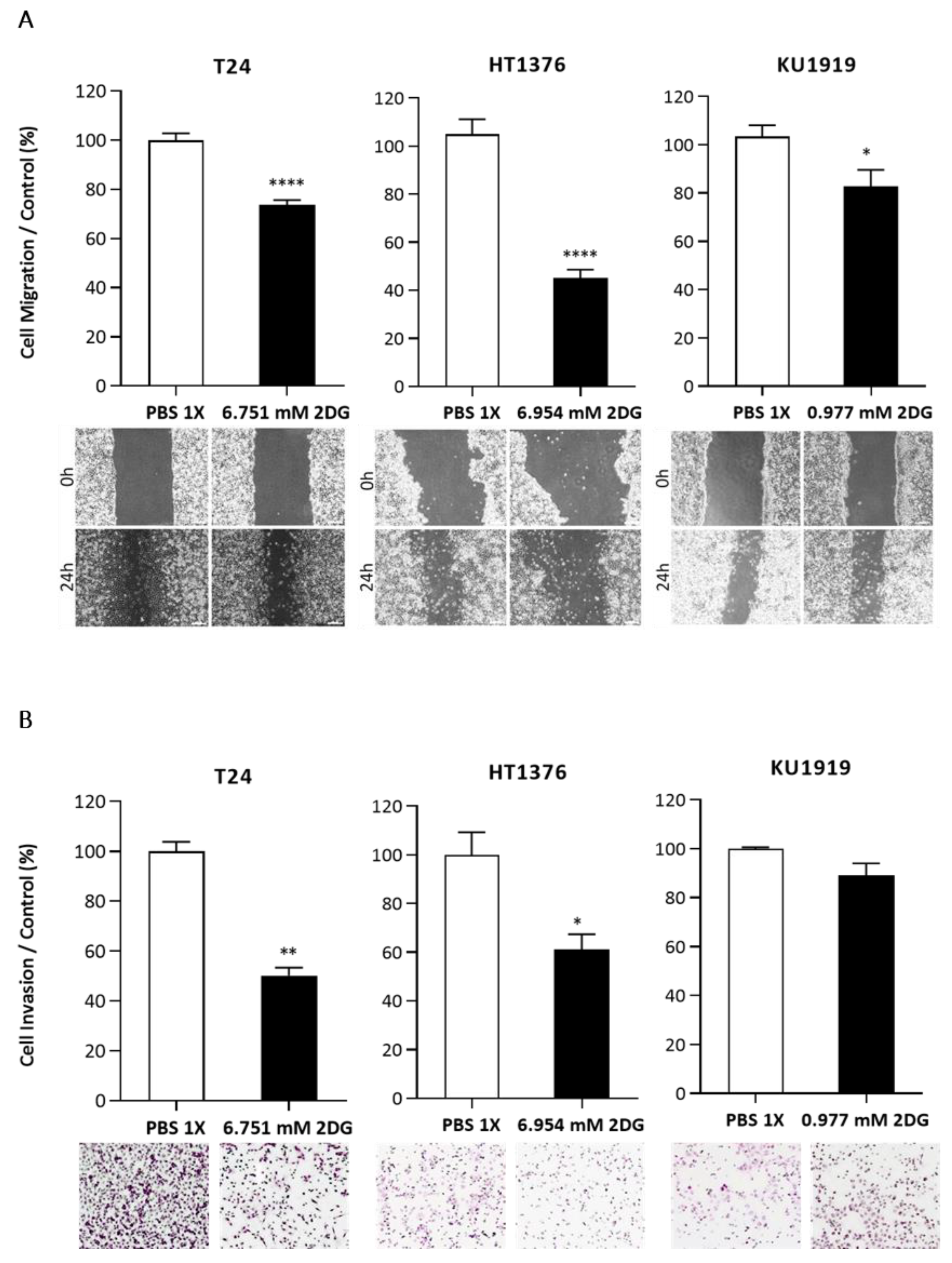
2.9. Wound-Healing Assay
2.10. Cell Invasion Assay
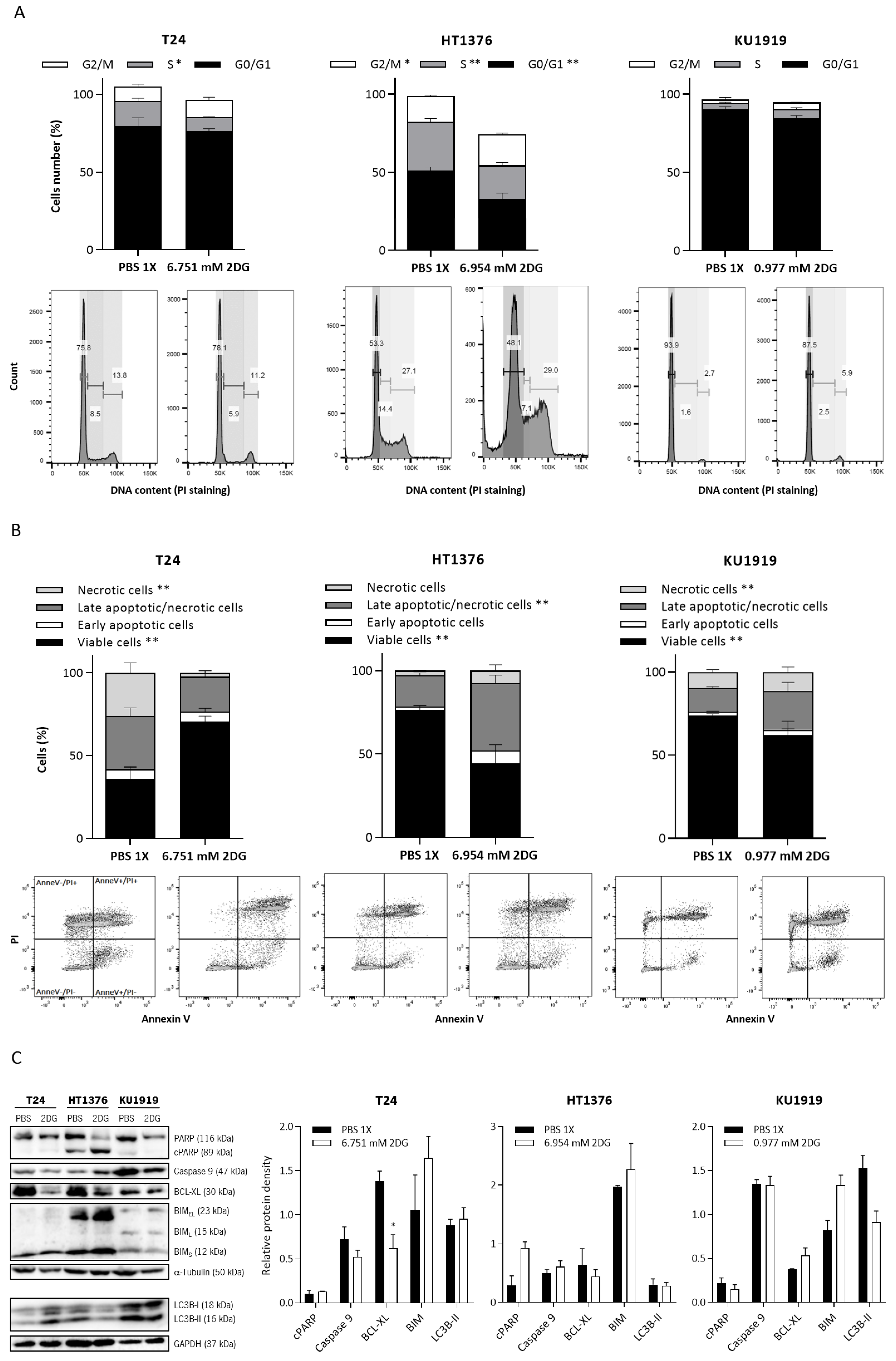
2.11. Cell Cycle Analysis
2.12. Cell Death Analysis
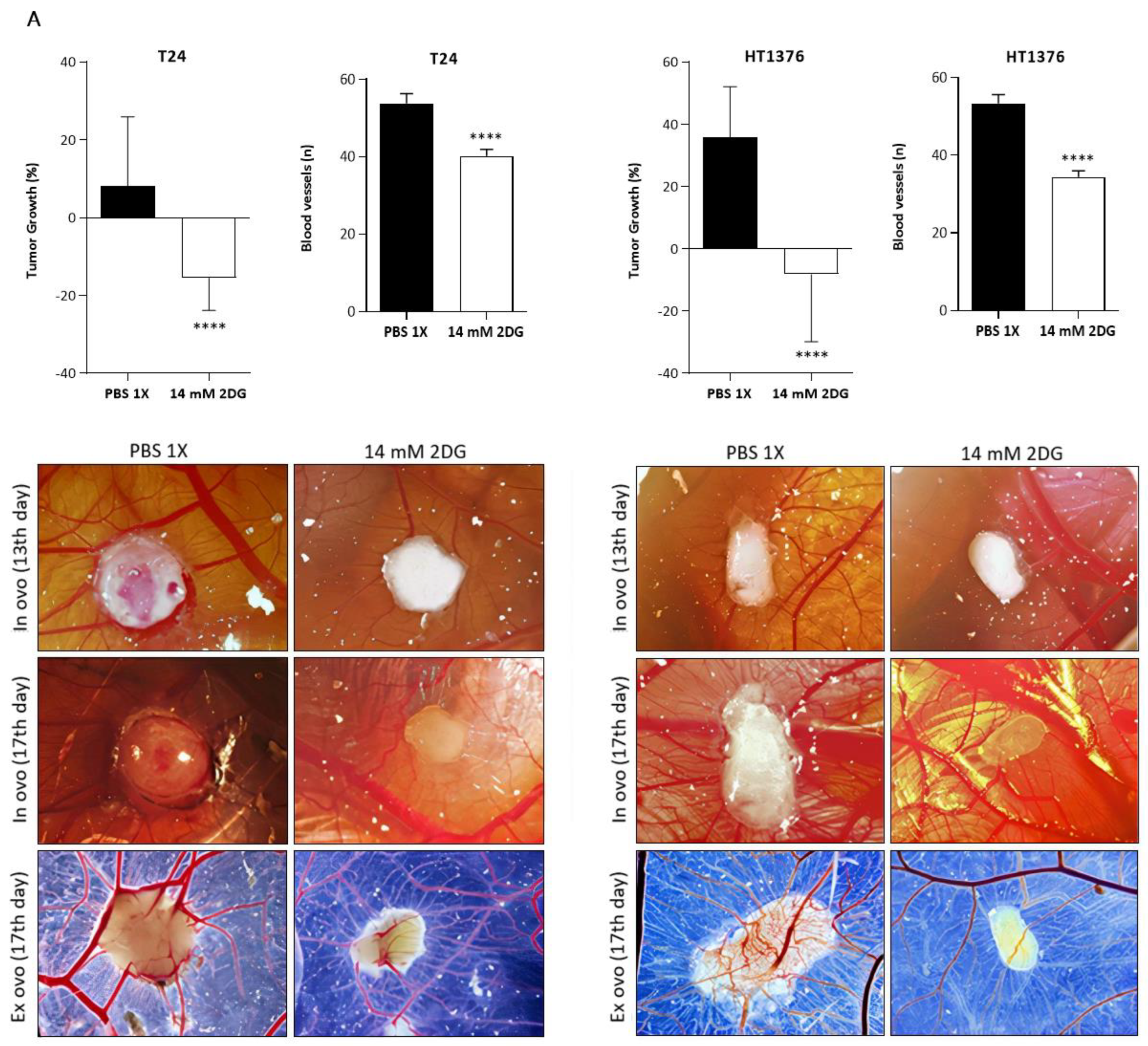
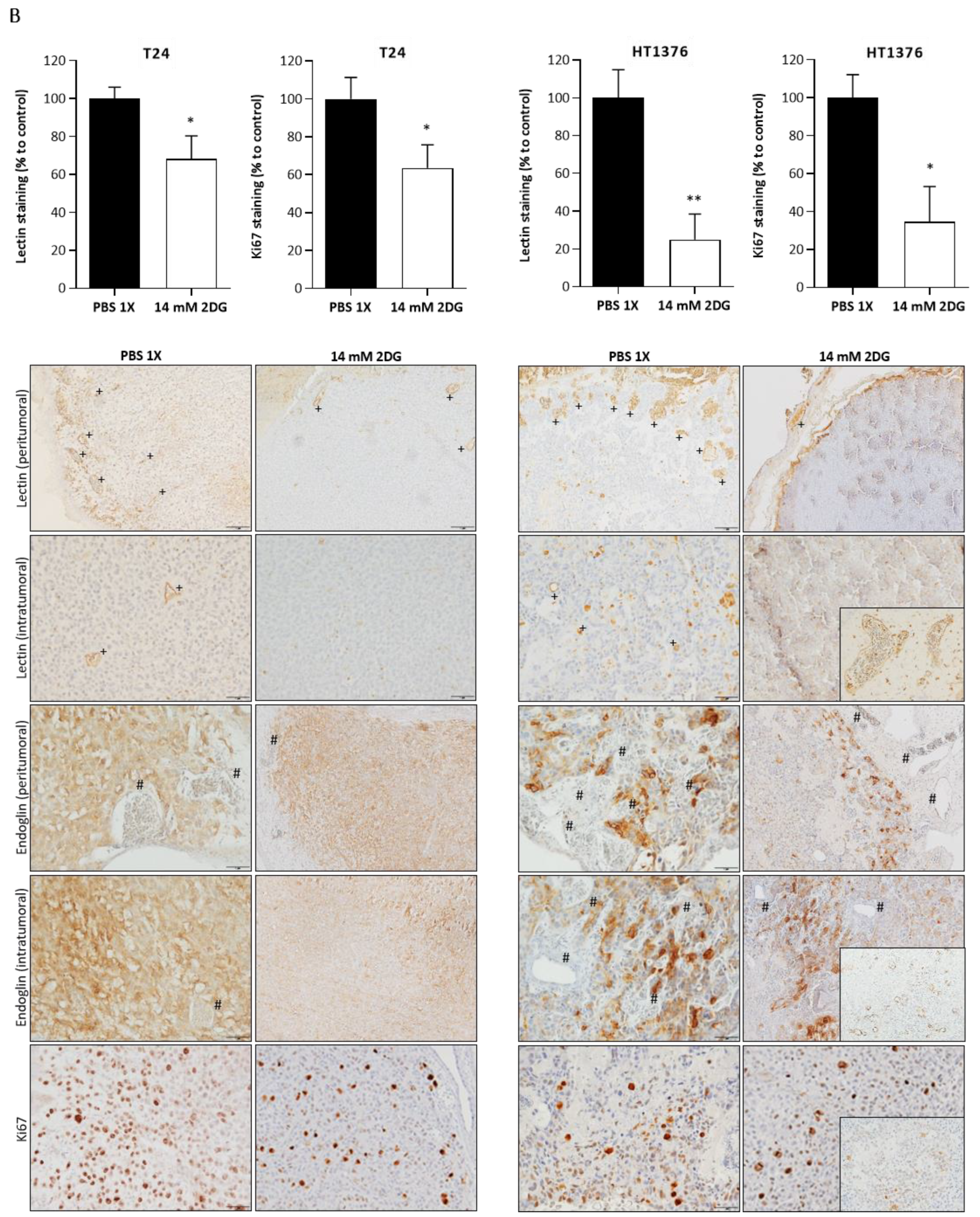
2.13. Chick Chorioallantoic Membrane (CAM) Assay
2.14. Statistical Analysis
3. Results
3.1. Prognostic Significance of Clinicopathological Parameters
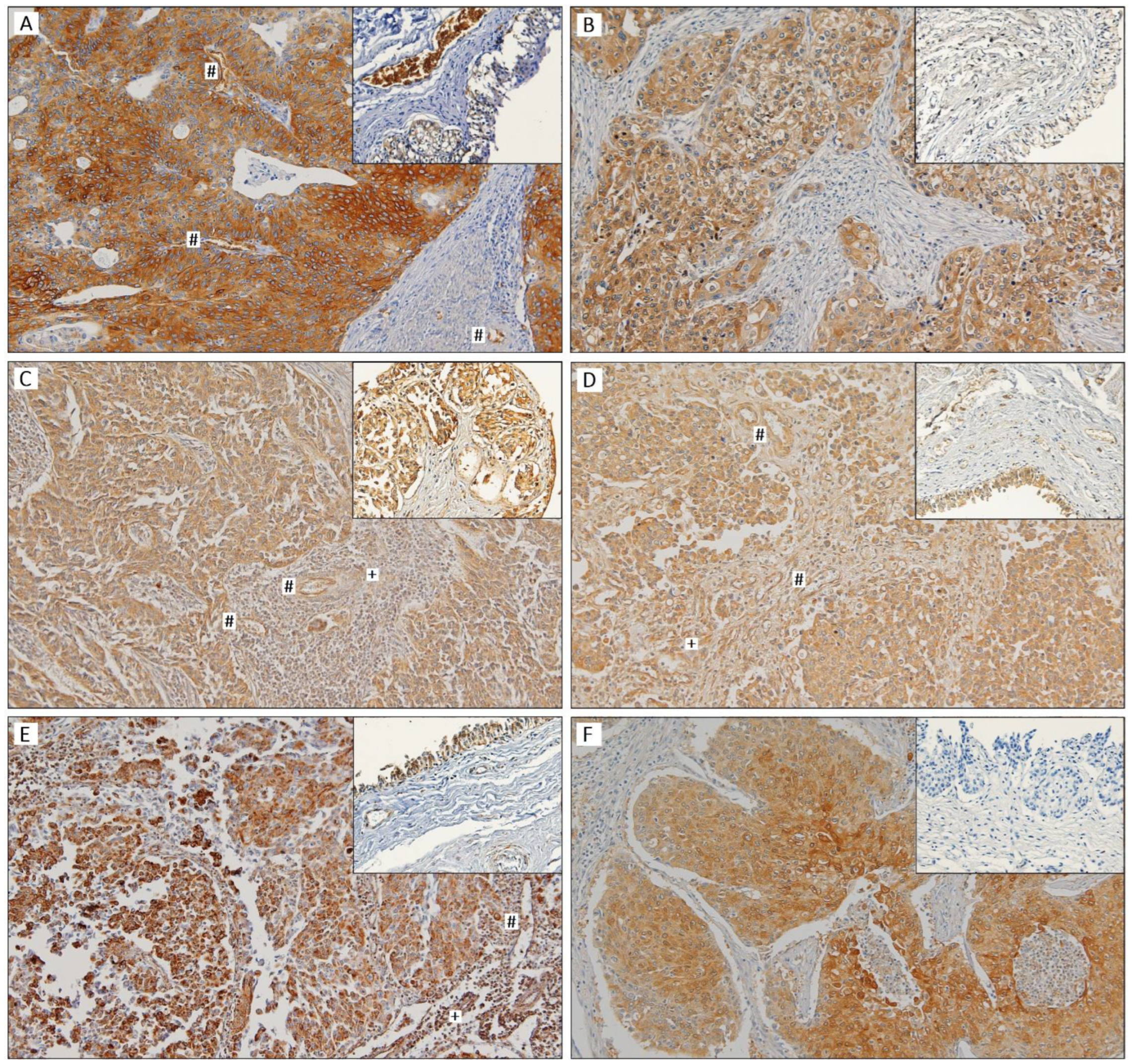
3.2. Immunoexpression of Glycometabolism-Related Biomarkers
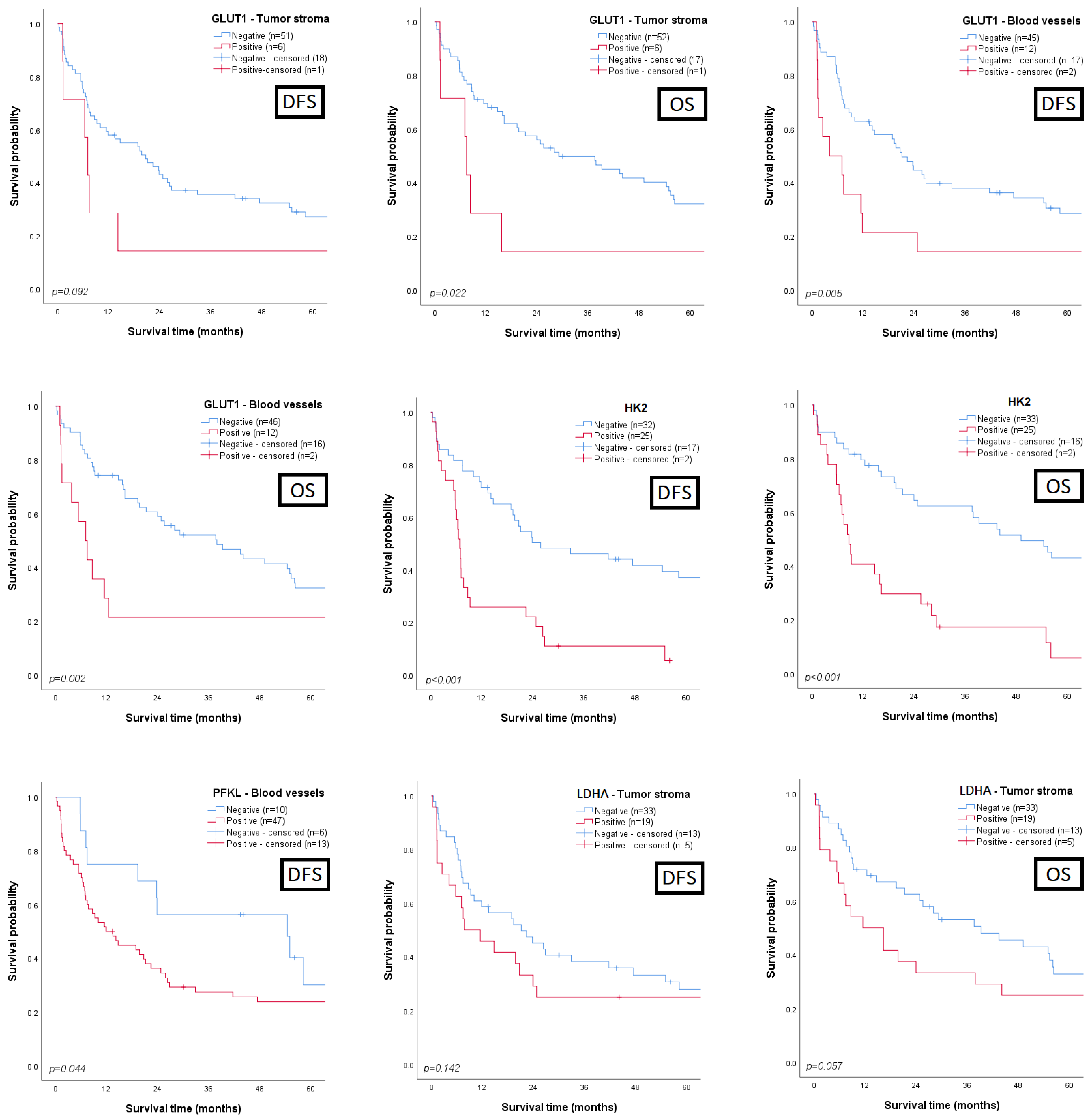
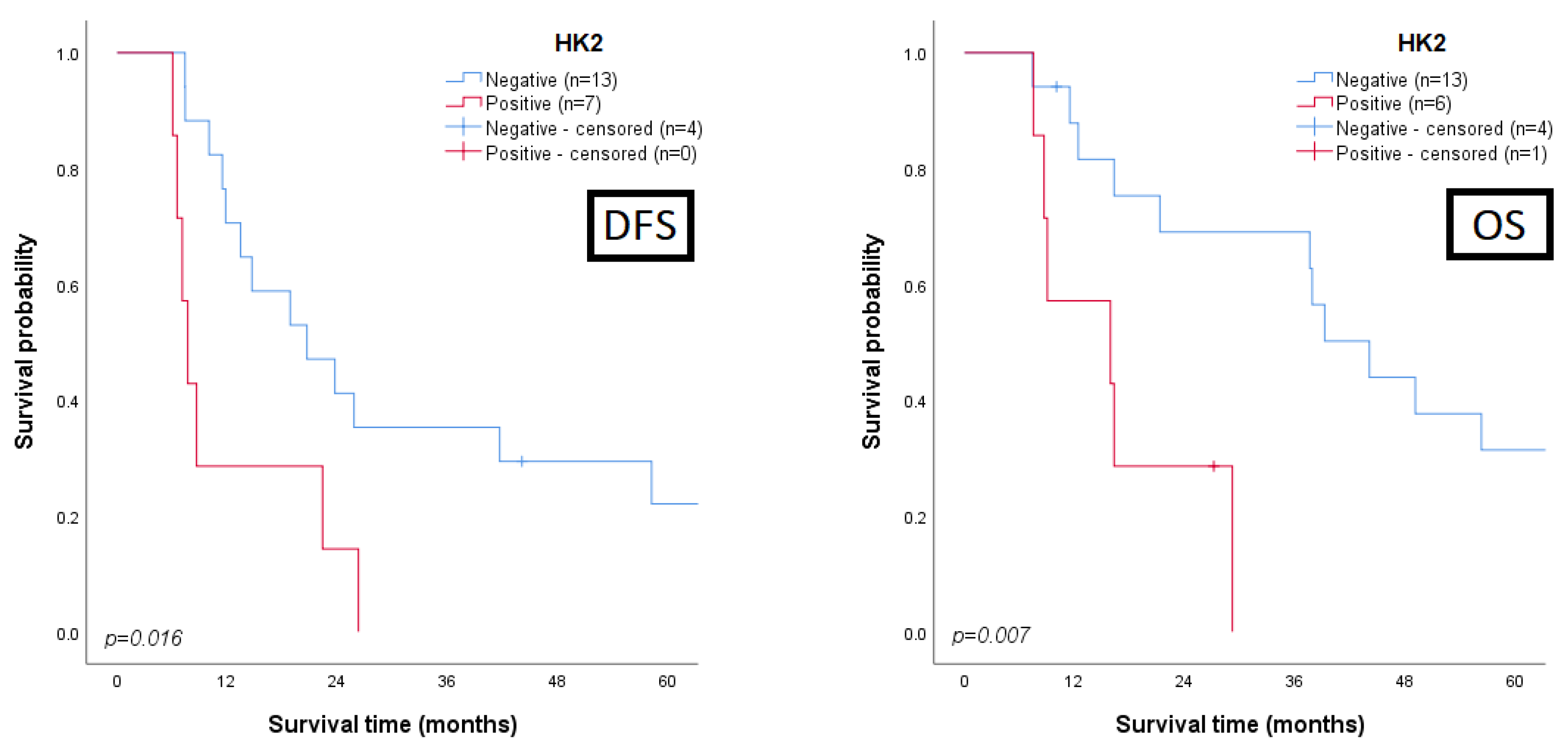
3.3. Clinicopathological and Prognostic Significance of Glycometabolism-Related Biomarkers
3.4. Univariate and Multivariate Analysis for Prognosis Prediction
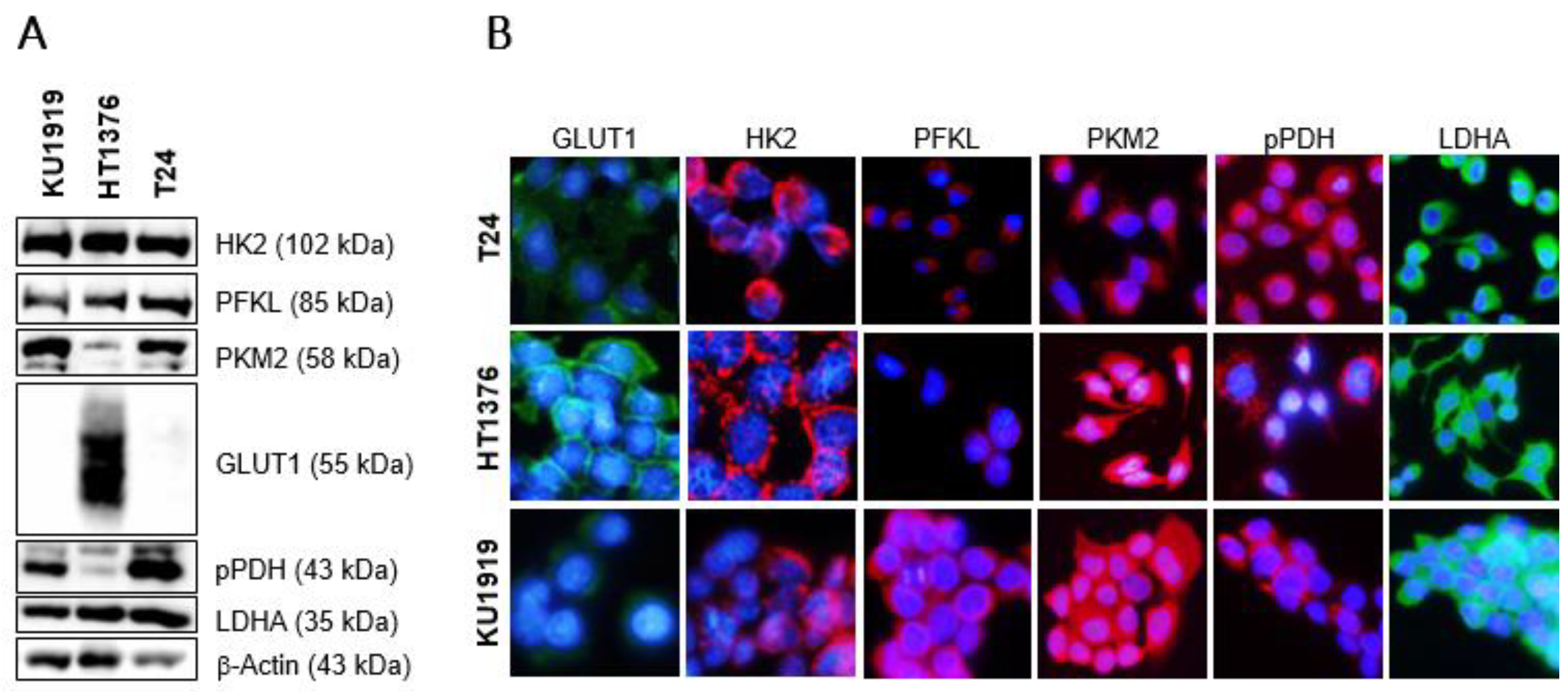
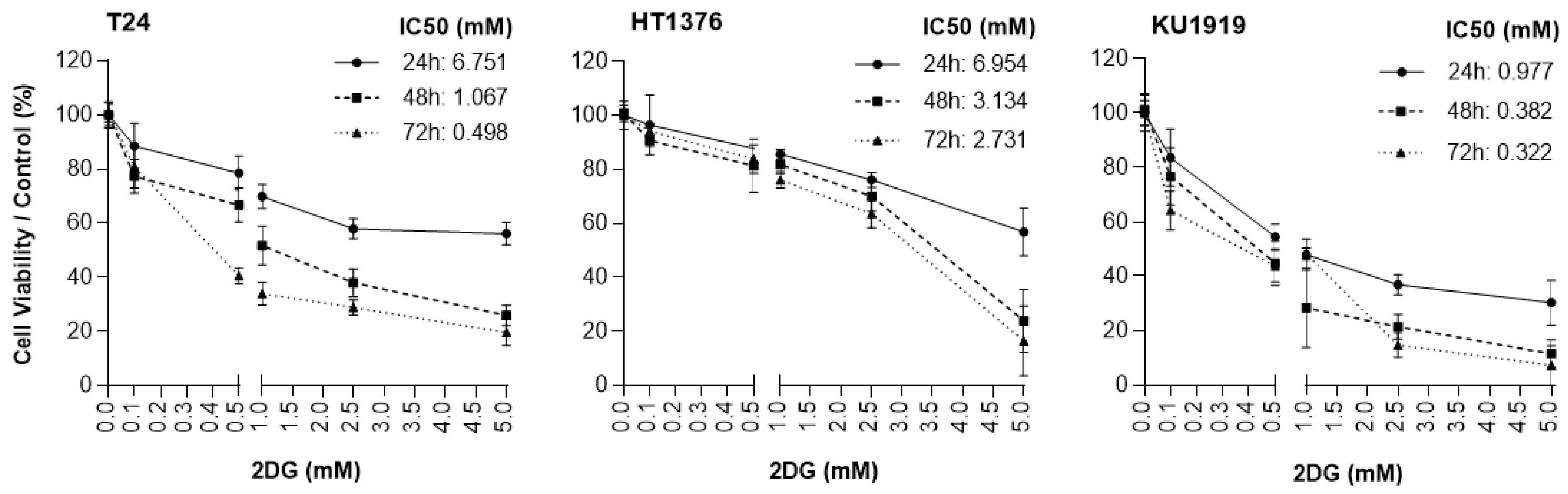
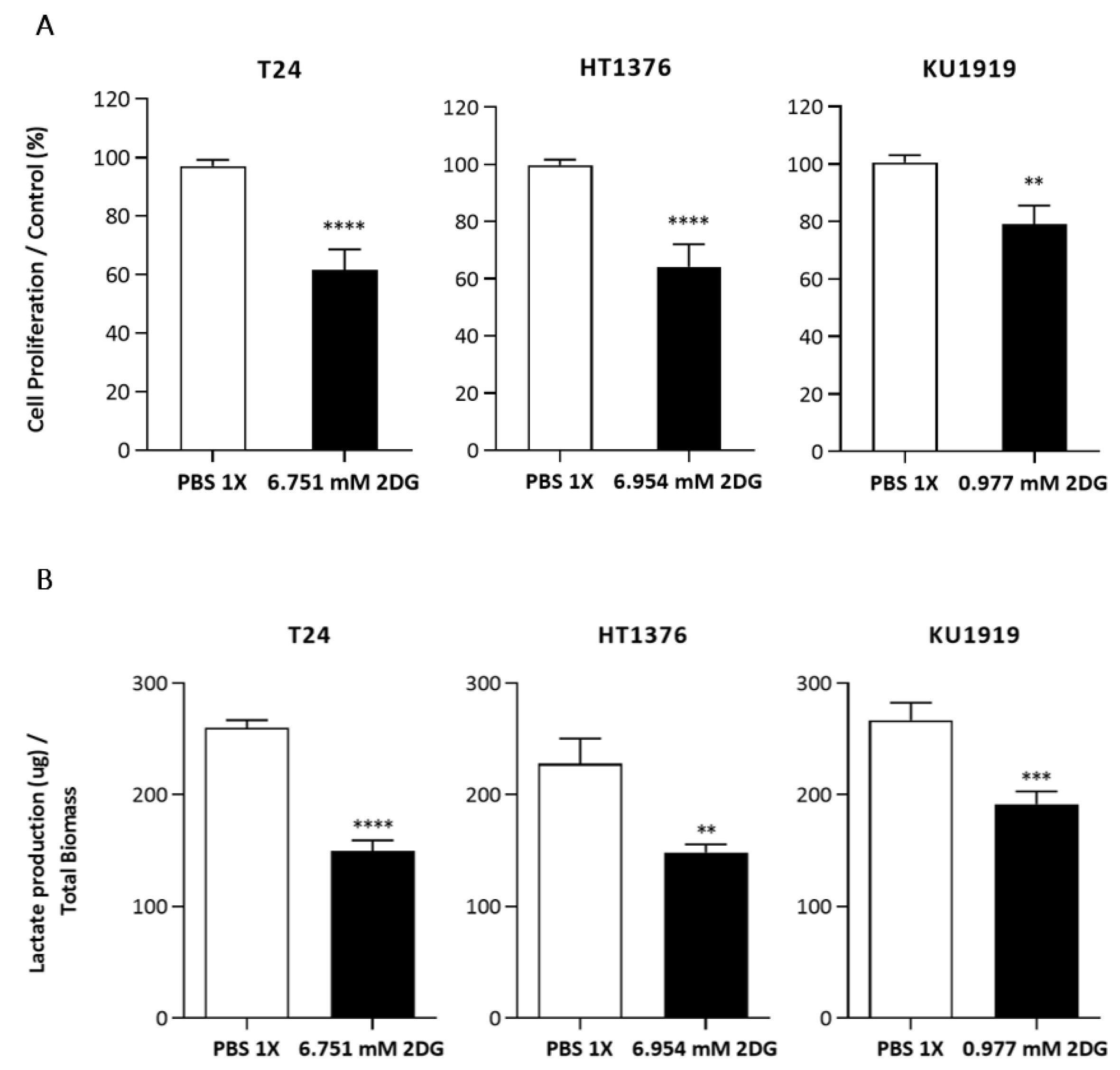
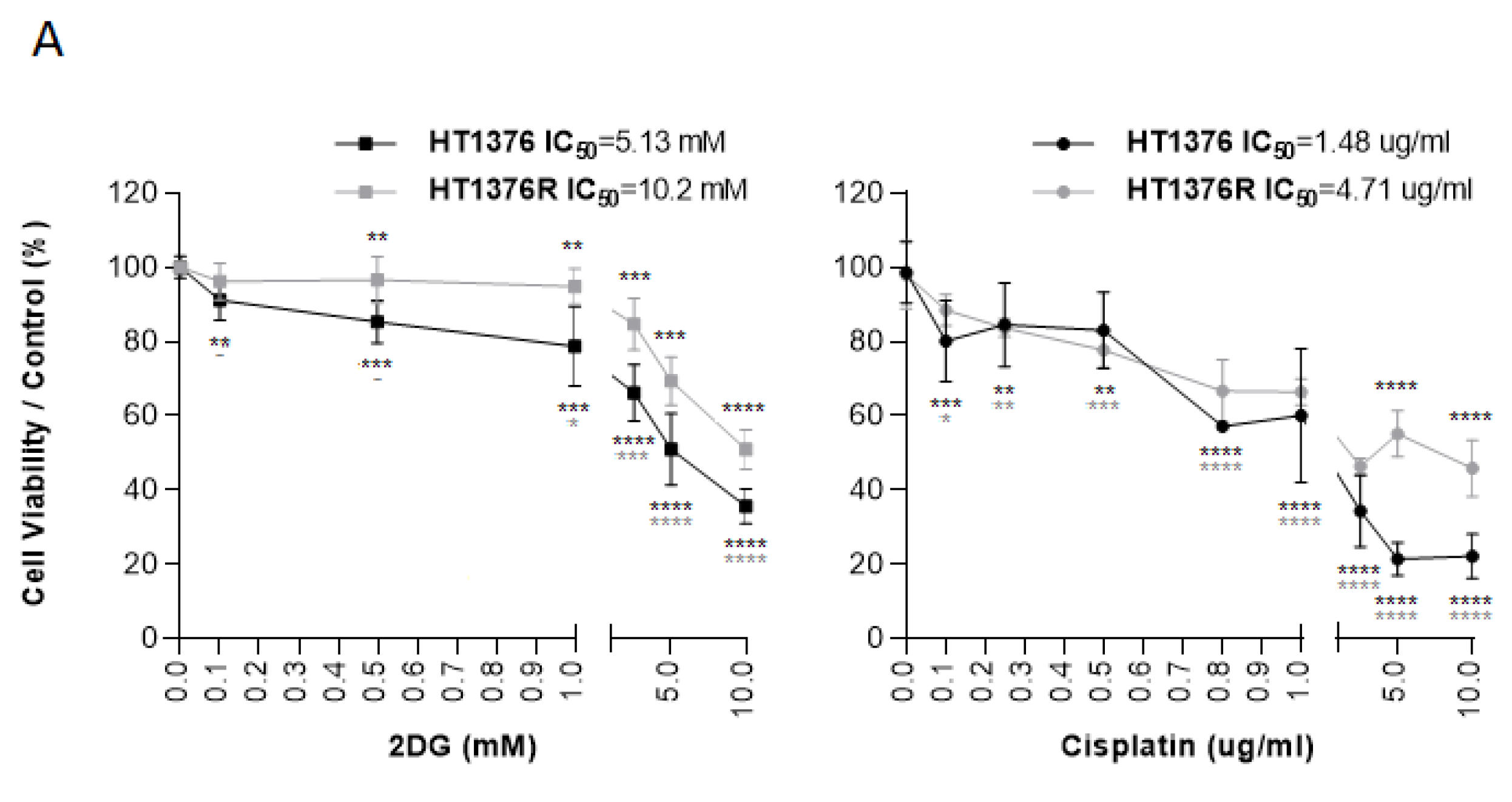
3.5. Effects of HK2 Inhibition by 2DG in Muscle-Invasive UBC Cell Lines
3.6. Effects of HK2 Inhibition by 2DG in an “In Vivo” Muscle-Invasive UBC Model
3.7. Effect of 2DG and Cisplatin Combination in an Isogenic Pair of Cisplatin-Sensitive/Resistant Muscle-Invasive UBC Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Sanli, O.; Dobruch, J.; Knowles, M.; Burger, M.; Alemozaffar, M.; Nielsen, M.; Lotan, Y. Bladder cancer. Nat. Reviews. Dis. Prim. 2017, 3, 1–19. [Google Scholar] [CrossRef]
- Hurst, C.; Rosenberg, J.; Knowles, M. SnapShot: Bladder Cancer. Cancer Cell 2018, 34, 350–350.e1. [Google Scholar] [CrossRef]
- Bellmunt, J.; Powles, T.; Henriksson, R.; Steinberg, G.D.; Batyrbekova, N.; Schain, F.; Fleming, S.; Shalaby, W.; Siefker-Radtke, A.O. Clinical outcomes and economic burden for bladder cancer patients: An analysis from a Swedish cancer registry. J. Clin. Oncol. 2020, 38, 5026. [Google Scholar] [CrossRef]
- Ripoll, J.; Ramos, M.; Montano, J.; Pons, J.; Ameijide, A.; Franch, P. Cancer-specific survival by stage of bladder cancer and factors collected by Mallorca Cancer Registry associated to survival. BMC Cancer 2021, 21, 676. [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Cooley, L.F.; Glaser, A.P.; Meeks, J.J. Mutation signatures to Pan-Cancer Atlas: Investigation of the genomic landscape of muscle-invasive bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2022, 40, 279–286. [Google Scholar] [CrossRef]
- Cooley, L.F.; McLaughlin, K.A.; Meeks, J.J. Genomic and Therapeutic Landscape of Non-muscle-invasive Bladder Cancer. Urol. Clin. N. Am. 2020, 47, 35–46. [Google Scholar] [CrossRef]
- Franza, A.; Pirovano, M.; Giannatempo, P.; Cosmai, L. Erdafitinib in locally advanced/metastatic urothelial carcinoma with certain FGFR genetic alterations. Futur. Oncol. 2022, 18, 2455–2464. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Cimadamore, A.; Blanca, A.; Massari, F.; Vau, N.; Scarpelli, M.; Cheng, L.; Montironi, R. Immune Checkpoint Inhibitors for the Treatment of Bladder Cancer. Cancers 2021, 13, 131. [Google Scholar] [CrossRef]
- Jiang, D.M.; Gupta, S.; Kitchlu, A.; Meraz-Munoz, A.; North, S.A.; Alimohamed, N.S.; Blais, N.; Sridhar, S.S. Defining cisplatin eligibility in patients with muscle-invasive bladder cancer. Nat. Rev. Urol. 2021, 18, 104–114. [Google Scholar] [CrossRef]
- Drayton, R.M.; Catto, J.W. Molecular mechanisms of cisplatin resistance in bladder cancer. Expert Rev. Anticancer. Ther. 2012, 12, 271–281. [Google Scholar] [CrossRef]
- Crispen, P.L.; Kusmartsev, S. Mechanisms of immune evasion in bladder cancer. Cancer Immunol. Immunother. 2020, 69, 3–14. [Google Scholar] [CrossRef]
