Simple Summary
The PI3K/AKT pathway is one of the most important signaling nodes in cancer. While class I PI3K roles in cancer are well known, class II and III functions in physiology and physiopathology are poorly studied. Moreover, the interactions between pathways controlled by all three classes are not fully understood. The understanding of the mechanisms behind their cooperative functions could be key for efficient PI3K combination targeting in cancer therapy. This review will focus on the recent advances on the roles of the different classes of PI3K in cancer biology, their cross-regulations and their targeting in preclinical models.
Abstract
Phosphatidylinositol-3-kinase (PI3K) enzymes, producing signaling phosphoinositides at plasma and intracellular membranes, are key in intracellular signaling and vesicular trafficking pathways. PI3K is a family of eight enzymes divided into three classes with various functions in physiology and largely deregulated in cancer. Here, we will review the recent evidence obtained during the last 5 years on the roles of PI3K class I, II and III isoforms in tumor biology and on the anti-tumoral action of PI3K inhibitors in preclinical cancer models. The dependency of tumors to PI3K isoforms is dictated by both genetics and context (e.g., the microenvironment). The understanding of class II/III isoforms in cancer development and progression remains scarce. Nonetheless, the limited available data are consistent and reveal that there is an interdependency between the pathways controlled by all PI3K class members in their role to promote cancer cell proliferation, survival, growth, migration and metabolism. It is unknown whether this feature contributes to partial treatment failure with isoform-selective PI3K inhibitors. Hence, a better understanding of class II/III functions to efficiently inhibit their positive and negative interactions with class I PI3Ks is needed. This research will provide the proof-of-concept to develop combination treatment strategies targeting several PI3K isoforms simultaneously.
1. Introduction
In humans, phosphatidylinositol-3-kinases (PI3Ks) comprise eight members that are divided into three classes. All PI3Ks catalyze a similar reaction, phosphorylation of phosphatidylinositols (PI) in position 3 of the inositol ring. However, they have different substrates, products and functions [1,2]. Class I PI3Ks are predominantly studied and play a critical role in cancer. The selective roles of those isoforms were exhaustively reviewed elsewhere [1,2,3]. Several inhibitors targeting class I PI3K are under development or have been clinically approved for cancer treatment. The current clinical application of PI3K inhibitors was recently reviewed by us and others [4,5]. On the contrary, class II and III PI3K physiopathological roles are still poorly understood. In the era of targeted therapies in cancer, it remains crucial to identify the specific PI3K isoform to target in each tumor to achieve an efficient clinical response (Figure 1). In this review, we will focus on the recent advances on the roles of the different classes of PI3K in preclinical models of cancer, their targeting and their cross-regulations. This knowledge is necessary and will inform our discussion on strategies to enhance the clinical efficacy of PI3K inhibitors, targeting one or several PI3K isoforms.
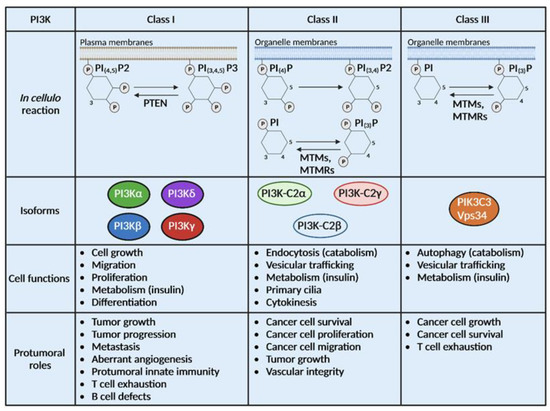
Figure 1.
PI3Ks are divided into 3 classes with different substrates and functions. Class I, II and III PI3Ks catalyze the same biochemical reaction on distinct substrates localized at the plasma membrane or the organelle membrane. Some phosphatases can catalyze the opposite reaction of PI3Ks such as Phosphatase and Tensin homolog (PTEN), myotubularin (MTM) and MTM-related protein (MTMR). Class I is composed of 4 isoforms (PI3Kα, β, δ and γ), class II of 3 isoforms (PI3K-C2α, β and γ) and class III has only one isoform, Vps34. Physiological and pro-tumoral functions of class I PI3Ks are well described while limited information is available on the roles of class II and III PI3Ks in cancer.
2. General Background on Clinical Use of Class I PI3K Isoform Inhibitors in Cancer and Rational of the Review
Class I PI3Ks comprise PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ. Their respective catalytic subunits are encoded by PIK3CA, PIK3CB, PIK3CG or PIK3CD. PI3Kα and PI3Kβ are ubiquitously expressed while PI3Kγ and PI3Kδ are highly expressed in immune cells [1,2,3]. Low levels of PI3Kγ and PI3Kδ play selective roles in some cell types (e.g., vascular and lymphatic endothelial cells, cardiomyocytes). All class I PI3Ks phosphorylate phosphatidylinositol-4,5-bisphosphate (PIP2) into PIP3. The activity of class I PI3Ks can be reverted by the tumor suppressor PTEN, a phosphatase which hydrolyses PIP3 into PIP2. PIP3 next acts as a second messenger in the cell that activates the AKT/mTOR pathways [1].
The PI3K/AKT/mTOR pathway regulates multiple functions such as proliferation, migration, differentiation or metabolism [1]. It is now considered as one of the most mutated pathways in cancer, justifying the development of inhibitors of its upstream members, such as PI3Ks [6].
The first clinical trials using inhibitors targeting all PI3K isoforms (pan-PI3K inhibitors) in monotherapy showed disappointing results due to toxicities and resistance to treatment [4]. Indeed, the use of the pan-PI3K inhibitors LY-294002, PX-866, BKM-120, GDC-0941 and NVP-BEZ235 triggers an AKT reactivation after transient inhibition in colorectal tumor cells [7] or in pancreatic cancer cell lines [8]. In non-small cell lung cancer cell lines, the pan-PI3K inhibitors BKM-120, LY294002 and NVP-BEZ235 are responsible for the increase of MET tyrosine kinase expression and activation. This leads to the activation of signal transducer and activator of transcription 3 (STAT3) and treatment failure [9]. Moreover, combining pan-PI3K inhibitors with classical chemotherapies or other targeted therapies heightens the risk of multiple adverse effects [4].
To decrease feedback responses and toxicity events, isoform-specific PI3K inhibitors have been and are being developed. They may be tolerated at doses resulting in more complete target inhibition with less resistance and fewer adverse effects. This therapeutic strategy was supported by the fact that, even though all class I PI3Ks catalyze the same reaction, they show distinct relevance in each organ and in each tumor type [2]. The first two inhibitors that were approved for clinical use were PI3Kδ and PI3Kα-specific/selective inhibitors [10,11]. Later, pharmacological research allowed the development of dual- or triple-selective inhibitors; the latter being called isoform-sparing inhibitors. The rationale behind this development is based on findings demonstrating that while each PI3K isoform has selective roles, they also have synergistic actions in a tumor-intrinsic or extrinsic manner and/or can be re-activated as an adaptive response to PI3K isoform-specific inhibition. Hence, it becomes clear that PI3K targeting strategies should aim to precisely target PI3K isoforms in combination depending on their relative importance in each tumor. To reach this aim, recent efforts have been put forward to understand which mutational or expression profiles could lead the choice of using PI3Kα and PI3Kβ inhibitors.
The aim of the review is to provide state-of-the art knowledge on those interconnections between isoforms focusing mainly on preclinical models.
3. Mutation Profiles of Cancer Cells and Sensitivity to PI3Kα or PI3Kβ Inhibitors, Related Issues
PI3Kα and PI3Kβ are ubiquitously expressed but have different functions and relative importance in organs and cancers [3]. The importance of genetic alterations to trigger PI3Kα or PI3Kβ dependency is the most described and understood mechanism of selective PI3K activation. However, they do not predict nor fully explain the sensitivity to PI3K inhibitors (e.g., for PI3Kα inhibitors: [12,13]), suggesting that this simple model might not be enough to explain the sensitivity to isoform-selective inhibitors.
3.1. Oncogenic Mutation of PIK3CA and Sensitivity to PI3Kα Inhibitors
The PI3Kα-specific inhibitor alpelisib (PIQRAY or BYL-719) recently obtained approval for PIK3CA-mutated hormone receptor-positive advanced breast cancer in combination with fulvestrant, a hormonotherapy [11]. A new generation of PI3Kα-specific inhibitors has been developed with GDC-0077 (inavolisib) [14]. This drug shows strong anti-tumor activity in breast cancer by inducing the degradation of the specific mutant-p110α (a catalytic subunit of PI3Kα) in an HER2-dependent manner [14]. This result, based on years of preclinical research [15], demonstrates that PI3K inhibitors are efficient in the clinic when the feedback mechanism upon their inhibition is targeted and is a main driver of oncogenesis. The genetic mutation of the PI3Kα encoding gene is not enough to drive tumor progression; a context-dependent activation of PI3Kα is needed.
Preclinical models and early phase clinical trials demonstrate that PI3Kα-specific inhibitors exhibit anti-tumor efficacy in cancers with frequent PIK3CA oncogenic mutations, such as lung and colon cancers, but also in hematological cancers such as juvenile myelomonocytic leukemia, acute myeloid leukemia (AML) or chronic lymphocytic leukemia (CLL) [4]. Likewise, PI3Kα-specific inhibition induces cell death in PI3KCA mutated endometrial cell lines; the clinical relevance of this finding is still unclear [16].
3.2. Context-Dependent Sensitivity to PI3Kα Inhibitors
Preclinical investigations showed that PI3Kα inhibitors have higher target inhibition in cell lines and xenografts with PIK3CA alterations; these findings were confirmed in the clinic in metastatic breast cancer (MBC) patients undergoing hormonotherapy [11,15]. Our recent study suggests that in contrast to MBC, the PI3K pathway could drive pancreatic cancer metastatic progression despite the absence of PIK3CA oncogenic mutations. Patients with a PI3Kα activation gene signature are enriched in the most aggressive subtype of pancreatic ductal adenocarcinoma (PDAC) patients. Pharmacological and genetic PI3Kα inactivation demonstrate that PI3Kα is crucial for the development, progression and metastasis of pancreatic cancer [17]. This cancer presents a low frequency of PIK3CA mutations (5%) but a high percentage of KRAS oncogenic alterations (90%), responsible for the global activation of the PI3K/AKT pathway [18].
Interestingly, the detection of a PIK3CA mutation in circulating tumor DNA in MBC patients treated with alpelisib was linked to significantly improved progression-free survival under treatment, suggesting a context-dependent activation of PI3K in metastatic processes [19].
Additionally, other mutations/genetic alterations might need to be targeted to improve the efficiency of PI3Kα-specific inhibitors. Indeed, in most cases, PI3Kα inhibitors are more efficient in combination strategies. In prostate cancer, PIK3CA oncogenic mutations were found to cooperate with PTEN loss to accelerate tumor growth and facilitate castration resistance [20]. In pancreatic cancer, we found that co-treatment with PI3Kγ inhibitors prevents the feedback re-activation of AKT after PI3Kα inhibition [8]. In rhabdomyosarcoma cells, the combined inhibition of PI3Kα and δ, using alpelisib and idelalisib, respectively, synergistically inhibited cell survival, highlighting the potential of targeting several class I PI3K isoforms [21]. In the case of multi-isoform inhibitors, genomic analyses of the PI3Kβ-sparing taselisib-treated patients identified mutations potentially associated with an upfront resistance to PI3K inhibition (TP53 and PTEN) and post-progression through the reactivation of the PI3K pathway (PTEN, STK11 and PIK3R1) [12], possibly leading to an activation of PI3Kβ in this context. In Philadelphia chromosome-positive CLL, the PI3Kα-specific inhibitor alpelisib exhibited a synergy with Bcr-Abl-targeted drugs [22]. Alpelisib also showed synergistic anti-tumor activity with paclitaxel in gastric cancer with higher effects in PIK3CA-mutant cells [23]. A phase I clinical trial showed an indication of a synergy between the PI3Kα-specific inhibitor alpelisib and the PARP inhibitor olaparib in epithelial ovarian cancer [24].
PI3Kα inhibition not only affects tumor cells but also modulates the tumor microenvironment. The PI3Kα-specific inhibitor CYH33 enhances CD4+ and CD8+ T cell infiltration, inhibits the proliferation of M2 pro-tumoral macrophages and activates the differentiation of macrophages into an M1 phenotype. Likewise, CYH33 accelerates fatty acid metabolism in the tumor microenvironment which activates CD8+ T cell activity. The combined inhibition of fatty acid synthase (FASN) and PI3Kα triggers a synergistic anti-tumor effect in 4T1 breast cancer cells, further reinforcing the need to use isoform-specific PI3K inhibitors in combination with other targeted therapies in cancer [25].
3.3. Loss of PTEN and Limited Sensitivity to PI3Kβ Inhibitors when Used in Monotherapy
PTEN-deficient tumors rely mainly on PI3Kβ rather than PI3Kα [3], yet the underlying mechanisms of such selectivity are still unclear. The inhibition of PI3Kβ by AZD8186 in PTEN-null tumors modifies metabolic pathways by inhibiting the expression of enzymes from the cholesterol biosynthesis pathway [26]. These data suggest that the codependency of pro-tumoral actions due to PIK3β activation and PTEN loss could be attributed to their common activities in metabolic pathways.
