
Mycosis Fungoides and Sézary Syndrome: Microenvironment and Cancer Progression


{kind=link}
{kind=link}
Simple Summary
Abstract
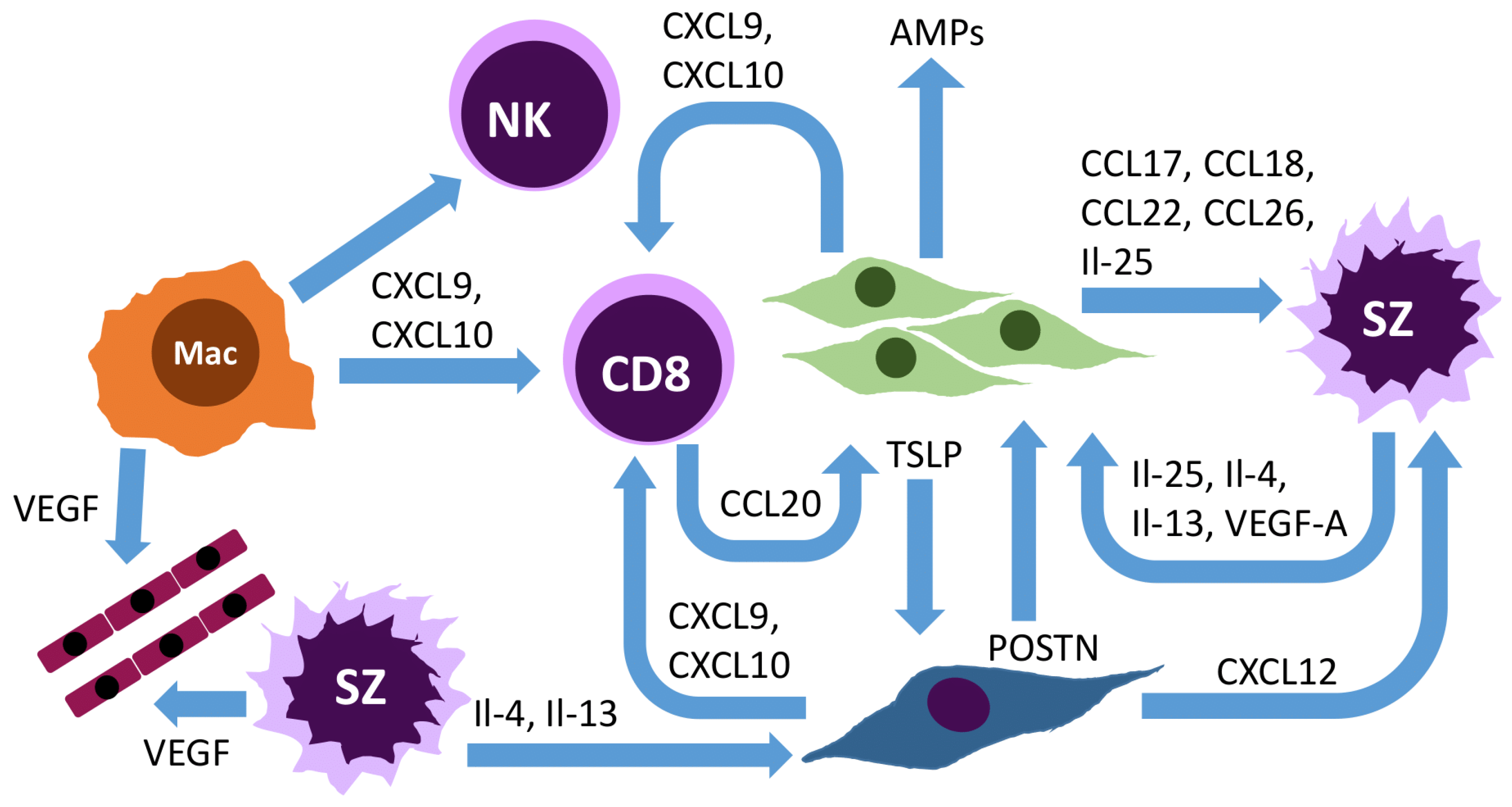
1. Introduction
2. Overall T-helper Type 2 (Th2) Polarization of the Mycosis Fungoides/Sézary Syndrome Tumor Microenvironment
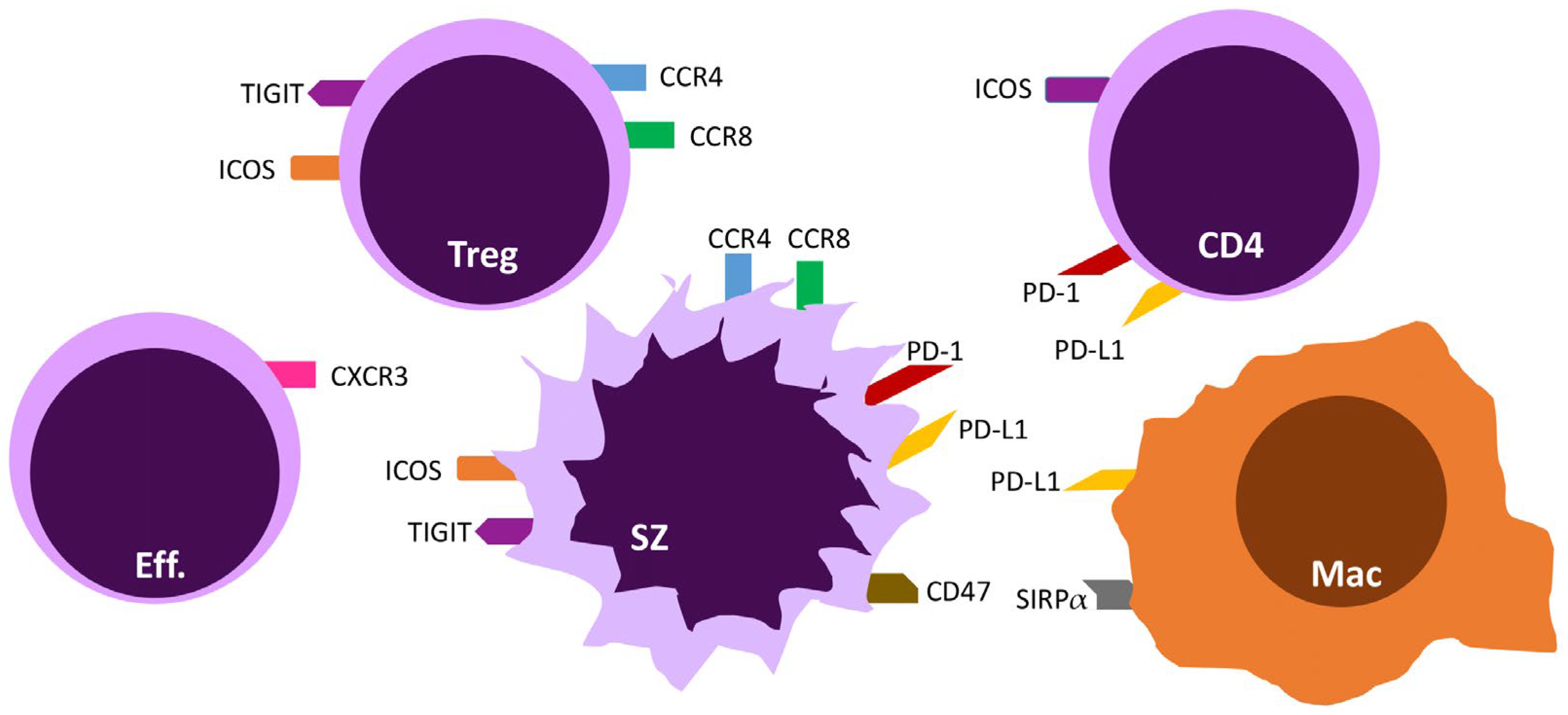
3. Targeting T-Cell Exhaustion via Immune Checkpoints
3.1. The PD-1/PD-L1 and PD-L2 Axis
3.2. ICOS/ICOS-L
3.3. TIGIT
4. Chemokine Receptors
4.1. CCR4
4.2. CCR8
5. Role of the Connective Tissue Cells and Other Cells of the Tumor Microenvironment in Mycosis Fungoides and Sézary Syndrome
5.1. Keratinocytes, Corneocytes, and the Microbiome
5.2. Fibroblasts
5.3. Endothelial Cells
5.4. Macrophages
5.5. Dendritic Cells
5.6. Mast Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Masson, A.; O’Malley, J.T.; Elco, C.P.; Garcia, S.S.; Divito, S.J.; Lowry, E.L.; Tawa, M.; Fisher, D.C.; Devlin, P.M.; Teague, J.E.; et al. High-Throughput Sequencing of the T Cell Receptor β Gene Identifies Aggressive Early-Stage Mycosis Fungoides. Sci. Transl. Med. 2018, 10, eaar5894. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, X.; Hwang, S.T.; Liu, J. The Role of Tumor Microenvironment in Mycosis Fungoides and Sézary Syndrome. Ann. Dermatol. 2021, 33, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Pileri, A.; Guglielmo, A.; Grandi, V.; Violetti, S.A.; Fanoni, D.; Fava, P.; Agostinelli, C.; Berti, E.; Quaglino, P.; Pimpinelli, N. The Microenvironment’s Role in Mycosis Fungoides and Sézary Syndrome: From Progression to Therapeutic Implications. Cells 2021, 10, 2780. [Google Scholar] [CrossRef]
- Quaglino, P.; Fava, P.; Pileri, A.; Grandi, V.; Sanlorenzo, M.; Panasiti, V.; Guglielmo, A.; Alberti-Violetti, S.; Novelli, M.; Astrua, C.; et al. Phenotypical Markers, Molecular Mutations, and Immune Microenvironment as Targets for New Treatments in Patients with Mycosis Fungoides and/or Sézary Syndrome. J. Invest. Dermatol. 2021, 141, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Rubio Gonzalez, B.; Zain, J.; Rosen, S.T.; Querfeld, C. Tumor Microenvironment in Mycosis Fungoides and Sézary Syndrome. Curr. Opin. Oncol. 2016, 28, 88–96. [Google Scholar] [CrossRef]