- Yue, S.; Li, Y.; Chen, X.; Wang, J.; Li, M.; Chen, Y.; Wu, D. FGFR-TKI resistance in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 23. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Läsche, M.; Emons, G.; Gründker, C. Shedding New Light on Cancer Metabolism: A Metabolic Tightrope between Life and Death. Front. Oncol. 2020, 10, 409. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Massari, F.; Ciccarese, C.; Santoni, M.; Iacovelli, R.; Mazzucchelli, R.; Piva, F.; Scarpelli, M.; Berardi, R.; Tortora, G.; Lopez-Beltran, A.; et al. Metabolic phenotype of bladder cancer. Cancer Treat. Rev. 2016, 45, 46–57. [Google Scholar] [CrossRef]
- Afonso, J.; Santos, L.L.; Longatto-Filho, A.; Baltazar, F. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat. Rev. Urol. 2020, 17, 77–106. [Google Scholar] [CrossRef]
- Burns, J.E.; Hurst, C.D.; Knowles, M.A.; Phillips, R.M.; Allison, S.J. The Warburg effect as a therapeutic target for bladder cancers and intratumoral heterogeneity in associated molecular targets. Cancer Sci. 2021, 112, 3822–3834. [Google Scholar] [CrossRef]
- Afonso, J.; Santos, L.L.; Miranda-Gonçalves, V.; Morais, A.; Amaro, T.; Longatto-Filho, A.; Baltazar, F. CD147 and MCT1-potential partners in bladder cancer aggressiveness and cisplatin resistance. Mol. Carcinog. 2015, 54, 1451–1466. [Google Scholar] [CrossRef]
- Afonso, J.; Santos, L.L.; Morais, A.; Amaro, T.; Longatto-Filho, A.; Baltazar, M.D.F.M. Metabolic coupling in urothelial bladder cancer compartments and its correlation to tumor aggressiveness. Cell Cycle 2016, 15, 368–380. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Chuang, H.-W.; Kuo, W.-T.; Lin, B.-S.; Chang, Y.-C. Current Development and Application of Anaerobic Glycolytic Enzymes in Urothelial Cancer. Int. J. Mol. Sci. 2021, 22, 10612. [Google Scholar] [CrossRef]
- Zhan, T.; Digel, M.; Küch, E.-M.; Stremmel, W.; Füllekrug, J. Silybin and dehydrosilybin decrease glucose uptake by inhibiting GLUT proteins. J. Cell. Biochem. 2011, 112, 849–859. [Google Scholar] [CrossRef]
- Sun, Y.; Guan, Z.; Zhao, W.; Jiang, Y.; Li, Q.; Cheng, Y.; Xu, Y. Silibinin suppresses bladder cancer cell malignancy and chemoresistance in an NF-kappaB signal-dependent and signal-independent manner. Int. J. Oncol. 2017, 51, 1219–1226. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor, J.A. The Role of Pyruvate Dehydrogenase Kinase-4 (PDK4) in Bladder Cancer and Chemoresistance. Mol. Cancer Ther. 2018, 17, 2004–2012. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. Gynecol. Oncol. 2017, 109, djx071. [Google Scholar] [CrossRef]
- Kim, J.; You, Y.-J. Regulation of organelle function by metformin. IUBMB Life 2017, 69, 459–469. [Google Scholar] [CrossRef]
- Peng, M.; Huang, Y.; Tao, T.; Peng, C.-Y.; Su, Q.; Xu, W.; Darko, K.O.; Tao, X.; Yang, X. Metformin and gefitinib cooperate to inhibit bladder cancer growth via both AMPK and EGFR pathways joining at Akt and Erk. Sci. Rep. 2016, 6, 28611. [Google Scholar] [CrossRef]
- Kozal, K.; Jozwiak, P.; Krzeslak, A. Contemporary Perspectives on the Warburg Effect Inhibition in Cancer Therapy. Cancer Control. J. Moffitt Cancer Cent. 2021, 28, 10732748211041243. [Google Scholar] [CrossRef]
- Amin, M.; Srigley, J.; Grignon, D.; Reuter, V.; Humphrey, P.; Cohen, M.; Hammond, M. Urinary Bladder Cancer Protocols and Checklists; College of American Pathologists: Northfield, IL, USA, 2005. [Google Scholar]
- Edge, S.; Byrd, D.; Compton, C.; Fritz, A.; Greene, F.; Trotti, A. AJCC Cancer Staging Manual; Springer: New York, NY, USA, 2010. [Google Scholar]
- Eble, J.; Sauter, G.; Epstein, J.; Sesterhenn, I. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs; IARC Press: Lyon, France, 2004. [Google Scholar]