Clinical research related to the use of PI3Kβ inhibitors is more advanced in prostate or breast cancers; nonetheless, PI3Kβ-specific inhibitors are promising therapies for other PTEN-deficient cancers such as melanoma, glioblastoma and bladder. Similar to PI3Kα targeting, PI3Kβ targeting seems to be more clinically efficient when used in combination treatments. Indeed, a phase I/II clinical trial evaluating the PI3Kβ inhibitor AZD8186 as a monotherapy or in combination with abiraterone acetate, an androgen inhibitor, showed preliminary data of anti-tumor activity in metastatic castrate-resistance prostate cancer [27]. In preclinical models of PTEN-deficient tumors, PI3Kβ inhibitors (AZD8186, AZD6482) yielded a higher efficacy when administered in combination with other therapeutic agents such as the mTOR inhibitor (prostate and other cancers [28]), PI3Kα inhibitor and fulvestrant (ER-positive breast cancers, [29]), paclitaxel and anti-PD1 treatment (triple negative breast cancer, [30]), epidermal growth factor receptor (EGFR) inhibitors (triple negative breast cancer, [31]), inhibitor of mixed-lineage protein kinase 3 (MLK3) (glioblastoma, [32,33]), MEK inhibitor selumetinib (malignant mesotheliomas, [34]) or antibodies directed against OX40, a T-cell costimulatory receptor melanoma [35].
3.4. Context-Dependent Sensitivity to PI3Kβ Inhibitors
PTEN genetic alterations are not solely responsible for the dependency on PI3Kβ inhibitors. Other teams have demonstrated the relevance of PI3Kβ in breast cancer metastasis, independently from its PTEN status [3,36]. Genetic alterations in PIK3CB, the gene encoding for the PI3Kβ catalytic subunit, could lead to a dependency on PI3Kβ signaling. A phase I clinical trial assessing the highly selective PI3Kβ-specific inhibitor GSK2636771 in PTEN-deficient advanced solid tumors showed a manageable safety profile with evidence of clinical activity in tumors harboring activating mutations in PIK3CB [37].
Though genetic mutations and epigenetic alterations of class I PI3Ks can result in their overactivation, the tumor context might also be permissive for its activation in the absence of oncogenic PI3K mutants. Recently, we highlighted the role of PI3K in mechanotransduction, a process in which mechanical forces (tensile, shear and compression) are transduced in biochemical signals [38]. Knowledge on PI3K isoform selectivity in mechanotransduction will allow the development of clinical applications using small molecule inhibitors in selective mechanical contexts. For example, the increase of YAP/TAZ gene signature transcriptional activity (a classical marker of tensile stress) is predictive of PI3Kβ isoform-selective KIN-193 efficacy in cancer [39]. Similarly, E-cadherin is a critical component of normal epithelial cell/cell adhesion and is known to inhibit YAP/TAZ, while the loss of E-cadherin expression in cancer increases sensitivity to PI3Kβ inhibition [40]. PI3Kα-driven YAP/TAZ activation, possibly through EGF-EGFR-PI3K or FAK–Src–PI3K activation sequences [41,42], is insufficient to promote tumor formation, whereas PI3Kβ-driven YAP/TAZ activation, likely via pertussis toxin-sensitive GPCRs-PI3K activation [43], allows tumor formation [44]. Overall, these findings could pave a way to novel leads of the predictive markers of PI3Kβ inhibitor efficiency.
In sum, PI3Kα and PI3Kβ have different roles in organs or in cell types (e.g., in endothelial cells [45] and in germinal cells [46]), but also in tumors with the same genetic alteration (PTEN heterozygous loss [47]). Their roles in cancer depending on the mutational context are now better understood (Figure 2). PIK3CA mutations or alterations in the tyrosine kinase receptor EGFR or KRAS signaling pathway would direct the treatment strategy towards drugs targeting PI3Kα. Meanwhile, the loss of PTEN would lead to approaches using PI3Kβ-specific inhibitors. Similarly, PTEN loss in breast cancer is responsible for the resistance to the PI3Kβ-sparing-inhibitor taselisib and CDK4/6 inhibitors [48]. At the moment, these proposed rules are insufficient to determine the appropriate administration of PI3K isoform-selective inhibitors. In a recent study, we find that even PTEN-depleted PDAC cell lines require a minimal PI3Kα activity to migrate [17]. The tumorigenic action of the knock-out of PTEN in the thyroid is protected by PI3Kα but not by PI3Kβ genetic inactivation [47]. The context-specific activation of PI3Kα or PI3Kβ in cancer requires further studies and characterization (Figure 2).
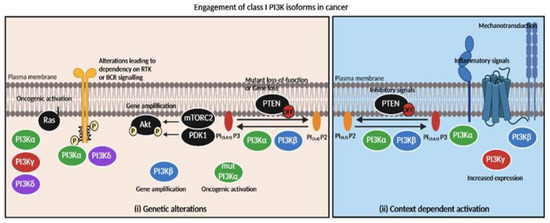
Figure 2.
Engagement of class I PI3K isoforms in cancer depending on genetic alterations or context-dependent activation. Tumors’ specific dependency on PI3K inhibitors is mainly based on (i) genetic alterations and (ii) context-dependent activation of isoforms. (i) Oncogenic activation of PI3Kα catalytic domain leads to PI3Kα sensitivity while PI3Kβ gene amplification or PTEN loss is responsible for PI3Kβ inhibitors’ efficacy. RAS oncogenic activation directly couples with PI3Kα,γ or δ isoforms (but not PI3Kβ) [49,50]. (ii) GSK3β-dependent inhibitory phosphorylation of PTEN could possibly increase sensitivity to PI3Kα or PI3Kβ inhibitors. Inflammatory context (e.g., TNFα [17], GPCR ligand S1P [43]) activates PI3Kα or PI3Kβ/PI3Kγ, respectively. Mechanical signals possibly promote PI3Kβ activation [40]. Increased expression of PI3Kγ promotes PI3K activation [51].
While the choice of using PI3Kα or PI3Kβ inhibitors was lately made depending on the mutational context, PI3Kγ and PI3Kδ remain the target of choice in hematologic cancers due to their high expression in these cell types.
4. PI3Kδ and PI3Kγ, Their Key Tumor Intrinsic and Extrinsic Roles in Hematologic Cancers
The first PI3K inhibitor to be clinically approved was idelalisib, a PI3Kδ-specific inhibitor that showed anti-tumor activity alone in relapsed indolent lymphoma, or in combination with rituximab (CD20 inhibitor) in chronic lymphocytic leukemia [5,10,52]. Idelalisib, which targets PI3Kδ in the BCR pathway, only generates a partial response in CLL patients, though the concomitant inactivation of PI3Kβ reduces further downstream activation in patient-derived CLL cells [53]. Recent data in murine models of CLL show that PI3Kδ-specific inhibition with idelalisib induces treatment resistance which could be overcome by targeting the insulin-like growth factor 1 receptor (IGF1R) [54]. Similarly, the combination of idelalisib and arsenic trioxide showed a synergy in human acute promyelocytic leukemia NB4 cells when compared to each treatment alone, accentuating the interest to use this PI3Kδ-specific inhibitor in combination in hematological diseases [55].
Other PI3Kδ-specific inhibitors, such as umbralisib, have thus been developed for hematological cancers and are currently being tested in clinical trials. In a phase I clinical trial, umbralisib showed promising signs of clinical efficacy in relapsed or refractory chronic lymphocytic leukemia, small lymphocytic lymphoma, B-cell and T–cell non-Hodgkin’s lymphoma, Hodgkin’s lymphoma and relapsed or refractory indolent lymphoma [56,57]. This inhibitor (also named TGR-1202) synergizes with carfilzomib, a proteasome inhibitor, by reducing c-Myc translation [58]. Umbralisib approval has been recently withdrawn due to high toxicities outweighing the benefits of the treatment, raising the question of its appropriate dosage in patients.
Inhibitors of multiple PI3K isoforms have been developed to mitigate toxicity issues and improve efficacy. In a phase II clinical trial, copanlisib, a PI3K inhibitor targeting preferentially α and δ isoforms, showed clinical efficacy in patients with indolent or aggressive malignant lymphoma [59].
Besides directly targeting lymphoma cells, PI3Kδ/γ inhibitors could act through immunomodulatory actions. A phase I clinical trial showed the promising clinical activity of duvelisib, a PI3Kδ/γ inhibitor in T-cell lymphoma, by reprogramming macrophages (cells enriched in PI3Kγ) and inducing tumor cell-autonomous death [59,60]. In CAR T cell therapy, the ex vivo treatment of T cells with a PI3Kγ/δ inhibitor reduced anti-tumor efficacy, while the single inhibition of each isoform generated cells with higher anti-tumoral activity [61]. These data clearly emphasize the relevance of targeting PI3Kγ or PI3Kδ individually in this context.
Even though PI3Kγ and PI3Kδ roles were first described in immune cells, their overexpression in solid tumors and their potential role in tumor stroma make them promising isoforms to target in all cancers.
5. PI3Kδ and PI3Kγ, Unexpected Roles in Solid Tumors
PI3Kδ is mainly expressed in white blood cells in physiological conditions; nonetheless, its expression has been found in other solid tumors such as liver or breast cancer. In hepatocellular carcinoma, the genetic or pharmacological inhibition of PI3Kδ by idelalisib reduces tumor progression and induces apoptosis via Bim [62,63]. In breast cancer, the pharmacological targeting of PI3Kδ inhibits tumor cell migration and prevents tumor progression by acting on cancer cells and macrophages [64].
A recent clinical trial demonstrated a novel clinical application of the PI3Kδ inhibitor AMG319 in solid cancer (head and neck), through its immunoregulation role. This clinical trial also showed that a modified treatment regimen with the intermittent dosing of a PI3Kδ inhibitor in mouse models led to a significant decrease in tumor growth without inducing autoimmune colitis through the depletion of regulatory T cells in colonic tissue [65], suggesting that alternative dosing regimens might limit toxicity.
Similarly, PI3Kγ might harbor tumor-intrinsic and tumor-extrinsic roles in cancer. PI3Kγ expression was described as specific to the immune cell lineage. Despite that, several laboratories including ours suggest that the expression of PI3Kγ in tumor cells, albeit not strong, plays a critical role in KRASG12D,G12V pancreatic tumor cells [8,51], but also in KRASG12R pancreatic cells (a rarer form of mutant KRAS found in PDAC) [66]. In this latter genetic context, PI3Kγ is not directly activated by KRAS but sustains macropinocytosis, a cellular process that promotes the non-selective uptake of extra-cellular material such as solute molecules, nutrients and antigens in harsh conditions. In pancreatic cancer, the long-term inhibition of PI3Kγ with specific inhibitors induces compensation by other class I PI3K isoforms. In vivo, the targeting of PI3Kα and PI3Kγ using the specific inhibitors BYL-719 and IPI-549, respectively, showed synergistic anti-tumor effects compared to each inhibitor alone. These results confirm the importance of PI3Kγ in pancreatic cancers [51] and highlight the interactions existing between several class I PI3K isoforms [8]. Furthermore, PI3Kγ has recently been identified as an indirect or direct target for retinoblastoma. Indeed, the direct inhibition of its expression using siRNA or indirect inhibition via CANT1 long non-coding RNAs reduced retinoblastoma tumor growth [67].
Targeting PI3Kγ in solid tumors due to its roles in the immune microenvironment has shown promising results. In pancreatic cancer, the combined inhibition of PI3Kγ and colony-stimulating factor-1 receptor (CSF-1R) modified the macrophage balance by decreasing M2 pro-tumor macrophages and increasing M1 anti-tumor macrophages, thus reducing the tumor volume in mice [68]. Such a strategy currently tested in a phase I clinical trial in advanced solid tumors using the PI3Kγ-specific inhibitor IPI-549 in monotherapy or in combination with nivolumab, a programmed cell death protein 1 (PD1) inhibitor, showed promising clinical results with favorable tolerability [69]. In poorly immunogenic head and neck squamous cell carcinoma mice models, the genetic inhibition of Pik3cg, the gene encoding for the PI3Kγ catalytic subunit, did not alter tumor growth nor lymph node metastasis. However, those mice showed an increased CD8+ T-cell tumor infiltration associated with increased IFN-γ, IL-17 and PD-1 expression, suggesting that PI3Kγ inhibition could synergize with immunotherapies targeting PD-L1 in this cancer [70].
Lastly, PI3Kγ inhibition protects from cardiotoxicity induced by doxorubicin by inducing mitophagy triggered by this anthracycline. Moreover, the inhibition of PI3Kγ with the specific inhibitor AS605240 synergizes with the doxorubicin anti-tumor effect by reducing the recruitment of pro-tumoral macrophages in mice models of breast cancer [71].
PI3Kδ and PI3Kγ are now considered the targets of choice in solid tumors, alone or in combination with existing treatments because of their capacity to modulate the immune tumor microenvironment and direct tumor-cell intrinsic actions (Figure 1).
The roles of class I PI3Ks in cancer have been extensively studied and are now better understood, while class II and III PI3Ks remain unexplored.