- Roelens, M.; Delord, M.; Ram-Wolff, C.; Marie-Cardine, A.; Alberdi, A.; Maki, G.; Homyrda, L.; Bensussan, A.; Bagot, M.; Toubert, A.; et al. Circulating and Skin-Derived Sézary Cells: Clonal but with Phenotypic Plasticity. Blood 2017, 130, 1468–1471. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Tabib, T.; Geskin, L.J.; Bayan, C.-A.; Conway, J.F.; Lafyatis, R.; Fuschiotti, P. Single-Cell Lymphocyte Heterogeneity in Advanced Cutaneous T-Cell Lymphoma Skin Tumors. Clin. Cancer Res. 2019, 25, 4443–4454. [Google Scholar] [CrossRef]
- Guenova, E.; Watanabe, R.; Teague, J.E.; Desimone, J.A.; Jiang, Y.; Dowlatshahi, M.; Schlapbach, C.; Schaekel, K.; Rook, A.H.; Tawa, M.; et al. TH2 Cytokines from Malignant Cells Suppress TH1 Responses and Enforce a Global TH2 Bias in Leukemic Cutaneous T-Cell Lymphoma. Clin. Cancer Res. 2013, 19, 3755–3763. [Google Scholar] [CrossRef] [PubMed]
- Tracey, L.; Villuendas, R.; Dotor, A.M.; Spiteri, I.; Ortiz, P.; García, J.F.; Peralto, J.L.R.; Lawler, M.; Piris, M.A. Mycosis Fungoides Shows Concurrent Deregulation of Multiple Genes Involved in the TNF Signaling Pathway: An Expression Profile Study. Blood 2003, 102, 1042–1050. [Google Scholar] [CrossRef]
- Fujii, K. New Therapies and Immunological Findings in Cutaneous T-Cell Lymphoma. Front. Oncol. 2018, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Krejsgaard, T.; Lindahl, L.M.; Mongan, N.P.; Wasik, M.A.; Litvinov, I.V.; Iversen, L.; Langhoff, E.; Woetmann, A.; Odum, N. Malignant Inflammation in Cutaneous T-cell Lymphoma—A Hostile Takeover. Semin. Immunopathol. 2017, 39, 269–282. [Google Scholar] [CrossRef]
- Stolearenco, V.; Namini, M.R.J.; Hasselager, S.S.; Gluud, M.; Buus, T.B.; Willerslev-Olsen, A.; Ødum, N.; Krejsgaard, T. Cellular Interactions and Inflammation in the Pathogenesis of Cutaneous T-Cell Lymphoma. Front. Cell Dev. Biol. 2020, 8, 851. [Google Scholar] [CrossRef]
- Johnson, V.E.; Vonderheid, E.C.; Hess, A.D.; Eischen, C.M.; McGirt, L.Y. Genetic Markers Associated with Progression in Early Mycosis Fungoides. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1431–1435. [Google Scholar] [CrossRef] [PubMed]
- Saulite, I.; Hoetzenecker, W.; Weidinger, S.; Cozzio, A.; Guenova, E.; Wehkamp, U. Sézary Syndrome and Atopic Dermatitis: Comparison of Immunological Aspects and Targets. BioMed Res. Int. 2016, 2016, 9717530. [Google Scholar] [CrossRef]
- Mollanazar, N.K.; Savage, K.T.; Pousti, B.T.; Jariwala, N.; Del Guzzo, C.; Haun, P.; Vittorio, C.C.; Rook, A.H.; Kim, E.J. Cutaneous T-Cell Lymphoma and Concomitant Atopic Dermatitis Responding to Dupilumab. Cutis 2020, 106, 131–132. [Google Scholar] [CrossRef]