- Ansari, A.A.; Park, I.; Kim, I.; Park, S.; Ahn, S.-M.; Lee, J.-L. Genomics of drug sensitivity in bladder cancer: An integrated resource for pharmacogenomic analysis in bladder cancer. BMC Med. Genom. 2018, 11, 88. [Google Scholar] [CrossRef]
- Pinto-Leite, R.; Carreira, I.; Melo, J.; Ferreira, S.I.; Ribeiro, I.; Ferreira, J.; Filipe, M.; Bernardo, C.; Arantes-Rodrigues, R.; Oliveira, P.; et al. Genomic characterization of three urinary bladder cancer cell lines: Understanding genomic types of urinary bladder cancer. Tumor Biol. 2014, 35, 4599–4617. [Google Scholar] [CrossRef]
- Warrick, J.I.; Walter, V.; Yamashita, H.; Shuman, L.; Amponsa, V.O.; Zheng, Z.; Chan, W.; Whitcomb, T.L.; Yue, F.; Iyyanki, T.; et al. FOXA1, GATA3 and PPARɣ Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Sci. Rep. 2016, 6, 38531. [Google Scholar] [CrossRef]
- Michaelis, M.; Wass, M.N.; Cinatl, J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resist 2019, 2, 447–456. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Löschmann, N.; Voges, Y.; Breitling, R.; A von Deimling, A.; Rödel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef]
- Jilani, S.M.; Murphy, T.J.; Thai, S.N.; Eichmann, A.; Alva, J.A.; Iruela-Arispe, M.L. Selective Binding of Lectins to Embryonic Chicken Vasculature. J. Histochem. Cytochem. 2003, 51, 597–604. [Google Scholar] [CrossRef]
- Seon, B.; Haba, A.; Matsuno, F.; Takahashi, N.; Tsujie, M.; She, X.; Harada, N.; Uneda, S.; Tsujie, T.; Toi, H.; et al. Endoglin-targeted cancer therapy. Curr. Drug Deliv. 2011, 8, 135–143. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, B.; Zha, Z.; Qu, W.; Zhao, H.; Yuan, J. Clinicopathological factors in bladder cancer for cancer-specific survival outcomes following radical cystectomy: A systematic review and meta-analysis. BMC Cancer 2019, 19, 716. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Y.; He, W.; Yang, B.; Gou, X. Nomogram for predicting overall survival of patients with bladder cancer: A population-based study. Int. J. Biol. Markers 2020, 35, 29–39. [Google Scholar] [CrossRef]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef] [PubMed]
- Reis, H.; Tschirdewahn, S.; Szarvas, T.; Rübben, H.; Schmid, K.W.; Grabellus, F. Expression of GLUT1 is associated with increasing grade of malignancy in non-invasive and invasive urothelial carcinomas of the bladder. Oncol. Lett. 2011, 2, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Boström, P.; Thoms, J.; Sykes, J.; Ahmed, O.; Evans, A.; van Rhijn, B.G.; Mirtti, T.; Stakhovskyi, O.; Laato, M.; Margel, D.; et al. Hypoxia Marker GLUT-1 (Glucose Transporter 1) is an Independent Prognostic Factor for Survival in Bladder Cancer Patients Treated with Radical Cystectomy. Bl. Cancer 2016, 2, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Al-Maghrabi, J.A.; Qureshi, I.A.; Khabaz, M.N. Immunhistochemical expression of GLUT1 is associated with low grade and low stage of urinary bladder cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 3049–3057. [Google Scholar] [PubMed]
- Whyard, T.; Waltzer, W.C.; Waltzer, D.; Romanov, V. Metabolic alterations in bladder cancer: Applications for cancer imaging. Exp. Cell Res. 2016, 341, 77–83. [Google Scholar] [CrossRef]
- Chung, F.-Y.; Huang, M.-Y.; Yeh, C.-S.; Chang, H.-J.; Cheng, T.-L.; Yen, L.-C.; Wang, J.-Y.; Lin, S.-R. GLUT1 gene is a potential hypoxic marker in colorectal cancer patients. BMC Cancer 2009, 9, 241. [Google Scholar] [CrossRef]
- Hoskin, P.J.; Sibtain, A.; Daley, F.M.; Wilson, G. GLUT1 and CAIX as intrinsic markers of hypoxia in bladder cancer: Relationship with vascularity and proliferation as predictors of outcome of ARCON. Br. J. Cancer 2003, 89, 1290–1297. [Google Scholar] [CrossRef]