6. Class II and III PI3Ks, Novel Possibilities to Target Cancer Progression
6.1. Class II PI3Ks and Organismal Functions
Class II PI3Ks consist of three isoforms, PI3K-C2α, PI3K-C2β and PI3K-C2γ, which synthetize in vitro and in cellulo two different products, PIP and PIP2, depending on the substrates. Class II PI3Ks have been largely neglected and their specific functions (Table 1), especially in cancer, are still poorly understood [72] (Figure 1).
Class II PI3Ks are monomeric enzymes consisting of a PI3K catalytic core extended by an N-terminus, a C-terminal phox homology (PX) domain and their characteristic C2 domain at the carboxyl terminus. Class II PI3K-C2α and PI3K-C2β are ubiquitously expressed, while PI3K-C2γ expression is restricted to the liver, breast, prostate, salivary glands and exocrine pancreas [73,74]. The lipid products of class II PI3Ks include PI(3)P and PI(3,4)P2 which are found in intracellular vesicles [75]. Class II PI3Ks mainly regulate intracellular dynamics, membrane trafficking, instead of acting as signal transducers like class I PI3Ks [1]. Knowledge on signaling through agonist dependent pools generated by class II PI3Ks is currently scarce and has only been described in specific settings such as insulin signaling: the role of PI3K-C2α in insulin signaling [76,77] and the role of PI3K-C2γ as a Rab5 effector [74]. Limited data on the modus operandi of class II PI3K has delayed the development of specific inhibitors; consequently, most of the information on their physiological functions has been obtained through genetic studies involving the knockdown of gene expression [78]. The use of genetically engineered mouse models (GEMMs) for class II PI3Ks has been crucial to determine their physiological roles. While PI3KC2α is essential in mice, human patients with inherited homozygous null mutations in the PIK3C2A gene display congenital syndromic features, including kidney failure and cataracts, due to early senescence of cells in the associated tissues [79,80]. This suggests that in humans, PIK3CA loss could be compensated with PI3KC2β encoded by PIK3C2B. Current published GEMMs are described in the following table (Table 1). Present GEMMs and genetic knockdown models have contributed to characterizing specific functions for class II PIKs and allowed the identification of further scaffolding properties of certain class II PI3Ks.

Table 1.
Genetically engineered mouse models of class II PI3Ks (only systemic KO and KI mouse models were included in the table).
Table 1.
Genetically engineered mouse models of class II PI3Ks (only systemic KO and KI mouse models were included in the table).
| PI3K | Genotype | Phenotype | Reference |
|---|---|---|---|
| PIK3C2A PI3K-C2α | PI3K-C2α−/− KO | Embryonic lethal E10.5 Defective vascularity | [81] |
| PIK3C2A PI3K-C2α | PI3K-C2α−/− KO | Embryonic lethal E10.5 Delayed development from E8 Defective vascularity Defective sonic hedgehog signaling | [82] |
| PIK3C2A PI3K-C2α | PI3K-C2αD1268A/wt KI | Only heterozygous are viable Metabolic defects in males: early onset leptin resistance and age-dependent obesity | [83] |
| PIK3C2B PI3K-C2β | PI3K-C2β−/− KO | Viable No overt phenotype | [84] |
| PIK3C2B PI3K-C2β | PI3K-C2βD1212A/D1212A KI | Viable and fertile Enhanced insulin sensitivity and glucose tolerance Resistance to liver steatosis under high-fat diet | [85] |
| PIK3C2G PI3K-C2γ | PI3K-C2γ−/− KO | Viable and fertile Age-dependent insulin resistance Defective insulin response Increased obesity and fatty liver | [74] |
6.2. Class II PI3Ks and Their Role in Cancer
PI3K-C2α is mainly described as regulating cancer cell death and mitosis [86,87], especially by controlling spindle stability [88]. PI3K-C2β is predominantly involved in cancer cell migration and invasion [89,90] but can also control proliferation via action on cyclin B1 expression [91]. The miRNA miR-362-5p can inhibit neuroblastoma cell proliferation and migration by targeting the 3′UTR of PI3K-C2β mRNA [92]. Current findings show that PI3K-C2β accelerates the disassembly of focal adhesion [93], a process that might be critical to drive metastatic dissemination. Likewise, data indicate that the inhibition of PI3K-C2β expression by siRNA delays mitosis in prostate cancer PC3 cells and potentiates the anti-clonogenic effect of docetaxel [94]. To date, PI3K-C2γ roles in cancer remain poorly described: the low expression of PIK3C2G is associated with an increased risk of recurrence and death in colorectal patients treated with oxaliplatin [95]. In pancreatic cancer, PI3K-C2γ expression is reduced in approximately 30% of pancreatic cancer cases. In mice models with pancreatic cancer, PI3K-C2γ loss is associated with an aggressive phenotype and an increased sensitivity to mTOR and glutaminase inhibitors [96]. In cancer settings, so far, the data from mouse models and human samples are concordant. In ovarian cancers, a nonsense mutation of PIK3C2G was identified but no functional analysis was performed [97]. Overall, these data suggest tissue-specific tumor suppressor functions of PI3K-C2γ that need be further confirmed. Notwithstanding, most of these works mostly describe in vitro experiments. More experimental work in integrated models or in more relevant cell models need to be performed to discern the specific roles of class II PI3K in different cancers.
As class II PI3Ks regulate key membrane-based cellular processes (through endocytosis, lysosomal activity or focal adhesion turnover), they might harbor pro- and anti-tumoral functions depending on the stage of the cancer process (the initiation vs. the progression stage). Playing a role in endothelial cell integrity [98], they might have key roles in tumoral vasculature. Their role in other cellular components of the tumor microenvironment also needs to be elucidated.
Ultimately, more in vivo data on the inactivation of those enzymes in a tissue- and time-specific manner and the development of class II-targeting drugs are needed. The latter is necessary to determine whether those compounds could strengthen the anti-tumoral PI3K-targeting arsenal.
6.3. Class III PI3K and Organismal Functions
Vacuole protein sorting 34 (Vps34) is the sole member of the class III PI3K and phosphorylates PI into PI-3-P. Vps34 is ubiquitously expressed and constitutes a heterodimer with Vps15 which has multiple physiological functions (Table 2). The Vps34/Vps15 complex is involved in complex I with Beclin-1 and ATG14, or complex II with Beclin-1 and UVRAG, playing a role in autophagy and endocytic sorting, respectively [1]. Vps34 participates in the regulation of autophagy, endocytosis and phagocytosis, which all converge at the lysosome degradation. During these processes, the activity of Vps34 is essential at the initial stages to produce the PI(3)P pools required for the biogenesis of autophagosomes, endosomes and phagosomes. Later on, it controls their maturation by recruiting effector proteins that promote their fusion with lysosomes [99,100]. Of note, in some physiological contexts such mechanical shear stress, autophagic pools of PI(3)P may also be synthesized by class II PI3Ks, including PI3KC2α [101]. Added to its functions in vesicle traffic, Vps34 has scaffolding properties. In several studies using Vps34-deficient mouse models, the animals presented a reduced expression of Vps34-binding partners, supporting the importance of the scaffolding function of Vps34 in maintaining the stability of the complexes [102]. Importantly, the phenotypes of Vps34 KO or of Vps15 KO do not fully match with the results obtained with the inactive mutant of Vps34 [1]. These findings accentuate the complexity when interpreting these phenotypes which might not be solely attributed to the catalytic activity of the enzyme, but also to its scaffolding properties.
The following table (Table 2) recapitulates the available global KO and KI mouse models, as well as some examples of the main conditional mouse models of Vps34. It is important to mention that only the full deletion of Vps34 leads to important phenotypes in vivo, including severe cardiac effects [102]. For instance, the clinical use of Vps34 inhibitors requires a cautious approach to achieve a suitable therapeutic response.
Table 2.
Genetically engineered mouse models of class III PI3Ks.
Table 2.
Genetically engineered mouse models of class III PI3Ks.
| Genotype | Phenotype | Reference | ||
|---|---|---|---|---|
| Global | Models | PIK3C3−/− KO | Homozygous mice—embryonic lethal E7.5–E8.5 Heterozygous mice are viable and healthy, no overt phenotype | [103] |
| Global | Models | Vps34D761A/+ KI | Homozygous mice—Embryonic lethal E6.5–E8.5 Heterozygous mice are viable and fertile Heterozygous mice display enhanced insulin sensitivity and glucose tolerance | [104] |
| Conditional Model | Deletion of exon 21 (kinase domain-24 amino acids) | PF4-Cre;Vps34fl/fl platelets | Viable mice Abnormalities in platelets Impaired thrombus formation and granule secretion | [105] |
| Conditional Model | Deletion of exon 21 (kinase domain-24 amino acids) | Pax8-Cre;Vps34fl/fl proximal tubular cells (PTC) | Fanconi-like syndrome Vacuolation of PTCs | [106] |
| Conditional model | Deletion of ATP-binding domain | Advilin-Cre;PIK3C3fl/fl neurons | Post-natal lethality after 2 weeks Neurodegeneration—vacuole formation in sensory neurons | [107] |
| Conditional model | Deletion of ATP-binding domain | CaMKII-Cre;PIK3C3fl/fl pyramidal neurons | Loss of synapses Neurodegeneration Extensive gliosis | [108] |
| Conditional model | Deletion of ATP-binding domain | TgCKmm-Cre;PIK3C3fl/fl cardiac and skeletal muscle | Post-natal lethality after 4 weeks Muscular dystrophy Cardiomyopathy | [109] |
| Conditional model | Deletion of ATP-binding domain | Pcp2-Cre;PIK3C3fl/fl bipolar and Purkinje cells | Progressive degeneration of retinal bipolar cells and cerebellar Purkinje cells Reduced cerebella Progressive ataxia | [110] |
| Conditional model | Deletion of ATP-binding domain | Cone-Cre;PIK3C3fl/fl retina cone cells | Progressive retinal degradation (onset 12 weeks) Loss of cone structure and degradation (1.5 months) | [111] |
| Conditional model | Deletion of exon 4 (N-terminus, any functional domain) | Mck-Cre;PIK3C3fl/fl heart | Post-natal lethality after 5 weeks Cardiomegaly | [102] |
| Conditional model | Deletion of exon 4 (N-terminus, any functional domain) | Alb-Cre;PIK3C3fl/fl liver | Post-natal lethality after 1 year Hepatomegaly Hepatic steatosis | [102] |
| Conditional model | Deletion of exon 4 (N-terminus, any functional domain) | Cd4-Cre;PIK3C3fl/fl CD4 and CD8 cells | Viable mice T cell lymphopenia, reduced T cell count Impaired autophagy in T cells | [112] |
6.4. Class III PI3K and Its Role in Cancer
Most of the research on the role of Vps34 in cancer is restricted to studies in cell lines. In cancer cells, this enzyme promotes cell survival and proliferation by inducing autophagy. In human breast cancer cells, Vps34 activates the transcription and the activating phosphorylation of the autophagosome cargo protein p62, contributing to cell oncogenicity [113]. Vps34 inhibitors also improve the sensitivity of breast cancer cells to tyrosine kinase inhibitors sunitinib and erlotinib [114]. In renal tumor cells, the pharmacological inhibition of Vps34 with SAR405 can synergize with everolimus, an mTOR inhibitor that promotes autophagy induction, to reduce cell proliferation [115]. Vps34 regulates iron metabolism (possibly inhibiting ferroptosis and promoting autophagy-induced cell protection) which can modulate RKO colon cancer cell line proliferation/survival [116]. Vps34 contributes to EGFR translocation to the nucleus, inhibiting the pro-apoptotic activity of the Arf promoter in lung tumor cells [117].
In vivo, the genetic or pharmacological inhibition of Vps34 in mice reprograms melanoma and colorectal immune-deprived cold tumors into inflammatory tumors that secrete, amongst others, CCL5 and CXCL10 chemokines [118,119]. This process could be dependent on autophagy regulation. These tumors are then highly infiltrated with natural killer cells and CD8+ T lymphocytes which increase the efficacy of anti-PD-1/PD-L1 immunotherapies [118,119]. Thus, Vps34 activates a tumor-intrinsic immune escape process during tumor development [118,120].
The opposite functions (protection) of autophagy in cancer initiation have also been described [121], yet a clear demonstration of this protective role remains to be provided. Events hinting at such a protective role have been described: Beclin-1 overexpression in breast cancer cell line MCF-7 decreases tumor formation in nude mice, while the mutation in the domain of Beclin-1 interacting with the Vps34 complex reverses these tumor suppression functions [122]. In addition, Bif1 can activate autophagy and act as a tumor suppressor protein by interacting with Beclin-1 and UVRAG [123].
Further in vivo data are required to define the exact functions of tumor-intrinsic and -extrinsic Vps34 on tumor initiation and progression in tissue-specific models. The current published results emphasize the need to follow a cautious strategy when targeting the class III PI3K in cancer, as there are potential severe toxic effects on cardiac and hepatic functions (Table 2). Nevertheless, the partial inactivation of Vps34 by pharmacological means seems to yield beneficial actions [104].
After years of neglect, the functions of class II and III PI3Ks in physiology are now being unraveled. The analysis of genetically engineered mouse models mimicking their pharmacological inhibition and their validation in human samples could uncover the potential toxic action of their respective inhibitors (Table 1 and Table 2).