- Lazaridou, I.; Ram-Wolff, C.; Bouaziz, J.; Bégon, E.; Battistella, M.; Rivet, J.; Jachiet, M.; Bagot, M.; Masson, A. Dupilumab Treatment in Two Patients with Cutaneous T-Cell Lymphomas. Acta Derm. Venereol. 2020, 100, adv00271. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Wong, L.; Lang, A.; Kraus, C.; Anderson, N.; Elsensohn, A. Cutaneous T-cell Lymphoma Following Dupilumab Use: A Systematic Review. Int. J. Dermatol. 2022, ijd.16388. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Greenberg, P.D. Tolerance and Exhaustion: Defining Mechanisms of T Cell Dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Leung, S.; Myskowski, P.L.; Curran, S.A.; Goldman, D.A.; Heller, G.; Wu, X.; Kil, S.H.; Sharma, S.; Finn, K.J.; et al. Primary T Cells from Cutaneous T-Cell Lymphoma Skin Explants Display an Exhausted Immune Checkpoint Profile. Cancer Immunol. Res. 2018, 6, 900–909. [Google Scholar] [CrossRef]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 Inhibitors as a Form of Cancer Immunotherapy: A Comprehensive Review of Registration Trials and Future Considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers, and Combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Samimi, S.; Benoit, B.; Evans, K.; Wherry, E.J.; Showe, L.; Wysocka, M.; Rook, A.H. Increased Programmed Death-1 Expression on CD4+ T Cells in Cutaneous T-Cell Lymphoma: Implications for Immune Suppression. Arch. Dermatol. 2010, 146, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Cetinözman, F.; Jansen, P.M.; Vermeer, M.H.; Willemze, R. Differential Expression of Programmed Death-1 (PD-1) in Sézary Syndrome and Mycosis Fungoides. Arch. Dermatol. 2012, 148, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Basis of PD1/PD-L1 Therapies. J. Clin. Med. 2019, 8, 2168. [Google Scholar] [CrossRef] [PubMed]
- Kantekure, K.; Yang, Y.; Raghunath, P.; Schaffer, A.; Woetmann, A.; Zhang, Q.; Odum, N.; Wasik, M. Expression Patterns of the Immunosuppressive Proteins PD-1/CD279 and PD-L1/CD274 at Different Stages of Cutaneous T-Cell Lymphoma/Mycosis Fungoides. Am. J. Dermatopathol. 2012, 34, 126–128. [Google Scholar] [CrossRef]
- Di Raimondo, C.; Rubio-Gonzalez, B.; Palmer, J.; Weisenburger, D.D.; Zain, J.; Wu, X.; Han, Z.; Rosen, S.T.; Song, J.Y.; Querfeld, C. Expression of Immune Checkpoint Molecules Programmed Death Protein 1, Programmed Death-Ligand 1 and Inducible T-Cell Co-Stimulator in Mycosis Fungoides and Sézary Syndrome: Association with Disease Stage and Clinical Outcome. Br. J. Dermatol. 2022, 187, 234–243. [Google Scholar] [CrossRef]