- Pragallapati, S.; Manyam, R. Glucose transporter 1 in health and disease. J. Oral. Maxillofac. Pathol. 2019, 23, 443–449. [Google Scholar] [CrossRef]
- Ancey, P.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef]
- Chen, J.; Cao, L.; Li, Z.; Li, Y. SIRT1 promotes GLUT1 expression and bladder cancer progression via regulation of glucose uptake. Hum. Cell 2019, 32, 193–201. [Google Scholar] [CrossRef]
- Li, P.; Yang, X.; Cheng, Y.; Zhang, X.; Yang, C.; Deng, X.; Li, P.; Tao, J.; Yang, H.; Wei, J.; et al. MicroRNA-218 Increases the Sensitivity of Bladder Cancer to Cisplatin by Targeting Glut1. Cell. Physiol. Biochem. 2017, 41, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, H.; Rui, W.; Zhang, N.; Zhu, Y.; Xie, X. TRIM38 triggers the uniquitination and degradation of glucose transporter type 1 (GLUT1) to restrict tumor progression in bladder cancer. J. Transl. Med. 2021, 19, 508. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Nunes, A.; Afonso, J.; Granja, S.; Baltazar, F. Lactate and lactate transporters as key players in the maintenance of the warburg effect. Adv. Exp. Med. Biol. 2020, 1219, 51–74. [Google Scholar] [PubMed]
- Wu, J.; Hong, Y.; Wu, T.; Wang, J.; Chen, X.; Wang, Z.; Cheng, B.; Xia, J. Stromal-epithelial lactate shuttle induced by tumorderived interleukin1beta promotes cell proliferation in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 41, 687–696. [Google Scholar]
- Cruz-Bermudez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.; Garcia-Grande, A.; Coronado, M.; Laine-Menendez, S.; Alfaro, C.; Sanchez, J.; Franco, F.; Calvo, V.; et al. Cancer-associated fibroblasts modify lung cancer metabolism involving ROS and TGF-beta signaling. Free. Radic. Biol. Med. 2019, 130, 163–173. [Google Scholar] [CrossRef]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal Metabolic Reprogramming through Lactate Shuttle Coordinately Influences Tumor-Stroma Interplay. Cancer Res 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Jiang, H.; Wang, L.; Cao, Y.; Liu, P.; Xu, X.; Wang, Y.; Sun, L.; Niu, H. Overexpression of monocarboxylate anion transporter 1 and 4 in T24-induced cancer-associated fibroblasts regulates the progression of bladder cancer cells in a 3D microfluidic device. Cell Cycle 2015, 14, 3058–3065. [Google Scholar] [CrossRef] [PubMed]
- Mezheyeuski, A.; Segersten, U.; Leiss, L.W.; Malmström, P.-U.; Hatina, J.; Östman, A.; Strell, C. Fibroblasts in urothelial bladder cancer define stroma phenotypes that are associated with clinical outcome. Sci. Rep. 2020, 10, 281. [Google Scholar] [CrossRef]
- Falkenberg, K.D.; Rohlenova, K.; Luo, Y.; Carmeliet, P. The metabolic engine of endothelial cells. Nat. Metab. 2019, 1, 937–946. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Hayashi, H.; Sakima, M.; Sato, M. Adenosine triphosphate is a critical determinant for VEGFR signal during hypoxia. Am. J. Physiol. Physiol. 2016, 311, C985–C995. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Hayashi, H.; Yamamura, A.; Nayeem, J.; Sato, M. Hypoxia induces the translocation of glucose transporter 1 to the plasma membrane in vascular endothelial cells. J. Physiol. Sci. 2020, 70, 44. [Google Scholar] [CrossRef] [PubMed]
- Merchan, J.R.; Kovács, K.; Railsback, J.W.; Kurtoglu, M.; Jing, Y.; Piña, Y.; Gao, N.; Murray, T.G.; Lehrman, M.A.; Lampidis, T.J. Antiangiogenic Activity of 2-Deoxy-D-Glucose. PLoS ONE 2010, 5, e13699. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; De Bock, K.; Cantelmo, A.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef]