7. Future Directions
7.1. Understanding Isoform Specificity in Cancer
Although the understanding of PI3K roles and their molecular mechanisms has vastly progressed since their discovery 30 years ago, much remains to be learned. There are several gaps in our knowledge regarding the interaction and organismal roles of PI3Ks. Emerging evidence suggests that lipid pools produced at different cellular locations might not have the same physiological functions. Likewise, there is growing evidence that timing and tissue context greatly impact the type of response that a certain stimuli might elicit [124].
The PI3K isoform specificity is still not fully understood but, so far, it is attributed to different PI3K expression levels, localizations and mutations. Another hypothesis sustains that each PI3K isoform could also act on specific PIP2 sub-species. Indeed, mutations on TP53, the gene encoding for the tumor suppressor protein p53, are responsible for the modification of the PIP3 (produced by PI3Ks) acylation state [125,126]; this could affect the phospholipid membrane localization [127] and could be responsible for differential phosphorylation by each PI3K, explaining the differential effect of isoforms [17,128]. Indeed, isoform selective inhibitors differently modify the distribution of PIP3 species.
Given the key importance of tumor-cell intrinsic and extrinsic metabolism for controlling cancer progression, one key hypothesis that we are pursuing is that the selective role of class I PI3Ks is explained by their selective roles in controlling cell metabolism. This is based on the isoform-selective physiological functions of PI3K identified in metabolic organs [3] but also on the identified selective molecular effectors or cell processes controlled by each isoform [8,66].
7.2. Cooperation between Class I PI3K Isoforms
Cooperation and compensations between class I PI3K isoforms have been widely described; for example, compensations between PI3Kα and PI3Kβ, a redundancy between PI3Kα and PI3Kδ through receptor tyrosine kinase (RTK) or between PI3Kγ and PI3Kβ downstream G-protein coupled receptor (GPCR) [3,4] (Figure 3). Hence, we and others have described: 1—immediate compensations/redundancy between isoforms that are activated through similar mechanisms (e.g., PI3Kα and PI3Kδ downstream RTK [129], or PI3Kγ and PI3Kβ downstream GPCR [43]), or 2—delayed (in a time of 1–2 weeks) compensation/redundancy between the two ubiquitously expressed PI3Ks, PI3Kα and PI3Kβ (e.g., an overexpression of RTK, a mutation of PTEN) [130,131,132]. We recently identified a possible new mode of resistance based on a rapid rewiring network. As described above, recent data show compensatory signals via the AKT pathway upon treatment with PI3Kα or PI3Kγ-specific inhibitors in pancreatic cancer cell lines. The combined inhibition of these two isoforms in subcutaneous mice models of pancreatic cancers showed a synergistic anti-tumor effect compared to each treatment alone [8].
The cooperation between isoforms has implications in the clinical application of PI3K inhibitors.
7.3. Cooperation between Class I at Plasma Membrane and Class II and III at Intracellular Membranes
The different PI3K classes are mainly studied as an individual class of enzymes regulating the phosphorylation of distinct phospholipids. However, the production of those lipids is interconnected, the production of each lipid potentially impacting the others. Therefore, altering one lipid kinase might trigger knock-on effects for the other classes of PI3Ks. This cross-regulation of PI3K classes may be considered to prevent PI3K inhibitor resistance. To our knowledge, this has not been analyzed. In any case, the indirect interactions between pathways and processes controlled by distinct PI3K isoforms is recognized as a line of research that goes beyond the mere regulation of mTORC1 activity.
Undeniably, the most described interconnection between those pathways is indirect and involves the regulation of the mammalian target of rapamycin complex 1 (mTORC1). PIP3 generated by a class I PI3K allows the recruitment of phosphoinositide-dependent kinase-1 (PDK1), mammalian target of rapamycin complex 2 (mTORC2) and AKT to the plasma membrane. PDK1 phosphorylates and activates AKT which subsequently enhances mTORC1 activity. AKT and the downstream effector mTORC1 are already known to inhibit autophagy, for example by phosphorylating and reducing the expression of UVRAG, a protein contained in the Vps34 complex II. Indeed, the pharmacological inhibition of Vps34 by SAR405 reduces autophagy induced by mTOR inhibition [104]. The SUMOylation of PDK1 enhances AKT/mTORC1 activation, which inhibits the autophagic flux. However, Vps34 was known to inhibit PDK1 SUMOylation, and non-SUMOylated PDK1 directly positively regulates an autophagosome’s formation, tethering LC3 to the endoplasmic reticulum to initiate autophagy [133]. The interconnection between class I and class III-regulated mTORC1 activation was also identified upon the long-term inhibition of class I PI3K, demonstrating their interdependence. The prolonged treatment of breast cancer cells with class I PI3K or AKT inhibitors leads to the increased expression and activation of a kinase termed SGK3 which is related to AKT. Under these conditions, SGK3 is controlled by Vps34 that generates PI(3)P, which binds to the PX domain of SGK3 promoting phosphorylation and activation by its upstream PDK1 activator; SGK3 next substitutes AKT by phosphorylating TSC2 to activate mTORC1 [134].
Data also described indirect links between class II PI3K and pathways regulated by class I PI3K with PIK3-C2α activating mTORC1 via Raptor in adipocytes [135] and PI3K-C2β repressing mTORC1 activity via PI(3,4)P2 production [136]. Interestingly, the combined pharmacological inhibition of PI3Kβ with PI3K-C2α downregulation significantly reduced PC3 cell migration, better than with each treatment alone [135]. This suggests that these two isoforms could regulate cell proliferation by distinct molecular pathways.
Previous data showed that PI3Kβ and Vps34 can interact with each other directly or indirectly via Rab5 [137]. On the other hand, PTEN, which preferentially inhibits PI3Kβ products, can be localized in intracellular vesicles and become inactive via its binding to PI(3)P whose level is regulated by Vps34 [138]. Hence, PI3Kβ inhibitor efficiency might be linked to Vps34 basal levels of activity (Figure 3).
In spite of the scarce knowledge, emerging data indicate that class I, II and class III-controlled pathways tightly regulate each other and can act as a rheostat for their respective pathways and functions (i.e., autophagy and AKT/mTOR pathway, but maybe other processes such as macropinocytosis of MHC-I surface receptors) (Figure 3). Although, it is important to note that those cross-regulations were demonstrated in different cell types and contexts. Limited information is available in cancer cells and in in vivo preclinical cancer models. If those regulations are confirmed in cancer settings, further combination treatments could be designed such as the targeting of Vps34 and class I PI3K to control tumoral immunity. Indeed, Vps34 inhibition triggers PD-L1 expression [118] while PI3Kδ induces the expression of PD-1 in CD8+ cells [139]. The combination targeting of class I and II or class I and class III PI3Ks has never been achieved in preclinical models. Further in vitro and in vivo studies are required to determine how different PI3K classes interact in the context of an organ or a tumor. These data are crucial for better targeting of isoforms’ specific functions (Figure 3).

Figure 3.
PI3K cross-regulations and possible class I, II and III PI3Ks combination targeting in cancer. (A). PI3K class I isoform cross-regulations upon inhibition of one isoform. Thick lines = primary inhibition, dashed arrows = subsequent regulations. (B). PI3K cross-regulations among different classes are still poorly understood but based on this knowledge, new therapeutic strategies arise to overcome treatment resistance in cancer. (i) All PI3K classes directly or indirectly regulate mTORC1 activity. Class I PI3Kα, β, γ and δ phosphorylate PIP2 into PIP3 which allow the recruitment of PDK1 to the plasma membrane. PDK1 then activates AKT which subsequently enhances mTORC1 activity. The latter can inhibit PI3K in a negative feedback. Class II PI3K-C2α activates mTORC1 and favors its translocation to the plasma membrane via Raptor association to PIP2 [135,140]. On the contrary, PI3K-C2β produces PI(3,4)P2, allowing the link of 14-3-3 which represses mTORC1 [136]. Loss of PI3K-C2γ is responsible for an mTOR activation [96]. Vps34 allows the amino acid activation of the phospholipase D1 (PLD1) which then translocates to the lysosome and stimulates mTORC1 activity [141] but can also inhibit PDK1 sumoylation which results in less AKT and mTOR activation. (ii) Vps34 regulates autophagy while class I PI3Ks modulate proliferation. Both mechanisms are important for cancer cell survival. (iii) Class III PI3K Vps34 can indirectly modulate PI3Kβ activity by allowing PTEN relocalization to intracellular PI(3)P vesicles whose level is regulated by Vps34 [138]. (iv) Vps34 inhibition triggers PD-L1 expression [118] while PI3Kδ induces the expression of PD-1 in CD8+ cells [139]. “ON” means that the protein is activated while “OFF” means it is inactivated or absent. Red inhibition or activation lines correspond to possible combination therapies that could be inhibitory or activatory. Of note, all these pathways were demonstrated in different cell types and contexts; the large significance of these data is currently unknown.
Figure 3.
PI3K cross-regulations and possible class I, II and III PI3Ks combination targeting in cancer. (A). PI3K class I isoform cross-regulations upon inhibition of one isoform. Thick lines = primary inhibition, dashed arrows = subsequent regulations. (B). PI3K cross-regulations among different classes are still poorly understood but based on this knowledge, new therapeutic strategies arise to overcome treatment resistance in cancer. (i) All PI3K classes directly or indirectly regulate mTORC1 activity. Class I PI3Kα, β, γ and δ phosphorylate PIP2 into PIP3 which allow the recruitment of PDK1 to the plasma membrane. PDK1 then activates AKT which subsequently enhances mTORC1 activity. The latter can inhibit PI3K in a negative feedback. Class II PI3K-C2α activates mTORC1 and favors its translocation to the plasma membrane via Raptor association to PIP2 [135,140]. On the contrary, PI3K-C2β produces PI(3,4)P2, allowing the link of 14-3-3 which represses mTORC1 [136]. Loss of PI3K-C2γ is responsible for an mTOR activation [96]. Vps34 allows the amino acid activation of the phospholipase D1 (PLD1) which then translocates to the lysosome and stimulates mTORC1 activity [141] but can also inhibit PDK1 sumoylation which results in less AKT and mTOR activation. (ii) Vps34 regulates autophagy while class I PI3Ks modulate proliferation. Both mechanisms are important for cancer cell survival. (iii) Class III PI3K Vps34 can indirectly modulate PI3Kβ activity by allowing PTEN relocalization to intracellular PI(3)P vesicles whose level is regulated by Vps34 [138]. (iv) Vps34 inhibition triggers PD-L1 expression [118] while PI3Kδ induces the expression of PD-1 in CD8+ cells [139]. “ON” means that the protein is activated while “OFF” means it is inactivated or absent. Red inhibition or activation lines correspond to possible combination therapies that could be inhibitory or activatory. Of note, all these pathways were demonstrated in different cell types and contexts; the large significance of these data is currently unknown.

7.4. Multi-Isoform Targeting in Cancer
To date, multi-isoform targeting tools are under development but are limited to the targeting of several class I isoforms. Taselisib, a β-sparing inhibitor (targeting all class I isoforms but PI3Kβ) has been recently tested in a phase I clinical trial in PIK3CA-mutated cancers [12]. Only proof-of-concept for the simultaneous targeting of PI3Kδ and Vps34 in AML, CLL and Burkitt lymphoma cell lines was demonstrated [142].
A solution to increase the possibilities of combinations could be to design combi/hybrid molecules, a combination of several existing drugs attached with a linker and designed to target several proteins with less adverse effects and a controlled pharmacokinetic [143,144,145] (Figure 4). Combi/hybrid molecules have been developed to target both PI3Ks and histone deacetylase (HDAC) such as CUDC-907 with relevant anti-tumor activity in neuroblastoma or AML [146,147]. However, no combi/hybrid molecule has yet been developed to target simultaneously several PI3K isoforms among different classes. Those molecules harbor several advantages, such as the possibility to choose linkers whose cleavage can be specifically induced by the intrinsic tumor metabolism, possibly reducing toxicity. Compared to normal cells, tumor cells overactivate class I PI3Ks. Therefore, combi/hybrid molecules targeting class I PI3K and class II or III could also act as tumor cell hunters with higher selectivity and lower toxicities. We would then be able to exclusively target the relevant isoforms and improve the efficacy of PI3K therapies in cancer by using novel methods to determine the potency of combination targeted therapies.
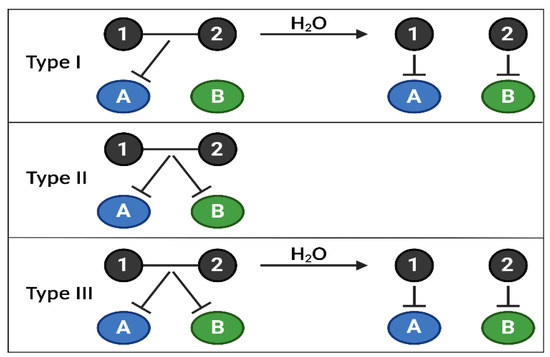
Figure 4.