- Park, J.-H.; Han, J.H.; Kang, H.Y.; Lee, E.-S.; Kim, Y.C. Expression of Follicular Helper T-Cell Markers in Primary Cutaneous T-Cell Lymphoma. Am. J. Dermatopathol. 2014, 36, 465–470. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Rook, A.H.; Porcu, P.; Foss, F.; Moskowitz, A.J.; Shustov, A.; Shanbhag, S.; Sokol, L.; Fling, S.P.; Ramchurren, N.; et al. Pembrolizumab in Relapsed and Refractory Mycosis Fungoides and Sézary Syndrome: A Multicenter Phase II Study. J. Clin. Oncol. 2020, 38, 20–28. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients with Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef]
- Stadler, R.; Romero, P.O.; Bagot, M.; Quaglino, P.; Guenova, E.; Jonak, C.; Papadavid, E.; Stranzenbach, R.; Sartori, D.; Musoro, J.Z.; et al. Phase II Trial of Atezolizumab (Anti-PD-L1) in the Treatment of Stage IIb-IVB Mycosis Fungoides/Sézary Syndrome Patients Relapsed/Refractory after a Previous Systemic Treatment (PARCT). Eur. J. Cancer 2021, 156 (Suppl. 1), S22–S23. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Tsai, N.-C.; Palmer, J.; Martinez, X.U.; Abdulla, F.; Wu, X.; Rosen, S.T.; Zain, J. Phase 1 Results of Anti-PD-Ligand 1 (Durvalumab) & Lenalidomide in Patients with Cutaneous T Cell Lymphoma and Correlation with Programmed Death Ligand 1 Expression and Gene Expression Profile. Blood 2020, 136, 20. [Google Scholar] [CrossRef]
- Phillips, D.; Matusiak, M.; Gutierrez, B.R.; Bhate, S.S.; Barlow, G.L.; Jiang, S.; Demeter, J.; Smythe, K.S.; Pierce, R.H.; Fling, S.P.; et al. Immune Cell Topography Predicts Response to PD-1 Blockade in Cutaneous T Cell Lymphoma. Nat. Commun. 2021, 12, 6726. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, K.H.; Kang, J.; Borcoman, E.; Saada-Bouzid, E.; Kronbichler, A.; Hong, S.H.; de Rezende, L.F.M.; Ogino, S.; Keum, N.; et al. Hyperprogressive Disease during Anti-PD-1 (PDCD1)/PD-L1 (CD274) Therapy: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 1699. [Google Scholar] [CrossRef]
- Gao, Y.; Hu, S.; Li, R.; Jin, S.; Liu, F.; Liu, X.; Li, Y.; Yan, Y.; Liu, W.; Gong, J.; et al. Hyperprogression of Cutaneous T Cell Lymphoma after Anti-PD-1 Treatment. JCI Insight 2023, e164793. [Google Scholar] [CrossRef]
- Rujas, E.; Cui, H.; Sicard, T.; Semesi, A.; Julien, J.-P. Structural Characterization of the ICOS/ICOS-L Immune Complex Reveals High Molecular Mimicry by Therapeutic Antibodies. Nat. Commun. 2020, 11, 5066. [Google Scholar] [CrossRef]