- El Sayed, S.M.; El-Magd, R.A.; Shishido, Y.; Yorita, K.; Chung, S.P.; Tran, D.H.; Sakai, T.; Watanabe, H.; Kagami, S.; Fukui, K. D-Amino acid oxidase-induced oxidative stress, 3-bromopyruvate and citrate inhibit angiogenesis, exhibiting potent anticancer effects. J. Bioenerg. Biomembr. 2012, 44, 513–523. [Google Scholar] [CrossRef]
- Hu, Y.; Lou, X.; Wang, R.; Sun, C.; Liu, X.; Liu, S.; Wang, Z.; Ni, C. Aspirin, a potential GLUT1 inhibitor in a vascular endothelial cell line. Open Med. 2019, 14, 552–560. [Google Scholar] [CrossRef]
- Sun, X.; Peng, Y.; Zhao, J.; Xie, Z.; Lei, X.; Tang, G. Discovery and development of tumor glycolysis rate-limiting enzyme inhibitors. Bioorganic Chem. 2021, 112, 104891. [Google Scholar] [CrossRef]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, Y.; Li, P.; Tao, J.; Deng, X.; Zhang, X.; Gu, M.; Lu, Q.; Yin, C. A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumor Biol. 2015, 36, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Yang, L.; Cao, M.; Yu, Y.; Zhang, R.; Quan, H.; Jiang, Q.; Hua, Y.; Wei, W.; et al. Single-Cell Sequencing-Enabled Hexokinase 2 Assay for Noninvasive Bladder Cancer Diagnosis and Screening by Detecting Rare Malignant Cells in Urine. Anal. Chem. 2020, 92, 16284–16292. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zeng, J.; Xie, R.; Schulz, M.J.; Tedesco, R.; Qu, J.; Erhard, K.F.; Mack, J.F.; Raha, K.; Rendina, A.R.; et al. Discovery of a Novel 2,6-Disubstituted Glucosamine Series of Potent and Selective Hexokinase 2 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 217–222. [Google Scholar] [CrossRef] [PubMed]
- A Lea, M.; Altayyar, M.; Desbordes, C. Inhibition of Growth of Bladder Cancer Cells by 3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one in Combination with Other Compounds Affecting Glucose Metabolism. Anticancer. Res. 2015, 35, 5889–5899. [Google Scholar]
- Li, Z.; Li, X.; Wu, S.; Xue, M.; Chen, W. Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci. 2014, 105, 951–955. [Google Scholar] [CrossRef]
- Li, T.; Sun, X.; Jiang, X. UCA1 involved in the metformin-regulated bladder cancer cell proliferation and glycolysis. Tumor Biol. 2017, 39, 1010428317710823. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, K.; Shi, L.; Xiang, F.; Tao, K.; Wang, G. Prognostic Significance of the Metabolic Marker Hexokinase-2 in Various Solid Tumors: A Meta-Analysis. PLoS ONE 2016, 11, e0166230. [Google Scholar] [CrossRef]
- Wu, J.; Hu, L.; Wu, F.; Zou, L.; He, T. Poor prognosis of hexokinase 2 overexpression in solid tumors of digestive system: A meta-analysis. Oncotarget 2017, 8, 32332–32344. [Google Scholar] [CrossRef]
- Yang, L.; Yan, X.; Chen, J.; Zhan, Q.; Hua, Y.; Xu, S.; Li, Z.; Wang, Z.; Dong, Y.; Zuo, D.; et al. Hexokinase 2 discerns a novel circulating tumor cell population associated with poor prognosis in lung cancer patients. Proc. Natl. Acad. Sci. USA 2021, 118, e2012228118. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, M.; Cong, Q.; Zhang, M.; Zhang, M.; Lu, Y.; Xu, C. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int. J. Biochem. Cell Biol. 2018, 95, 9–16. [Google Scholar] [CrossRef]
- Yang, H.; Hou, H.; Zhao, H.; Yu, T.; Hu, Y.; Hu, Y.; Guo, J. HK2 Is a Crucial Downstream Regulator of miR-148a for the Maintenance of Sphere-Forming Property and Cisplatin Resistance in Cervical Cancer Cells. Front. Oncol. 2021, 11, 794015. [Google Scholar] [CrossRef]
- Mansour, M.A.; Ibrahim, W.M.; Shalaan, E.S.; Salama, A.F. Combination of arsenic trioxide and cisplatin synergistically inhibits both hexokinase activity and viability of Ehrlich ascites carcinoma cells. J. Biochem. Mol. Toxicol. 2019, 33, e22350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [PubMed]