Combi/hybrid molecules, annihilating several targets at once. The use of targeted therapies in clinics has highlighted the appearance of frequent treatment resistance due to the activation of compensatory feedbacks, requiring inhibiting several signaling nodes at once. Combi- or hybrid molecules have been developed to overcome these limitations by targeting several biological targets (kinases, DNA, etc.) with one entity, allowing better control of the pharmacokinetics and pharmacodynamics of drug combinations. Currently, there are 3 types of combi/hybrid molecules, depending on their mechanism of action. Type I can block target A as an intact molecule but is required to be hydrolyzed to block A and B; Type II can block targets A and B without being hydrolyzed; Type III can block targets A and B as an intact molecule or after being hydrolyzed. Combi/hybrid molecules are one of the proposed novel therapeutic solutions to target specific PI3K isoforms among the 3 classes, depending on their relative importance in a type of cancer, using only one molecule.
8. Conclusions
The entire family of PI3Ks regulate key mechanisms in cancer. The pathways controlled by class I PI3Ks are altered in more than 50% of all cancers (solid and hematopoietic). Notwithstanding the fact that the specific roles of class I PI3K isoforms become better characterized in each cancer subtype [3], more research is needed to clearly decipher the functions of class II and class III PI3Ks in cancer, as there is emerging evidence that they play a crucial role in tumor biology.
The strategy consisting of targeting all class I PI3Ks with pan-PI3K inhibitors rapidly showed limitations due to toxicities in combination treatments and an increased resistance to treatment [8], emphasizing the need to specifically target the key PI3K isoforms in each cancer subtype [4]. Currently, PI3Kα-specific inhibitors are administered to PIK3CA-mutated tumors in combination with hormonotherapy; the clinical use of PI3Kα inhibitors could be extended to cancer with global PI3K/AKT activation (e.g., pancreatic cancer due to KRAS mutation) but not in single therapy and possibly with PI3Kγ inhibitors. PI3Kβ-specific inhibitors should not be restricted to PTEN-deficient cancers, but the key predictive markers of their use and the treatment that needs to be combined are not clearly defined.
Regarding the targeting of PI3Kδ, which is efficient to target hematopoietic cells but also as an immunotherapy, the main challenge is to handle the toxicity. Approaches such as intermittent doses [65], but also a better determination of the efficacy, need to be reviewed [148]. Analyzing the toxicity of the compounds and their metabolites when used in combination is necessary to lead those pharmaceutical compounds to successful clinical trials (Figure 5).
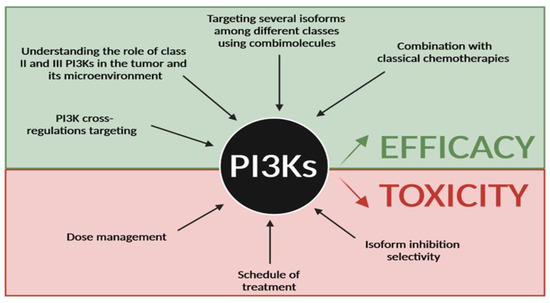
Figure 5.
Future direction for PI3K targeting in cancer. Efforts have to be made to improve efficacy of PI3K inhibitors and reduce their toxicity in cancer treatment. Improving efficacy will require better knowledge of PI3K cross-regulations and the role of class II and III in cancer, to target several PI3K isoforms among the 3 classes and to combine existing PI3K inhibitors with classical chemotherapies. Reducing toxicity will go through a better dose management, different schedules of treatment and a better understanding of isoform specificity in organs and in cancer subtypes.
Finally, given the emerging evidence that class I and II/III are cross-regulating their downstream cellular processes and cooperating to promote cancer cell proliferation, survival and migration, one underexplored strategy could be to specifically target class I, II and/or III PI3K isoforms through combination therapies (Figure 5). However, those combinations may increase the adverse effects in patients, possibly leading to the arrest of the treatment. While emerging evidence was found in pancreatic cancer [96], a basic knowledge of the roles of class II and III PI3Ks in tumor biology is still missing to enable those strategies.
In parallel to the clinical assessment of class I PI3K inhibitors, future basic and preclinical research should provide the proof-of-concept for developing combination treatment strategies targeting several PI3K isoforms amongst the three classes.
Author Contributions
Conceptualization, B.T. and J.G.-G.; writing—original draft preparation, B.T., F.R.-D. and J.G.-G.; writing—review and editing, B.T., F.R.-D. and J.G.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the ANR JCJC program Radiance (Projet-ANR-18-CE14-0013), MSCA-ITN/ETN PhD-PI3K (Project ID: 675392), IUCT-O/ICR translational program (Project: Cluster), Ligue Régionale Contre Le Cancer (2020, 2021), Fondation Arc (PJA2021060003932), the ANR (Toucan), Laboratoire d’Excellence.
Acknowledgments
The authors apologize to the authors whose work we could not cite in this review due to article format constraint. JGG’s laboratory belongs to Toucan, Laboratoire d’Excellence, ANR, an integrated research program on Signal-targeted Drug Resistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K Isoforms in Cell Signalling and Vesicle Trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Arcucci, S.; Ramos-Delgado, F.; Cayron, C.; Therville, N.; Gratacap, M.-P.; Basset, C.; Thibault, B.; Guillermet-Guibert, J. Organismal Roles for the PI3Kα and β Isoforms: Their Specificity, Redundancy or Cooperation Is Context-Dependent. Biochem. J. 2021, 478, 1199–1225. [Google Scholar] [CrossRef] [PubMed]
- Pons-Tostivint, E.; Thibault, B.; Guillermet-Guibert, J. Targeting PI3K Signaling in Combination Cancer Therapy. Trends Cancer 2017, 3, 454–469. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K Inhibitors Are Finally Coming of Age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef]
- Millis, S.Z.; Jardim, D.L.; Albacker, L.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Kurzrock, R. Phosphatidylinositol 3-Kinase Pathway Genomic Alterations in 60,991 Diverse Solid Tumors Informs Targeted Therapy Opportunities. Cancer 2019, 125, 1185–1199. [Google Scholar] [CrossRef]
- Dufour, M.; Dormond-Meuwly, A.; Pythoud, C.; Demartines, N.; Dormond, O. Reactivation of AKT Signaling Following Treatment of Cancer Cells with PI3K Inhibitors Attenuates Their Antitumor Effects. Biochem. Biophys. Res. Commun. 2013, 438, 32–37. [Google Scholar] [CrossRef]
- Cintas, C.; Douche, T.; Dantes, Z.; Mouton-Barbosa, E.; Bousquet, M.-P.; Cayron, C.; Therville, N.; Pont, F.; Ramos-Delgado, F.; Guyon, C.; et al. Phosphoproteomics Identifies PI3K Inhibitor–Selective Adaptive Responses in Pancreatic Cancer Cell Therapy and Resistance. Mol. Cancer Ther. 2021, 20, 2433–2445. [Google Scholar] [CrossRef]
- Bian, C.; Liu, Z.; Li, D.; Zhen, L. PI3K/AKT Inhibition Induces Compensatory Activation of the MET/STAT3 Pathway in Non-Small Cell Lung Cancer. Oncol. Lett. 2018, 15, 9655–9662. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kδ Inhibition by Idelalisib in Patients with Relapsed Indolent Lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.-S.; et al. Alpelisib plus Fulvestrant for PIK3CA-Mutated, Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor-2-Negative Advanced Breast Cancer: Final Overall Survival Results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Chang, M.T.; Juric, D.; Saura, C.; Gambardella, V.; Melnyk, A.; Patel, M.R.; Ribrag, V.; Ma, C.X.; Aljumaily, R.; et al. Phase I Basket Study of Taselisib, an Isoform-Selective PI3K Inhibitor, in Patients with PIK3CA -Mutant Cancers. Clin. Cancer Res. 2021, 27, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly Targeted Therapy Based on Tumour Molecular Profiling versus Conventional Therapy for Advanced Cancer (SHIVA): A Multicentre, Open-Label, Proof-of-Concept, Randomised, Controlled Phase 2 Trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Song, K.W.; Edgar, K.A.; Hanan, E.J.; Hafner, M.; Oeh, J.; Merchant, M.; Sampath, D.; Nannini, M.A.; Hong, R.; Phu, L.; et al. RTK-Dependent Inducible Degradation of Mutant PI3Kα Drives GDC-0077 (Inavolisib) Efficacy. Cancer Discov. 2022, 12, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K Inhibition Results in Enhanced Estrogen Receptor Function and Dependence in Hormone Receptor-Positive Breast Cancer. Sci. Transl. Med. 2015, 7, 283ra51. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, T.; Krakstad, C.; Tangen, I.L.; Hoivik, E.A.; Pollock, P.M.; Salvesen, H.B.; Lewis, A.E. Endometrial Cancer Cells Exhibit High Expression of P110β and Its Selective Inhibition Induces Variable Responses on PI3K Signaling, Cell Survival and Proliferation. Oncotarget 2017, 8, 3881–3894. [Google Scholar] [CrossRef]
- Thibault, B.; Ramos-Delgado, F.; Pons-Tostivint, E.; Therville, N.; Cintas, C.; Arcucci, S.; Cassant-Sourdy, S.; Reyes-Castellanos, G.; Tosolini, M.; Villard, A.V.; et al. Pancreatic Cancer Intrinsic PI3Kα Activity Accelerates Metastasis and Rewires Macrophage Component. EMBO Mol. Med. 2021, 13, e13502. [Google Scholar] [CrossRef]
- Diehl, A.; Hannan, L.M.; Chiorean, E.G. Prognostic Value of KRAS and PI3K Pathway Mutations for Advanced Pancreatic Ductal Adenocarcinoma (PDAC) Patients (Pts). JCO 2021, 39, 424. [Google Scholar] [CrossRef]
- Delaloge, S.; DeForceville, L. Targeting PI3K/AKT Pathway in Triple-Negative Breast Cancer. Lancet Oncol. 2017, 18, 1293–1294. [Google Scholar] [CrossRef]
- Pearson, H.B.; Li, J.; Meniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; Macpherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef]
- Piazzi, M.; Bavelloni, A.; Cenni, V.; Salucci, S.; Bartoletti Stella, A.; Tomassini, E.; Scotlandi, K.; Blalock, W.L.; Faenza, I. Combined Treatment with PI3K Inhibitors BYL-719 and CAL-101 Is a Promising Antiproliferative Strategy in Human Rhabdomyosarcoma Cells. Molecules 2022, 27, 2742. [Google Scholar] [CrossRef] [PubMed]
- Ultimo, S.; Simioni, C.; Martelli, A.M.; Zauli, G.; Evangelisti, C.; Celeghini, C.; McCubrey, J.A.; Marisi, G.; Ulivi, P.; Capitani, S.; et al. PI3K Isoform Inhibition Associated with Anti Bcr-Abl Drugs Shows in Vitro Increased Anti-Leukemic Activity in Philadelphia Chromosome-Positive B-Acute Lymphoblastic Leukemia Cell Lines. Oncotarget 2017, 8, 23213–23227. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Kim, J.-W.; Sung, J.H.; Suh, K.J.; Lee, J.Y.; Kim, S.H.; Lee, J.-O.; Kim, J.W.; Kim, Y.J.; Kim, J.H.; et al. PI3K-Targeting Strategy Using Alpelisib to Enhance the Antitumor Effect of Paclitaxel in Human Gastric Cancer. Sci. Rep. 2020, 10, 12308. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-Specific PI3K Inhibitor Alpelisib for Patients with Epithelial Ovarian Cancer: A Dose-Escalation and Dose-Expansion Phase 1b Trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Sun, P.; Zhang, X.; Wang, R.-J.; Ma, Q.-Y.; Xu, L.; Wang, Y.; Liao, H.-P.; Wang, H.-L.; Hu, L.-D.; Kong, X.; et al. PI3Kα Inhibitor CYH33 Triggers Antitumor Immunity in Murine Breast Cancer by Activating CD8 + T Cells and Promoting Fatty Acid Metabolism. J. Immunother. Cancer 2021, 9, e003093. [Google Scholar] [CrossRef]