- Solinas, C.; Gu-Trantien, C.; Willard-Gallo, K. The Rationale behind Targeting the ICOS-ICOS Ligand Costimulatory Pathway in Cancer Immunotherapy. ESMO Open 2020, 5, e000544. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.; Schürch, C.M.; Khodadoust, M.S.; Kim, Y.H.; Nolan, G.P.; Jiang, S. Highly Multiplexed Phenotyping of Immunoregulatory Proteins in the Tumor Microenvironment by CODEX Tissue Imaging. Front. Immunol. 2021, 12, 687673. [Google Scholar] [CrossRef]
- Geskin, L.J.; Akilov, O.E.; Kwon, S.; Schowalter, M.; Watkins, S.; Whiteside, T.L.; Butterfield, L.H.; Falo, L.D. Therapeutic Reduction of Cell-Mediated Immunosuppression in Mycosis Fungoides and Sézary Syndrome. Cancer Immunol. Immunother. 2018, 67, 423–434. [Google Scholar] [CrossRef]
- Amatore, F.; Ortonne, N.; Lopez, M.; Orlanducci, F.; Castellano, R.; Ingen-Housz-Oro, S.; De Croos, A.; Salvado, C.; Gorvel, L.; Goubard, A.; et al. ICOS Is Widely Expressed in Cutaneous T-Cell Lymphoma, and Its Targeting Promotes Potent Killing of Malignant Cells. Blood Adv. 2020, 4, 5203–5214. [Google Scholar] [CrossRef]
- Chavez, J.C.; Foss, F.M.; William, B.M.; Brammer, J.E.; Smith, S.M.; Prica, A.; Zain, J.M.; Tuscano, J.M.; Glenn, M.; Mehta-Shah, N.; et al. A Phase I Study of Anti-ICOS Antibody MEDI-570 for Relapsed/Refractory (R/R) Peripheral T-Cell Lymphoma (PTCL) and Angioimmunoblastic T-Cell Lymphoma (AITL) (NCI-9930). Blood 2020, 136, 5–6. [Google Scholar] [CrossRef]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New Checkpoint Receptor Targets for Cancer Immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Johnston, R.J.; Yu, X.; Grogan, J.L. The Checkpoint Inhibitor TIGIT Limits Antitumor and Antiviral CD8+ T Cell Responses. OncoImmunology 2015, 4, e1036214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the Checkpoint Receptor TIGIT Prevents NK Cell Exhaustion and Elicits Potent Anti-Tumor Immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Anzengruber, F.; Ignatova, D.; Schlaepfer, T.; Chang, Y.-T.; French, L.E.; Pascolo, S.; Contassot, E.; Bobrowicz, M.; Hoetzenecker, W.; Guenova, E. Divergent LAG-3 versus BTLA, TIGIT, and FCRL3 Expression in Sézary Syndrome. Leuk. Lymphoma 2019, 60, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Preillon, J.; Cuende, J.; Rabolli, V.; Garnero, L.; Mercier, M.; Wald, N.; Pappalardo, A.; Denies, S.; Jamart, D.; Michaux, A.