- Nogués, A.; Gallardo-Vara, E.; Zafra, M.P.; Mate, P.; Marijuan, J.L.; Alonso, A.; Botella, L.M.; Prieto, M.I. Endoglin (CD105) and VEGF as potential angiogenic and dissemination markers for colorectal cancer. World J. Surg. Oncol. 2020, 18, 99. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2017, 8, 56081–56094. [Google Scholar] [CrossRef]
- Meng, Y.-M.; Jiang, X.; Zhao, X.; Meng, Q.; Wu, S.; Chen, Y.; Kong, X.; Qiu, X.; Su, L.; Huang, C.; et al. Hexokinase 2-driven glycolysis in pericytes activates their contractility leading to tumor blood vessel abnormalities. Nat. Commun. 2021, 12, 6011. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Bénéteau, M.; Zunino, B.; Jacquin, M.A.; Meynet, O.; Chiche, J.; Pradelli, L.A.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.-E. Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response. Proc. Natl. Acad. Sci. USA 2012, 109, 20071–20076. [Google Scholar] [CrossRef]
- Jalota, A.; Kumar, M.; Das, B.C.; Yadav, A.K.; Chosdol, K.; Sinha, S. Synergistic increase in efficacy of a combination of 2-deoxy-d-glucose and cisplatin in normoxia and hypoxia: Switch from autophagy to apoptosis. Tumor Biol. 2016, 37, 12347–12358. [Google Scholar] [CrossRef]
- Singh, D.; Banerji, A.; Dwarakanath, B.; Tripathi, R.; Gupta, J.; Mathew, T.; Ravindranath, T.; Jain, V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Ishige, N. 2-Deoxyglucose and sorafenib synergistically suppress the proliferation and motility of hepatocellular carcinoma cells. Oncol. Lett. 2017, 13, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Xu, J.; Sun, J.; Huang, H.; Zhang, Z.; Ma, W.; Wan, Z.; Liu, Y.; Pardeshi, A.; Li, S. Co-delivery of 2-Deoxyglucose and a glutamine metabolism inhibitor V9302 via a prodrug micellar formulation for synergistic targeting of metabolism in cancer. Acta Biomater. 2020, 105, 239–252. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.; Porter, R.K.; McNamee, N.; Martinez, V.G.; O’Driscoll, L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci. Rep. 2019, 9, 3788. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; DiPaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Maximchik, P.; Abdrakhmanov, A.; Inozemtseva, E.; Tyurin-Kuzmin, P.; Zhivotovsky, B.; Gogvadze, V. 2-Deoxy-D-glucose has distinct and cell line-specific effects on the survival of different cancer cells upon antitumor drug treatment. FEBS J. 2018, 285, 4590–4601. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Mizushima, N.; Murphy, L.O. Autophagy Assays for Biological Discovery and Therapeutic Development. Trends Biochem. Sci. 2020, 45, 1080–1093. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Kim, S.W.; Park, K.C.; Yun, M. The bifunctional autophagic flux by 2-deoxyglucose to control survival or growth of prostate cancer cells. BMC Cancer 2015, 15, 623. [Google Scholar] [CrossRef]
- Al Hasawi, N.; Alkandari, M.F.; Luqmani, Y.A. Phosphofructokinase: A mediator of glycolytic flux in cancer progression. Crit. Rev. Oncol. 2014, 92, 312–321. [Google Scholar] [CrossRef]
- Zheng, J.; Luo, J.; Zeng, H.; Guo, L.; Shao, G. (125)I suppressed the Warburg effect viaregulating miR-338/PFKL axis in hepatocellular carcinoma. Biomed. Pharmacother. 2019, 119, 109402. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, J.; Le, Y.; Zhou, C.; Zhang, S.; Gong, Z. PFKL/miR-128 axis regulates glycolysis by inhibiting AKT phosphorylation and predicts poor survival in lung cancer. Am. J. Cancer Res. 2016, 6, 473–485. [Google Scholar] [PubMed]
- Zheng, C.; Yu, X.; Liang, Y.; Zhu, Y.; He, Y.; Liao, L.; Wang, D.; Yang, Y.; Yin, X.; Li, A.; et al. Targeting PFKL with penfluridol inhibits glycolysis and suppresses esophageal cancer tumorigenesis in an AMPK/FOXO3a/BIM-dependent manner. Acta Pharm. Sin. B 2021, 12, 1271–1287. [Google Scholar] [CrossRef]