- Lynch, J.T.; Polanska, U.M.; Delpuech, O.; Hancox, U.; Trinidad, A.G.; Michopoulos, F.; Lenaghan, C.; McEwen, R.; Bradford, J.; Polanski, R.; et al. Inhibiting PI3Kβ with AZD8186 Regulates Key Metabolic Pathways in PTEN-Null Tumors. Clin. Cancer Res. 2017, 23, 7584–7595. [Google Scholar] [CrossRef]
- de Bono, J.S.; Hansen, A.; Choudhury, A.D.; Cook, N.; Heath, E.I.; Higano, C.; Linch, M.; Martin-Liberal, J.; Rathkopf, D.E.; Wisinski, K.B.; et al. AZD8186, a Potent and Selective Inhibitor of PI3Kβ/δ, as Monotherapy and in Combination with Abiraterone Acetate plus Prednisone (AAP), in Patients (Pts) with Metastatic Castrate-Resistant Prostate Cancer (MCRPC). Ann. Oncol. 2018, 29, viii291–viii292. [Google Scholar] [CrossRef]
- Lynch, J.T.; Polanska, U.M.; Hancox, U.; Delpuech, O.; Maynard, J.; Trigwell, C.; Eberlein, C.; Lenaghan, C.; Polanski, R.; Avivar-Valderas, A.; et al. Combined Inhibition of PI3Kβ and MTOR Inhibits Growth of PTEN-Null Tumors. Mol. Cancer Ther. 2018, 17, 2309–2319. [Google Scholar] [CrossRef]
- Hosford, S.R.; Dillon, L.M.; Bouley, S.J.; Rosati, R.; Yang, W.; Chen, V.S.; Demidenko, E.; Morra, R.P.; Miller, T.W. Combined Inhibition of Both P110α and P110β Isoforms of Phosphatidylinositol 3-Kinase Is Required for Sustained Therapeutic Effect in PTEN-Deficient, ER + Breast Cancer. Clin. Cancer Res. 2017, 23, 2795–2805. [Google Scholar] [CrossRef]
- Owusu-Brackett, N.; Zhao, M.; Akcakanat, A.; Evans, K.W.; Yuca, E.; Dumbrava, E.I.; Janku, F.; Meric-Bernstam, F. Targeting PI3Kβ Alone and in Combination with Chemotherapy or Immunotherapy in Tumors with PTEN Loss. Oncotarget 2020, 11, 969–981. [Google Scholar] [CrossRef]
- Zecchin, D.; Moore, C.; Michailidis, F.; Horswell, S.; Rana, S.; Howell, M.; Downward, J. Combined Targeting of G Protein-Coupled Receptor and EGF Receptor Signaling Overcomes Resistance to PI3K Pathway Inhibitors in PTEN-Null Triple Negative Breast Cancer. EMBO Mol. Med. 2020, 12, e11987. [Google Scholar] [CrossRef]
- Xu, P.-F.; Yang, J.-A.; Liu, J.-H.; Yang, X.; Liao, J.-M.; Yuan, F.-E.; Liu, B.-H.; Chen, Q.-X. PI3Kβ Inhibitor AZD6482 Exerts Antiproliferative Activity and Induces Apoptosis in Human Glioblastoma Cells. Oncol. Rep. 2019, 41, 125–132. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, C.; Zhou, X.; Diao, P.; Xu, Y.; Liu, J.; Wang, J.; Huang, X.; Liu, W.; Chen, Z.; et al. Synergism between the Phosphatidylinositol 3-Kinase P110β Isoform Inhibitor AZD6482 and the Mixed Lineage Kinase 3 Inhibitor URMC-099 on the Blockade of Glioblastoma Cell Motility and Focal Adhesion Formation. Cancer Cell Int. 2021, 21, 24. [Google Scholar] [CrossRef]
- Marqués, M.; Tranchant, R.; Risa-Ebrí, B.; Suárez-Solís, M.L.; Fernández, L.C.; Carrillo-de-Santa-Pau, E.; del Pozo, N.; Martínez de Villarreal, J.; Meiller, C.; Allory, Y.; et al. Combined MEK and PI3K/P110β Inhibition as a Novel Targeted Therapy for Malignant Mesothelioma Displaying Sarcomatoid Features. Cancer Res. 2020, 80, 843–856. [Google Scholar] [CrossRef]
- Peng, W.; Williams, L.J.; Xu, C.; Melendez, B.; McKenzie, J.A.; Chen, Y.; Jackson, H.L.; Voo, K.S.; Mbofung, R.M.; Leahey, S.E.; et al. Anti-OX40 Antibody Directly Enhances The Function of Tumor-Reactive CD8 + T Cells and Synergizes with PI3Kβ Inhibition in PTEN Loss Melanoma. Clin. Cancer Res. 2019, 25, 6406–6416. [Google Scholar] [CrossRef]
- Heitz, S.D.; Hamelin, D.J.; Hoffmann, R.M.; Greenberg, N.; Salloum, G.; Erami, Z.; Khalil, B.D.; Shymanets, A.; Steidle, E.A.; Gong, G.Q.; et al. A Single Discrete Rab5-Binding Site in Phosphoinositide 3-Kinase β Is Required for Tumor Cell Invasion. J. Biol. Chem. 2019, 294, 4621–4633. [Google Scholar] [CrossRef]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Han, S.-W.; Swales, K.; Decordova, S.; DeYoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S.; et al. A First-Time-in-Human Study of GSK2636771, a Phosphoinositide 3 Kinase Beta-Selective Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5981–5992. [Google Scholar] [CrossRef]
- Di-Luoffo, M.; Ben-Meriem, Z.; Lefebvre, P.; Delarue, M.; Guillermet-Guibert, J. PI3K Functions as a Hub in Mechanotransduction. Trends Biochem. Sci. 2021, 46, 878–888. [Google Scholar] [CrossRef]
- Sethi, I.; Cai, Z.; Roberts, T.M.; Yuan, G.-C. Molecular Profiling Establishes Genetic Features Predictive of the Efficacy of the P110β Inhibitor KIN-193. Cancer Res. 2019, 79, 4524–4531. [Google Scholar] [CrossRef]
- Millán-Uclés, Á.; Zuluaga, S.; Marqués, M.; Vallejo-Díaz, J.; Sanz, L.; Cariaga-Martínez, A.E.; Real, F.X.; Carrera, A.C. E-Cadherin Downregulation Sensitizes PTEN-Mutant Tumors to PI3Kβ Silencing. Oncotarget 2016, 7, 84054–84071. [Google Scholar] [CrossRef]
- Fan, R.; Kim, N.-G.; Gumbiner, B.M. Regulation of Hippo Pathway by Mitogenic Growth Factors via Phosphoinositide 3-Kinase and Phosphoinositide-Dependent Kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef]
- Kim, N.-G.; Gumbiner, B.M. Adhesion to Fibronectin Regulates Hippo Signaling via the FAK-Src-PI3K Pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.H.; Okkenhaug, K.; Vanhaesebroeck, B. The P110beta Isoform of Phosphoinositide 3-Kinase Signals Downstream of G Protein-Coupled Receptors and Is Functionally Redundant with P110gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Montminy, T.; Azad, T.; Lightbody, E.; Hao, Y.; SenGupta, S.; Asselin, E.; Nicol, C.; Yang, X. PI3K Positively Regulates YAP and TAZ in Mammary Tumorigenesis Through Multiple Signaling Pathways. Mol. Cancer Res. 2018, 16, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.-K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis Selectively Requires the P110alpha Isoform of PI3K to Control Endothelial Cell Migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef]
- Guillermet-Guibert, J.; Smith, L.B.; Halet, G.; Whitehead, M.A.; Pearce, W.; Rebourcet, D.; León, K.; Crépieux, P.; Nock, G.; Strömstedt, M.; et al. Novel Role for P110β PI 3-Kinase in Male Fertility through Regulation of Androgen Receptor Activity in Sertoli Cells. PLoS Genet. 2015, 11, e1005304. [Google Scholar] [CrossRef]
- Berenjeno, I.M.; Guillermet-Guibert, J.; Pearce, W.; Gray, A.; Fleming, S.; Vanhaesebroeck, B. Both P110α and P110β Isoforms of PI3K Can Modulate the Impact of Loss-of-Function of the PTEN Tumour Suppressor. Biochem. J. 2012, 442, 151–159. [Google Scholar] [CrossRef]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of Ras to Phosphoinositide 3-Kinase P110α Is Required for Ras- Driven Tumorigenesis in Mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO Families of GTPases Directly Regulate Distinct Phosphoinositide 3-Kinase Isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef]
- Edling, C.E.; Selvaggi, F.; Buus, R.; Maffucci, T.; Di Sebastiano, P.; Friess, H.; Innocenti, P.; Kocher, H.M.; Falasca, M. Key Role of Phosphoinositide 3-Kinase Class IB in Pancreatic Cancer. Clin. Cancer Res. 2010, 16, 4928–4937. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef]
- Mamidi, M.K.; Mahmud, H.; Maiti, G.P.; Mendez, M.T.; Fernandes, S.M.; Vesely, S.K.; Holter-Chakrabarty, J.; Brown, J.R.; Ghosh, A.K. Idelalisib Activates AKT via Increased Recruitment of PI3Kδ/PI3Kβ to BCR Signalosome While Reducing PDK1 in Post-Therapy CLL Cells. Leukemia 2022, 36, 1806–1817. [Google Scholar] [CrossRef] [PubMed]
- Scheffold, A.; Jebaraj, B.M.C.; Tausch, E.; Bloehdorn, J.; Ghia, P.; Yahiaoui, A.; Dolnik, A.; Blätte, T.J.; Bullinger, L.; Dheenadayalan, R.P.; et al. IGF1R as Druggable Target Mediating PI3K-δ Inhibitor Resistance in a Murine Model of Chronic Lymphocytic Leukemia. Blood 2019, 134, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Bashash, D.; Safaroghli-Azar, A.; Dadashi, M.; Safa, M.; Momeny, M.; Ghaffari, S.H. Anti-Tumor Activity of PI3K-δ Inhibitor in Hematologic Malignant Cells: Shedding New Light on Resistance to Idelalisib. Int. J. Biochem. Cell Biol. 2017, 85, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Flinn, I.W.; Patel, M.R.; Fenske, T.S.; Deng, C.; Brander, D.M.; Gutierrez, M.; Essell, J.H.; Kuhn, J.G.; Miskin, H.P.; et al. Umbralisib, a Novel PI3Kδ and Casein Kinase-1ε Inhibitor, in Relapsed or Refractory Chronic Lymphocytic Leukaemia and Lymphoma: An Open-Label, Phase 1, Dose-Escalation, First-in-Human Study. Lancet Oncol. 2018, 19, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.H.; Samaniego, F.; Jurczak, W.; Ghosh, N.; Derenzini, E.; Reeves, J.A.; Knopińska-Posłuszny, W.; Cheah, C.Y.; Phillips, T.; Lech-Maranda, E.; et al. Umbralisib, a Dual PI3Kδ/CK1ε Inhibitor in Patients With Relapsed or Refractory Indolent Lymphoma. JCO 2021, 39, 1609–1618. [Google Scholar] [CrossRef]
- Deng, C.; Lipstein, M.R.; Scotto, L.; Jirau Serrano, X.O.; Mangone, M.A.; Li, S.; Vendome, J.; Hao, Y.; Xu, X.; Deng, S.-X.; et al. Silencing C-Myc Translation as a Therapeutic Strategy through Targeting PI3Kδ and CK1ε in Hematological Malignancies. Blood 2017, 129, 88–99. [Google Scholar] [CrossRef]
- Flinn, I.W.; Patel, M.; Oki, Y.; Horwitz, S.; Foss, F.F.; Allen, K.; Douglas, M.; Stern, H.; Sweeney, J.; Kharidia, J.; et al. Duvelisib, an Oral Dual PI3K-δ, γ Inhibitor, Shows Clinical Activity in Indolent Non-Hodgkin Lymphoma in a Phase 1 Study. Am. J. Hematol. 2018, 93, 1311–1317. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,γ Inhibitor Duvelisib in a Phase 1 Trial and Preclinical Models of T-Cell Lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef]
- Dwyer, C.J.; Arhontoulis, D.C.; Rangel Rivera, G.O.; Knochelmann, H.M.; Smith, A.S.; Wyatt, M.M.; Rubinstein, M.P.; Atkinson, C.; Thaxton, J.E.; Neskey, D.M.; et al. Ex Vivo Blockade of PI3K Gamma or Delta Signaling Enhances the Antitumor Potency of Adoptively Transferred CD8 + T Cells. Eur. J. Immunol. 2020, 50, 1386–1399. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.; Seo, H.-W.; Jung, E.S.; Ju, S.; Kim, B.-H.; Cho, H.; Kim, Y.J.; Park, Y.M.; Kim, J.-S.; Jung, G. PI3Kδ Is a Therapeutic Target in Hepatocellular Carcinoma. Hepatology 2018, 68, 2285–2300. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Sun, X. Idelalisib Promotes Bim-Dependent Apoptosis through AKT/FoxO3a in Hepatocellular Carcinoma. Cell Death Dis. 2018, 9, 935. [Google Scholar] [CrossRef] [PubMed]