-C.; et al. Restoration of T-Cell Effector Function, Depletion of Tregs, and Direct Killing of Tumor Cells: The Multiple Mechanisms of Action of a-TIGIT Antagonist Antibodies. Mol. Cancer Ther. 2021, 20, 121–131. [Google Scholar] [CrossRef]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus Atezolizumab versus Placebo plus Atezolizumab as a First-Line Treatment for PD-L1-Selected Non-Small-Cell Lung Cancer (CITYSCAPE): Primary and Follow-up Analyses of a Randomised, Double-Blind, Phase 2 Study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef]
- Campbell, J.J.; O’Connell, D.J.; Wurbel, M.-A. Cutting Edge: Chemokine Receptor CCR4 Is Necessary for Antigen-Driven Cutaneous Accumulation of CD4 T Cells under Physiological Conditions. J. Immunol. 2007, 178, 3358–3362. [Google Scholar] [CrossRef]
- Shono, Y.; Suga, H.; Kamijo, H.; Fujii, H.; Oka, T.; Miyagaki, T.; Shishido-Takahashi, N.; Sugaya, M.; Sato, S. Expression of CCR3 and CCR4 Suggests a Poor Prognosis in Mycosis Fungoides and Sézary Syndrome. Acta Derm. Venereol. 2019, 99, 809–812. [Google Scholar] [CrossRef]
- Kim, Y.H.; Bagot, M.; Pinter-Brown, L.; Rook, A.H.; Porcu, P.; Horwitz, S.M.; Whittaker, S.; Tokura, Y.; Vermeer, M.; Zinzani, P.L.; et al. Mogamulizumab versus Vorinostat in Previously Treated Cutaneous T-Cell Lymphoma (MAVORIC): An International, Open-Label, Randomised, Controlled Phase 3 Trial. Lancet Oncol. 2018, 19, 1192–1204. [Google Scholar] [CrossRef]
- De Masson, A.; Darbord, D.; Dobos, G.; Boisson, M.; Roelens, M.; Ram-Wolff, C.; Cassius, C.; Le Buanec, H.; de la Grange, P.; Jouenne, F.; et al. Macrophage-Derived CXCL9 and CXCL11, T-Cell Skin Homing, and Disease Control in Mogamulizumab-Treated CTCL Patients. Blood 2022, 139, 1820–1832. [Google Scholar] [CrossRef]
- Beygi, S.; Duran, G.E.; Fernandez-Pol, S.; Rook, A.H.; Kim, Y.H.; Khodadoust, M.S. Resistance to Mogamulizumab Is Associated with Loss of CCR4 in Cutaneous T-Cell Lymphoma. Blood 2022, 139, 3732–3736. [Google Scholar] [CrossRef] [PubMed]
- Roelens, M.; de Masson, A.; Andrillon, A.; Ram-Wolff, C.; Biard, L.; Boisson, M.; Mourah, S.; Battistella, M.; Toubert, A.; Bagot, M.; et al. Mogamulizumab Induces Long-Term Immune Restoration and Reshapes Tumour Heterogeneity in Sézary Syndrome. Br. J. Dermatol. 2022, 186, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Watanabe, R.; Teague, J.E.; Schlapbach, C.; Tawa, M.C.; Adams, N.; Dorosario, A.A.; Chaney, K.S.; Cutler, C.S.; Leboeuf, N.R.; et al. Skin Effector Memory T Cells Do Not Recirculate and Provide Immune Protection in Alemtuzumab-Treated CTCL Patients. Sci. Transl. Med. 2012, 4, 117ra7. [Google Scholar] [CrossRef]