- Sun, C.-M.; Xiong, D.-B.; Yan, Y.; Geng, J.; Liu, M.; Yao, X.-D. Genetic Alteration in Phosphofructokinase Family Promotes Growth of Muscle-Invasive Bladder Cancer. Int. J. Biol. Markers 2016, 31, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.-C.; Vandekeere, S.; Bouché, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7, 12240. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; Ma, Q.; Zhang, X.; Yang, Q.; Wang, L.; Cao, Y.; Xu, Z.; Tawfik, A.; Sun, Y.; et al. Glycolysis links reciprocal activation of myeloid cells and endothelial cells in the retinal angiogenic niche. Sci. Transl. Med. 2020, 12, eaay1371. [Google Scholar] [CrossRef]
- Israelsen, W.; Heiden, M.V. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef]
- Chhipa, A.S.; Patel, S. Targeting pyruvate kinase muscle isoform 2 (PKM2) in cancer: What do we know so far? Life Sci. 2021, 280, 119694. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, X.; Mo, L.; Liu, Y.; He, F.; Zhang, F.; Huang, K.-H.; Wu, X.-R. Role of isoenzyme M2 of pyruvate kinase in urothelial tumorigenesis. Oncotarget 2016, 7, 23947–23960. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wang, X.; Liu, Y.; Shapiro, E.; Lepor, H.; Tang, M.-S.; Sun, T.-T.; Wu, X.-R. PKM2 Is Essential for Bladder Cancer Growth and Maintenance. Cancer Res. 2022, 82, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Bluemlein, K.; Grüning, N.-M.; Feichtinger, R.G.; Lehrach, H.; Kofler, B.; Ralser, M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget 2011, 2, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Chiavarina, B.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Witkiewicz, A.K.; Birbe, R.; Howell, A.; Pestell, R.G.; Smith, J.; Daniel, R.; Sotgia, F.; et al. Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol. Ther. 2011, 12, 1101–1113. [Google Scholar] [CrossRef]
- Gómez-Escudero, J.; Clemente, C.; García-Weber, D.; Acin-Perez, R.; Millán, J.; Enríquez, J.A.; Bentley, K.; Carmeliet, P.; Arroyo, A.G. PKM2 regulates endothelial cell junction dynamics and angiogenesis via ATP production. Sci. Rep. 2019, 9, 15022. [Google Scholar] [CrossRef]
- Wołącewicz, M.; Hrynkiewicz, R.; Grywalska, E.; Suchojad, T.; Leksowski, T.; Roliński, J.; Niedźwiedzka-Rystwej, P. Immunotherapy in Bladder Cancer: Current Methods and Future Perspectives. Cancers 2020, 12, 1181. [Google Scholar] [CrossRef]
- Wang, X.; Shen, X.; Yan, Y.; Li, H. Pyruvate dehydrogenase kinases (PDKs): An overview toward clinical applications. Biosci. Rep. 2021, 41, BSR20204402. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A., 3rd. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef]
- Kuo, Y.-H.; Chan, T.-C.; Lai, H.-Y.; Chen, T.-J.; Wu, L.-C.; Hsing, C.-H.; Li, C.-F. Overexpression of Pyruvate Dehydrogenase Kinase-3 Predicts Poor Prognosis in Urothelial Carcinoma. Front. Oncol. 2021, 11, 749142. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, G.; Xu, H.; Jin, X.; Tang, T.; Wang, X. Expression and prognostic significance of pyruvate dehydrogenase kinase 1 in bladder urothelial carcinoma. Virchows Arch. 2020, 477, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Terado, T.; Tambe, Y.; Mukaisho, K.; Kageyama, S.; Kawauchi, A.; Inoue, H. Cryptotanshinone, a novel PDK 4 inhibitor, suppresses bladder cancer cell invasiveness via the mTOR/betacatenin/Ncadherin axis. Int. J. Oncol. 2021, 59, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, X.; Ji, Y.; Li, X.; Li, Y.; Yu, D.; Yuan, Y.; Liu, J.; Li, H.; Zhang, M.; et al. Pyruvate dehydrogenase expression is negatively associated with cell stemness and worse clinical outcome in prostate cancers. Oncotarget 2017, 8, 13344–13356. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Lv, J.; Zhou, Z.; Wang, J.; Yu, H.; Lu, H.; Yuan, B.; Han, J.; Zhou, R.; Zhang, X.; Yang, X.; et al. Prognostic Value of Lactate Dehydrogenase Expression in Different Cancers: A Meta-Analysis. Am. J. Med. Sci. 2019, 358, 412–421. [Google Scholar] [CrossRef]