- Goulielmaki, E.; Bermudez-Brito, M.; Andreou, M.; Tzenaki, N.; Tzardi, M.; de Bree, E.; Tsentelierou, E.; Makrigiannakis, A.; Papakonstanti, E.A. Pharmacological Inactivation of the PI3K P110δ Prevents Breast Tumour Progression by Targeting Cancer Cells and Macrophages. Cell Death Dis. 2018, 9, 678. [Google Scholar] [CrossRef] [PubMed]
- Eschweiler, S.; Ramírez-Suástegui, C.; Li, Y.; King, E.; Chudley, L.; Thomas, J.; Wood, O.; von Witzleben, A.; Jeffrey, D.; McCann, K.; et al. Intermittent PI3Kδ Inhibition Sustains Anti-Tumour Immunity and Curbs IrAEs. Nature 2022, 605, 741–746. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020, 10, 104–123. [Google Scholar] [CrossRef]
- Ni, H.; Chai, P.; Yu, J.; Xing, Y.; Wang, S.; Fan, J.; Ge, S.; Wang, Y.; Jia, R.; Fan, X. LncRNA CANT1 Suppresses Retinoblastoma Progression by Repellinghistone Methyltransferase in PI3Kγ Promoter. Cell Death Dis. 2020, 11, 306. [Google Scholar] [CrossRef]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling Tumor Immune Microenvironment via Targeted Blockade of PI3K-γ and CSF-1/CSF-1R Pathways in Tumor Associated Macrophages for Pancreatic Cancer Therapy. J. Control. Release 2020, 321, 23–35. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hong, D.S.; Tolcher, A.W.; Patnaik, A.; Shapiro, G.; Chmielowski, B.; Ribas, A.; Brail, L.H.; Roberts, J.; Lee, L.; et al. Initial Results from First-in-Human Study of IPI-549, a Tumor Macrophage-Targeting Agent, Combined with Nivolumab in Advanced Solid Tumors. JCO 2018, 36, 3013. [Google Scholar] [CrossRef]
- Anderson, K.; Ryan, N.; Alkhimovitch, A.; Siddiqui, A.; Oghumu, S. Inhibition of PI3K Isoform P110γ Increases Both Anti-Tumor and Immunosuppressive Responses to Aggressive Murine Head and Neck Squamous Cell Carcinoma with Low Immunogenicity. Cancers 2021, 13, 953. [Google Scholar] [CrossRef]
- Li, M.; Sala, V.; De Santis, M.C.; Cimino, J.; Cappello, P.; Pianca, N.; Di Bona, A.; Margaria, J.P.; Martini, M.; Lazzarini, E.; et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects from Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 2018, 138, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Gulluni, F.; De Santis, M.C.; Margaria, J.P.; Martini, M.; Hirsch, E. Class II PI3K Functions in Cell Biology and Disease. Trends Cell Biol. 2019, 29, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Maffucci, T. Regulation and Cellular Functions of Class II Phosphoinositide 3-Kinases. Biochem. J. 2012, 443, 587–601. [Google Scholar] [CrossRef]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ Is a Rab5 Effector Selectively Controlling Endosomal Akt2 Activation Downstream of Insulin Signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [PubMed]
- Marat, A.L.; Haucke, V. Phosphatidylinositol 3-Phosphates-at the Interface between Cell Signalling and Membrane Traffic. EMBO J. 2016, 35, 561–579. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Hughes, W.E.; Dominguez, V.; Sala, G.; Fostira, F.; Fang, M.Q.; Cazzolli, R.; Shepherd, P.R.; James, D.E.; Maffucci, T. The Role of Phosphoinositide 3-Kinase C2alpha in Insulin Signaling. J. Biol. Chem. 2007, 282, 28226–28236. [Google Scholar] [CrossRef]
- Leibiger, B.; Moede, T.; Uhles, S.; Barker, C.J.; Creveaux, M.; Domin, J.; Berggren, P.-O.; Leibiger, I.B. Insulin-Feedback via PI3K-C2alpha Activated PKBalpha/Akt1 Is Required for Glucose-Stimulated Insulin Secretion. FASEB J. 2010, 24, 1824–1837. [Google Scholar] [CrossRef]
- Mountford, S.J.; Zheng, Z.; Sundaram, K.; Jennings, I.G.; Hamilton, J.R.; Thompson, P.E. Class II but Not Second Class-Prospects for the Development of Class II PI3K Inhibitors. ACS Med. Chem. Lett. 2015, 6, 3–6. [Google Scholar] [CrossRef]
- Tiosano, D.; Baris, H.N.; Chen, A.; Hitzert, M.M.; Schueler, M.; Gulluni, F.; Wiesener, A.; Bergua, A.; Mory, A.; Copeland, B.; et al. Mutations in PIK3C2A Cause Syndromic Short Stature, Skeletal Abnormalities, and Cataracts Associated with Ciliary Dysfunction. PLoS Genet. 2019, 15, e1008088. [Google Scholar] [CrossRef]
- Gulluni, F.; Prever, L.; Li, H.; Krafcikova, P.; Corrado, I.; Lo, W.-T.; Margaria, J.P.; Chen, A.; De Santis, M.C.; Cnudde, S.J.; et al. PI(3,4)P2-Mediated Cytokinetic Abscission Prevents Early Senescence and Cataract Formation. Science 2021, 374, eabk0410. [Google Scholar] [CrossRef]
- Yoshioka, K.; Yoshida, K.; Cui, H.; Wakayama, T.; Takuwa, N.; Okamoto, Y.; Du, W.; Qi, X.; Asanuma, K.; Sugihara, K.; et al. Endothelial PI3K-C2α, a Class II PI3K, Has an Essential Role in Angiogenesis and Vascular Barrier Function. Nat. Med. 2012, 18, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Franco, I.; Gulluni, F.; Campa, C.C.; Costa, C.; Margaria, J.P.; Ciraolo, E.; Martini, M.; Monteyne, D.; De Luca, E.; Germena, G.; et al. PI3K Class II α Controls Spatially Restricted Endosomal PtdIns3P and Rab11 Activation to Promote Primary Cilium Function. Dev. Cell 2014, 28, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Alliouachene, S.; Bilanges, B.; Chaussade, C.; Pearce, W.; Foukas, L.C.; Scudamore, C.L.; Moniz, L.S.; Vanhaesebroeck, B. Inactivation of Class II PI3K-C2α Induces Leptin Resistance, Age-Dependent Insulin Resistance and Obesity in Male Mice. Diabetologia 2016, 59, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Truong, A.B.; Cai, T.; Khavari, P.A. The Class II Phosphoinositide 3-Kinase C2beta Is Not Essential for Epidermal Differentiation. Mol. Cell. Biol. 2005, 25, 11122–11130. [Google Scholar] [CrossRef]
- Alliouachene, S.; Bilanges, B.; Chicanne, G.; Anderson, K.E.; Pearce, W.; Ali, K.; Valet, C.; Posor, Y.; Low, P.C.; Chaussade, C.; et al. Inactivation of the Class II PI3K-C2β Potentiates Insulin Signaling and Sensitivity. Cell Rep. 2015, 13, 1881–1894. [Google Scholar] [CrossRef]
- Dai, J.; Lu, Y.; Wang, J.; Yang, L.; Han, Y.; Wang, Y.; Yan, D.; Ruan, Q.; Wang, S. A Four-Gene Signature Predicts Survival in Clear-Cell Renal-Cell Carcinoma. Oncotarget 2016, 7, 82712–82726. [Google Scholar] [CrossRef]
- Liu, T.; Zu, C.-H.; Wang, S.-S.; Song, H.-L.; Wang, Z.-L.; Xu, X.-N.; Liu, H.-S.; Wang, Y.-L.; Shen, Z.-Y. PIK3C2A MRNA Functions as a MiR-124 Sponge to Facilitate CD151 Expression and Enhance Malignancy of Hepatocellular Carcinoma Cells. Oncotarget 2016, 7, 43376–43389. [Google Scholar] [CrossRef]
- Gulluni, F.; Martini, M.; De Santis, M.C.; Campa, C.C.; Ghigo, A.; Margaria, J.P.; Ciraolo, E.; Franco, I.; Ala, U.; Annaratone, L.; et al. Mitotic Spindle Assembly and Genomic Stability in Breast Cancer Require PI3K-C2α Scaffolding Function. Cancer Cell 2017, 32, 444–459.e7. [Google Scholar] [CrossRef]
- Katso, R.M.; Pardo, O.E.; Palamidessi, A.; Franz, C.M.; Marinov, M.; De Laurentiis, A.; Downward, J.; Scita, G.; Ridley, A.J.; Waterfield, M.D.; et al. Phosphoinositide 3-Kinase C2β Regulates Cytoskeletal Organization and Cell Migration via Rac-Dependent Mechanisms. MBoC 2006, 17, 3729–3744. [Google Scholar] [CrossRef]
- Mavrommati, I.; Cisse, O.; Falasca, M.; Maffucci, T. Novel Roles for Class II Phosphoinositide 3-Kinase C2β in Signalling Pathways Involved in Prostate Cancer Cell Invasion. Sci. Rep. 2016, 6, 23277. [Google Scholar] [CrossRef]
- Chikh, A.; Ferro, R.; Abbott, J.J.; Piñeiro, R.; Buus, R.; Iezzi, M.; Ricci, F.; Bergamaschi, D.; Ostano, P.; Chiorino, G.; et al. Class II Phosphoinositide 3-Kinase C2β Regulates a Novel Signaling Pathway Involved in Breast Cancer Progression. Oncotarget 2016, 7, 18325–18345. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yang, L.; Chen, J.; Zhao, H.; Wang, J.; Xu, S.; Huang, Z. MiR-362-5p Inhibits Proliferation and Migration of Neuroblastoma Cells by Targeting Phosphatidylinositol 3-Kinase-C2β. FEBS Lett. 2015, 589, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Posor, Y.; Kampyli, C.; Bilanges, B.; Ganguli, S.; Koch, P.A.; Wallroth, A.; Morelli, D.; Jenkins, M.; Alliouachene, S.; Deltcheva, E.; et al. Local Synthesis of the Phosphatidylinositol-3,4-Bisphosphate Lipid Drives Focal Adhesion Turnover. Dev. Cell 2022, 57, 1694–1711.e7. [Google Scholar] [CrossRef] [PubMed]
- Cisse, O.; Quraishi, M.; Gulluni, F.; Guffanti, F.; Mavrommati, I.; Suthanthirakumaran, M.; Oh, L.C.R.; Schlatter, J.N.; Sarvananthan, A.; Broggini, M.; et al. Downregulation of Class II Phosphoinositide 3-Kinase PI3K-C2β Delays Cell Division and Potentiates the Effect of Docetaxel on Cancer Cell Growth. J. Exp. Clin. Cancer Res. 2019, 38, 472. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Chen, H.; Lin, M.; Zhang, C.; Tang, E.; Peng, J.; Wei, Q.; Li, H.; Yin, L. PIK3C2G Copy Number Is Associated with Clinical Outcomes of Colorectal Cancer Patients Treated with Oxaliplatin. Int. J. Clin. Exp. Med. 2015, 8, 1137–1143. [Google Scholar]
- De Santis, M.C.; Gozzelino, L.; Margaria, J.P.; Costamagna, A.; Ratto, E.; Gulluni, F.; Di Gregorio, E.; Mina, E.; Lorito, N.; Bacci, M.; et al. Lysosomal Lipid Switch Sensitises to Nutrient Deprivation and MTOR Targeting in Pancreatic Cancer. Gut 2022, 72, 360–371. [Google Scholar] [CrossRef]
- Chaudhry, S.R.; Lopes, J.; Levin, N.K.; Kalpage, H.; Tainsky, M.A. Germline Mutations in Apoptosis Pathway Genes in Ovarian Cancer; the Functional Role of a TP53I3 (PIG3) Variant in ROS Production and DNA Repair. Cell Death Discov. 2021, 7, 62. [Google Scholar] [CrossRef]
- Anquetil, T.; Solinhac, R.; Jaffre, A.; Chicanne, G.; Viaud, J.; Darcourt, J.; Orset, C.; Geuss, E.; Kleinschnitz, C.; Vanhaesebroeck, B.; et al. PI3KC2β Inactivation Stabilizes VE-Cadherin Junctions and Preserves Vascular Integrity. EMBO Rep. 2021, 22, e51299. [Google Scholar] [CrossRef]
- Backer, J.M. The Intricate Regulation and Complex Functions of the Class III Phosphoinositide 3-Kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef]
- Carpentier, S.; N’Kuli, F.; Grieco, G.; Van Der Smissen, P.; Janssens, V.; Emonard, H.; Bilanges, B.; Vanhaesebroeck, B.; Gaide Chevronnay, H.P.; Pierreux, C.E.; et al. Class III Phosphoinositide 3-Kinase/VPS34 and Dynamin Are Critical for Apical Endocytic Recycling. Traffic 2013, 14, 933–948. [Google Scholar] [CrossRef]
- Boukhalfa, A.; Nascimbeni, A.C.; Ramel, D.; Dupont, N.; Hirsch, E.; Gayral, S.; Laffargue, M.; Codogno, P.; Morel, E. PI3KC2α-Dependent and VPS34-Independent Generation of PI3P Controls Primary Cilium-Mediated Autophagy in Response to Shear Stress. Nat. Commun. 2020, 11, 294. [Google Scholar] [CrossRef] [PubMed]
- Jaber, N.; Dou, Z.; Chen, J.-S.; Catanzaro, J.; Jiang, Y.-P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J.; et al. Class III PI3K Vps34 Plays an Essential Role in Autophagy and in Heart and Liver Function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Takatoh, J.; Wang, F. The Mammalian Class 3 PI3K (PIK3C3) Is Required for Early Embryogenesis and Cell Proliferation. PLoS ONE 2011, 6, e16358. [Google Scholar] [CrossRef]