- Campbell, J.R.; McDonald, B.R.; Mesko, P.B.; Siemers, N.O.; Singh, P.B.; Selby, M.; Sproul, T.W.; Korman, A.J.; Vlach, L.M.; Houser, J.; et al. Fc-Optimized Anti-CCR8 Antibody Depletes Regulatory T Cells in Human Tumor Models. Cancer Res. 2021, 81, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, H.; Dombrecht, B.; Kiss, M.; Roose, H.; Allen, E.; Van Overmeire, E.; Kancheva, D.; Martens, L.; Murgaski, A.; Bardet, P.M.R.; et al. Therapeutic Depletion of CCR8+ Tumor-Infiltrating Regulatory T Cells Elicits Antitumor Immunity and Synergizes with Anti-PD-1 Therapy. J. Immunother. Cancer 2021, 9, e001749. [Google Scholar] [CrossRef]
- Giustiniani, J.; Dobos, G.; Moins-Teisserenc, H.; Eustaquio, T.; Battistella, M.; Ortonne, N.; Ram-Wolff, C.; Bouaziz, J.-D.; Marie-Cardine, A.; Mourah, S.; et al. CCR8 Is a New Therapeutic Target in Cutaneous T-Cell Lymphomas. Blood Adv. 2022, 6, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Jost, M.; Wehkamp, U. The Skin Microbiome and Influencing Elements in Cutaneous T-Cell Lymphomas. Cancers 2022, 14, 1324. [Google Scholar] [CrossRef] [PubMed]
- Miyagaki, T.; Sugaya, M.; Suga, H.; Kamata, M.; Ohmatsu, H.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. IL-22, but Not IL-17, Dominant Environment in Cutaneous T-Cell Lymphoma. Clin. Cancer Res. 2011, 17, 7529–7538. [Google Scholar] [CrossRef]
- Xu, M.; Dong, C. IL-25 in Allergic Inflammation. Immunol. Rev. 2017, 278, 185–191. [Google Scholar] [CrossRef]
- Nakajima, R.; Miyagaki, T.; Hirakawa, M.; Oka, T.; Takahashi, N.; Suga, H.; Yoshizaki, A.; Fujita, H.; Asano, Y.; Sugaya, M.; et al. Interleukin-25 Is Involved in Cutaneous T-Cell Lymphoma Progression by Establishing a T Helper 2-Dominant Microenvironment. Br. J. Dermatol. 2018, 178, 1373–1382. [Google Scholar] [CrossRef]
- Takahashi, N.; Sugaya, M.; Suga, H.; Oka, T.; Kawaguchi, M.; Miyagaki, T.; Fujita, H.; Sato, S. Thymic Stromal Chemokine TSLP Acts through Th2 Cytokine Production to Induce Cutaneous T-Cell Lymphoma. Cancer Res. 2016, 76, 6241–6252. [Google Scholar] [CrossRef]