- Cheng, H.; Hao, Y.; Gao, Y.; He, Y.; Luo, C.; Sun, W.; Yuan, M.; Wu, X. PLCepsilon promotes urinary bladder cancer cells proliferation through STAT3/LDHA pathwaymediated glycolysis. Oncol. Rep. 2019, 41, 2844–2854. [Google Scholar]
- Jiang, F.; Ma, S.; Xue, Y.; Hou, J.; Zhang, Y. LDH-A promotes malignant progression via activation of epithelial-to-mesenchymal transition and conferring stemness in muscle-invasive bladder cancer. Biochem. Biophys. Res. Commun. 2016, 469, 985–992. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UBC—Global Expression | Non-Neoplastic Sections | Tumour Stroma | Tumour Blood Vessels | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Biomarker | n | Positive (%) | n | Positive (%) | p1 | n | Positive (%) | p2 | n | Positive (%) | p3 |
| GLUT1 | 76 | 18 (23.7) | 8 | 0 (0.0) | 0.192 | 76 | 7 (9.2) | 0.346 | 76 | 14 (18.4) | 0.083 |
| HK2 | 76 | 27 (35.5) | 8 | 0 (0.0) | 0.050 | 76 | 2 (2.6) | 1.000 | 76 | 1 (1.3) | 0.355 |
| PFKL | 73 | 65 (89.0) | 8 | 8 (100.0) | 1.000 | 73 | 56 (76.7) | 0.014 | 73 | 57 (78.1) | 0.011 |
| PKM2 | 76 | 47 (61.8) | 8 | 5 (62.5) | 1.000 | 76 | 67 (88.2) | 0.023 | 76 | 76 (100.0) | 1.000 |
| pPDH | 73 | 64 (87.7) | 8 | 7 (87.5) | 1.000 | 73 | 58 (79.5) | 1.000 | 73 | 68 (93.2) | 1.000 |
| LDHA | 69 | 31 (44.9) | 8 | 0 (0.0) | 0.019 | 68 | 22 (32.4) | 0.305 | 69 | 45 (65.2) | 0.015 |
| Biomarker | Clinicopathological Associations * |
|---|---|
| GLUT1 | Global expression|T3/T4 stage (p = 0.010), MI tumours (p = 0.042), LVI occurrence (p = 0.003) Normoxic areas|T3/T4 stage (p = 0.026), LVI occurrence (p = 0.002) Hypoxic areas|T3/T4 stage (p = 0.011) Tumour stroma|LVI occurrence (p = 0.048) Blood vessels|LVI occurrence (p = 0.010) |
| HK2 | - |
| PFKL | Blood vessels|LVI occurrence (p = 0.044) |
| PKM2 | - |
| pPDH | Tumour stroma|T3/T4 stage (p = 0.022), MI tumours (p = 0.016), LVI occurrence (p = 0.002) |
| LDHA | Tumour stroma|T3/T4 stage (p = 0.019) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonso, J.; Gonçalves, C.; Costa, M.; Ferreira, D.; Santos, L.; Longatto-Filho, A.; Baltazar, F. Glucose Metabolism Reprogramming in Bladder Cancer: Hexokinase 2 (HK2) as Prognostic Biomarker and Target for Bladder Cancer Therapy. Cancers 2023, 15, 982. https://doi.org/10.3390/cancers15030982
Afonso J, Gonçalves C, Costa M, Ferreira D, Santos L, Longatto-Filho A, Baltazar F. Glucose Metabolism Reprogramming in Bladder Cancer: Hexokinase 2 (HK2) as Prognostic Biomarker and Target for Bladder Cancer Therapy. Cancers. 2023; 15(3):982. https://doi.org/10.3390/cancers15030982
Chicago/Turabian StyleAfonso, Julieta, Céline Gonçalves, Marta Costa, Débora Ferreira, Lúcio Santos, Adhemar Longatto-Filho, and Fátima Baltazar. 2023. "Glucose Metabolism Reprogramming in Bladder Cancer: Hexokinase 2 (HK2) as Prognostic Biomarker and Target for Bladder Cancer Therapy" Cancers 15, no. 3: 982. https://doi.org/10.3390/cancers15030982
APA StyleAfonso, J., Gonçalves, C., Costa, M., Ferreira, D., Santos, L., Longatto-Filho, A., & Baltazar, F. (2023). Glucose Metabolism Reprogramming in Bladder Cancer: Hexokinase 2 (HK2) as Prognostic Biomarker and Target for Bladder Cancer Therapy. Cancers, 15(3), 982. https://doi.org/10.3390/cancers15030982