- Bilanges, B.; Alliouachene, S.; Pearce, W.; Morelli, D.; Szabadkai, G.; Chung, Y.-L.; Chicanne, G.; Valet, C.; Hill, J.M.; Voshol, P.J.; et al. Vps34 PI 3-Kinase Inactivation Enhances Insulin Sensitivity through Reprogramming of Mitochondrial Metabolism. Nat. Commun. 2017, 8, 1804. [Google Scholar] [CrossRef] [PubMed]
- Valet, C.; Levade, M.; Chicanne, G.; Bilanges, B.; Cabou, C.; Viaud, J.; Gratacap, M.-P.; Gaits-Iacovoni, F.; Vanhaesebroeck, B.; Payrastre, B.; et al. A Dual Role for the Class III PI3K, Vps34, in Platelet Production and Thrombus Growth. Blood 2017, 130, 2032–2042. [Google Scholar] [CrossRef]
- Grieco, G.; Janssens, V.; Gaide Chevronnay, H.P.; N’Kuli, F.; Van Der Smissen, P.; Wang, T.; Shan, J.; Vainio, S.; Bilanges, B.; Jouret, F.; et al. Vps34/PI3KC3 Deletion in Kidney Proximal Tubules Impairs Apical Trafficking and Blocks Autophagic Flux, Causing a Fanconi-like Syndrome and Renal Insufficiency. Sci. Rep. 2018, 8, 14133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, R.; Wang, L.; Shen, S.; Okamoto, H.; Sugawara, A.; Xia, L.; Wang, X.; Noguchi, N.; Yoshikawa, T.; et al. Upregulation of REG Iα Accelerates Tumor Progression in Pancreatic Cancer with Diabetes. Int. J. Cancer 2010, 127, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Budolfson, K.; Wang, F. Pik3c3 Deletion in Pyramidal Neurons Results in Loss of Synapses, Extensive Gliosis and Progressive Neurodegeneration. Neuroscience 2011, 172, 427–442. [Google Scholar] [CrossRef]
- Reifler, A.; Li, X.; Archambeau, A.J.; McDade, J.R.; Sabha, N.; Michele, D.E.; Dowling, J.J. Conditional Knockout of Pik3c3 Causes a Murine Muscular Dystrophy. Am. J. Pathol. 2014, 184, 1819–1830. [Google Scholar] [CrossRef]
- He, F.; Nichols, R.M.; Kailasam, L.; Wensel, T.G.; Agosto, M.A. Critical Role for Phosphatidylinositol-3 Kinase Vps34/PIK3C3 in ON-Bipolar Cells. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2861–2874. [Google Scholar] [CrossRef]
- Rajala, A.; He, F.; Anderson, R.E.; Wensel, T.G.; Rajala, R.V.S. Loss of Class III Phosphoinositide 3-Kinase Vps34 Results in Cone Degeneration. Biology 2020, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Flavell, R.A. Canonical Autophagy Dependent on the Class III Phosphoinositide-3 Kinase Vps34 Is Required for Naive T-Cell Homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 8670–8675. [Google Scholar] [CrossRef]
- Jiang, X.; Bao, Y.; Liu, H.; Kou, X.; Zhang, Z.; Sun, F.; Qian, Z.; Lin, Z.; Li, X.; Liu, X.; et al. VPS34 Stimulation of P62 Phosphorylation for Cancer Progression. Oncogene 2017, 36, 6850–6862. [Google Scholar] [CrossRef] [PubMed]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting Autophagy by Small Molecule Inhibitors of Vacuolar Protein Sorting 34 (Vps34) Improves the Sensitivity of Breast Cancer Cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, B. SAR405, a PIK3C3/Vps34 Inhibitor That Prevents Autophagy and Synergizes with MTOR Inhibition in Tumor Cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef]
- Kobylarz, M.J.; Goodwin, J.M.; Kang, Z.B.; Annand, J.W.; Hevi, S.; O’Mahony, E.; McAllister, G.; Reece-Hoyes, J.; Wang, Q.; Alford, J.; et al. An Iron-Dependent Metabolic Vulnerability Underlies VPS34-Dependence in RKO Cancer Cells. PLoS ONE 2020, 15, e0235551. [Google Scholar] [CrossRef] [PubMed]
- Dayde, D.; Guerard, M.; Perron, P.; Hatat, A.-S.; Barrial, C.; Eymin, B.; Gazzeri, S. Nuclear Trafficking of EGFR by Vps34 Represses Arf Expression to Promote Lung Tumor Cell Survival. Oncogene 2016, 35, 3986–3994. [Google Scholar] [CrossRef]
- Noman, M.Z.; Parpal, S.; Van Moer, K.; Xiao, M.; Yu, Y.; Arakelian, T.; Viklund, J.; De Milito, A.; Hasmim, M.; Andersson, M.; et al. Inhibition of Vps34 Reprograms Cold into Hot Inflamed Tumors and Improves Anti–PD-1/PD-L1 Immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef]
- Janji, B.; Hasmim, M.; Parpal, S.; De Milito, A.; Berchem, G.; Noman, M.Z. Lighting up the Fire in Cold Tumors to Improve Cancer Immunotherapy by Blocking the Activity of the Autophagy-Related Protein PIK3C3/VPS34. Autophagy 2020, 16, 2110–2111. [Google Scholar] [CrossRef]
- Cayron, C.; Rigal, S.; Guillermet-Guibert, J. Is Targeting Autophagy a Promising Lead to Unveil the Cloak of Invisibility in Pancreatic Cancer? Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101622. [Google Scholar] [CrossRef]
- Verma, A.K.; Bharti, P.S.; Rafat, S.; Bhatt, D.; Goyal, Y.; Pandey, K.K.; Ranjan, S.; Almatroodi, S.A.; Alsahli, M.A.; Rahmani, A.H.; et al. Autophagy Paradox of Cancer: Role, Regulation, and Duality. Oxidative Med. Cell. Longev. 2021, 2021, 8832541. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The Evolutionarily Conserved Domain of Beclin 1 Is Required for Vps34 Binding, Autophagy, and Tumor Suppressor Function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 Interacts with Beclin 1 through UVRAG and Regulates Autophagy and Tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Madsen, R.R.; Vanhaesebroeck, B. Cracking the Context-Specific PI3K Signaling Code. Sci. Signal 2020, 13, eaay2940. [Google Scholar] [CrossRef] [PubMed]
- Naguib, A.; Bencze, G.; Engle, D.D.; Chio, I.I.C.; Herzka, T.; Watrud, K.; Bencze, S.; Tuveson, D.A.; Pappin, D.J.; Trotman, L.C. P53 Mutations Change Phosphatidylinositol Acyl Chain Composition. Cell Rep. 2015, 10, 8–19. [Google Scholar] [CrossRef]
- Rueda-Rincon, N.; Bloch, K.; Derua, R.; Vyas, R.; Harms, A.; Hankemeier, T.; Khan, N.A.; Dehairs, J.; Bagadi, M.; Binda, M.M.; et al. P53 Attenuates AKT Signaling by Modulating Membrane Phospholipid Composition. Oncotarget 2015, 6, 21240–21254. [Google Scholar] [CrossRef]
- Choy, C.H.; Han, B.-K.; Botelho, R.J. Phosphoinositide Diversity, Distribution, and Effector Function: Stepping Out of the Box. BioEssays 2017, 39, 1700121. [Google Scholar] [CrossRef]
- Barneda, D.; Cosulich, S.; Stephens, L.; Hawkins, P. How Is the Acyl Chain Composition of Phosphoinositides Created and Does It Matter? Biochem. Soc. Trans. 2019, 47, 1291–1305. [Google Scholar] [CrossRef]
- Foukas, L.C.; Berenjeno, I.M.; Gray, A.; Khwaja, A.; Vanhaesebroeck, B. Activity of Any Class IA PI3K Isoform Can Sustain Cell Proliferation and Survival. Proc. Natl. Acad. Sci. USA 2010, 107, 11381–11386. [Google Scholar] [CrossRef]
- Costa, C.; Ebi, H.; Martini, M.; Beausoleil, S.A.; Faber, A.C.; Jakubik, C.T.; Huang, A.; Wang, Y.; Nishtala, M.; Hall, B.; et al. Measurement of PIP3 Levels Reveals an Unexpected Role for P110β in Early Adaptive Responses to P110α-Specific Inhibitors in Luminal Breast Cancer. Cancer Cell 2015, 27, 97–108. [Google Scholar] [CrossRef]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback Suppression of PI3Kα Signaling in PTEN-Mutated Tumors Is Relieved by Selective Inhibition of PI3Kβ. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent Loss of PTEN Leads to Clinical Resistance to a PI(3)Kα Inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhang, Y.; Deng, T.; Gu, J.; Liu, J.; Yang, H.; Xu, Y.; Yan, Y.; Yang, F.; Zhang, H.; et al. PDPK1 Regulates Autophagosome Biogenesis by Binding to PIK3C3. Autophagy 2021, 17, 2166–2183. [Google Scholar] [CrossRef] [PubMed]
- Bago, R.; Sommer, E.; Castel, P.; Crafter, C.; Bailey, F.P.; Shpiro, N.; Baselga, J.; Cross, D.; Eyers, P.A.; Alessi, D.R. The HVps34-SGK3 Pathway Alleviates Sustained PI3K/Akt Inhibition by Stimulating MTORC1 and Tumour Growth. EMBO J. 2016, 35, 1902–1922. [Google Scholar] [CrossRef]
- Bridges, D.; Ma, J.-T.; Park, S.; Inoki, K.; Weisman, L.S.; Saltiel, A.R. Phosphatidylinositol 3,5-Bisphosphate Plays a Role in the Activation and Subcellular Localization of Mechanistic Target of Rapamycin 1. Mol. Biol. Cell 2012, 23, 2955–2962. [Google Scholar] [CrossRef]
- Marat, A.L.; Wallroth, A.; Lo, W.-T.; Müller, R.; Norata, G.D.; Falasca, M.; Schultz, C.; Haucke, V. MTORC1 Activity Repression by Late Endosomal Phosphatidylinositol 3,4-Bisphosphate. Science 2017, 356, 968–972. [Google Scholar] [CrossRef]
- Shin, H.-W.; Hayashi, M.; Christoforidis, S.; Lacas-Gervais, S.; Hoepfner, S.; Wenk, M.R.; Modregger, J.; Uttenweiler-Joseph, S.; Wilm, M.; Nystuen, A.; et al. An Enzymatic Cascade of Rab5 Effectors Regulates Phosphoinositide Turnover in the Endocytic Pathway. J. Cell Biol. 2005, 170, 607–618. [Google Scholar] [CrossRef]
- Naguib, A.; Bencze, G.; Cho, H.; Zheng, W.; Tocilj, A.; Elkayam, E.; Faehnle, C.R.; Jaber, N.; Pratt, C.P.; Chen, M.; et al. PTEN Functions by Recruitment to Cytoplasmic Vesicles. Mol. Cell 2015, 58, 255–268. [Google Scholar] [CrossRef]
- Isoyama, S.; Mori, S.; Sugiyama, D.; Kojima, Y.; Tada, Y.; Shitara, K.; Hinohara, K.; Dan, S.; Nishikawa, H. Cancer Immunotherapy with PI3K and PD-1 Dual-Blockade via Optimal Modulation of T Cell Activation Signal. J. Immunother. Cancer 2021, 9, e002279. [Google Scholar] [CrossRef]
- Posor, Y.; Jang, W.; Haucke, V. Phosphoinositides as Membrane Organizers. Nat. Rev. Mol. Cell Biol. 2022, 23, 797–816. [Google Scholar] [CrossRef]
- Yoon, M.-S.; Du, G.; Backer, J.M.; Frohman, M.A.; Chen, J. Class III PI-3-Kinase Activates Phospholipase D in an Amino Acid–Sensing MTORC1 Pathway. J. Cell Biol. 2011, 195, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, A.; Liang, X.; Liu, J.; Zou, F.; Chen, C.; Zhao, Z.; Deng, Y.; Wu, H.; Qi, Z.; et al. Simultaneous Inhibition of Vps34 Kinase Would Enhance PI3Kδ Inhibitor Cytotoxicity in the B-Cell Malignancies. Oncotarget 2016, 7, 53515–53525. [Google Scholar] [CrossRef] [PubMed]
- Barchéchath, S.; Williams, C.; Saade, K.; Lauwagie, S.; Jean-Claude, B. Rational Design of Multitargeted Tyrosine Kinase Inhibitors: A Novel Approach. Chem. Biol. Drug Des. 2009, 73, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Fortin, S.; Bérubé, G. Advances in the Development of Hybrid Anticancer Drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. [Google Scholar] [CrossRef]
- Chilamakuri, R.; Agarwal, S. Dual Targeting of PI3K and HDAC by CUDC-907 Inhibits Pediatric Neuroblastoma Growth. Cancers 2022, 14, 1067. [Google Scholar] [CrossRef]
- Li, X.; Su, Y.; Hege, K.; Madlambayan, G.; Edwards, H.; Knight, T.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; et al. The HDAC and PI3K Dual Inhibitor CUDC-907 Synergistically Enhances the Antileukemic Activity of Venetoclax in Preclinical Models of Acute Myeloid Leukemia. Haematologica 2020, 106, 1262–1277. [Google Scholar] [CrossRef]
- Rao, S.; Thibault, B.; Peyrard, L.; Larroque-Lombard, A.-L.; Rupp, M.; Thauvin, C.; Jean-Claude, B.J. Quantitative Analysis of the Potency of Equimolar Two-Drug Combinations and Combi-Molecules Involving Kinase Inhibitors In Vitro: The Concept of Balanced Targeting. Int. J. Mol. Sci. 2021, 22, 9569. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).