- Aronovich, A.; Moyal, L.; Gorovitz, B.; Amitay-Laish, I.; Naveh, H.P.; Forer, Y.; Maron, L.; Knaneh, J.; Ad-El, D.; Yaacobi, D.; et al. Cancer-Associated Fibroblasts in Mycosis Fungoides Promote Tumor Cell Migration and Drug Resistance through CXCL12/CXCR4. J. Invest. Dermatol. 2021, 141, 619–627.e2. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Suryawanshi, H.; Morozov, P.; Gay-Mimbrera, J.; Del Duca, E.; Kim, H.J.; Kameyama, N.; Estrada, Y.; Der, E.; Krueger, J.G.; et al. Single-Cell Transcriptome Analysis of Human Skin Identifies Novel Fibroblast Subpopulation and Enrichment of Immune Subsets in Atopic Dermatitis. J. Allergy Clin. Immunol. 2020, 145, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Vieyra-Garcia, P.; Crouch, J.D.; O’Malley, J.T.; Seger, E.W.; Yang, C.H.; Teague, J.E.; Vromans, A.M.; Gehad, A.; Win, T.S.; Yu, Z.; et al. Benign T Cells Drive Clinical Skin Inflammation in Cutaneous T Cell Lymphoma. JCI Insight 2019, 4, e124233. [Google Scholar] [CrossRef] [PubMed]
- Dobos, G.; Calugareanu, A.; Michel, L.; Battistella, M.; Ram-Wolff, C.; Bouaziz, J.-D.; Bensussan, A.; de Masson, A.; Bagot, M. Exploring the Role of the Skin Microenvironment in Cutaneous T-Cell Lymphoma Using Single Cell RNA-Sequencing. Eur. J. Cancer 2021, 156, S3–S4. [Google Scholar] [CrossRef]
- Jankowska-Konsur, A.; Kobierzycki, C.; Grzegrzolka, J.; Piotrowska, A.; Gomulkiewicz, A.; Glatzel-Plucinska, N.; Olbromski, M.; Podhorska-Okolow, M.; Szepietowski, J.C.; Dziegiel, P. Expression of CD31 in Mycosis Fungoides. Anticancer Res. 2016, 36, 4575–4582. [Google Scholar] [CrossRef]
- Jankowska-Konsur, A.; Kobierzycki, C.; Grzegrzółka, J.; Piotrowska, A.; Gomulkiewicz, A.; Glatzel-Plucinska, N.; Reich, A.; Podhorska-Okołów, M.; Dzięgiel, P.; Szepietowski, J. Podoplanin Expression Correlates with Disease Progression in Mycosis Fungoides. Acta Derm. Venereol. 2017, 97, 235–241. [Google Scholar] [CrossRef]
- Pedersen, I.H.; Willerslev-Olsen, A.; Vetter-Kauczok, C.; Krejsgaard, T.; Lauenborg, B.; Kopp, K.L.; Geisler, C.; Bonefeld, C.M.; Zhang, Q.; Wasik, M.A.; et al. Vascular Endothelial Growth Factor Receptor-3 Expression in Mycosis Fungoides. Leuk. Lymphoma 2013, 54, 819–826. [Google Scholar] [CrossRef]
- Sakamoto, M.; Miyagaki, T.; Kamijo, H.; Oka, T.; Takahashi, N.; Suga, H.; Yoshizaki, A.; Asano, Y.; Sugaya, M.; Sato, S. Serum Vascular Endothelial Growth Factor A Levels Reflect Itch Severity in Mycosis Fungoides and Sézary Syndrome. J. Dermatol. 2018, 45, 95–99. [Google Scholar] [CrossRef]
- Rasheed, H.; Tolba Fawzi, M.M.; Abdel-Halim, M.R.E.; Eissa, A.M.; Mohammed Salem, N.; Mahfouz, S. Immunohistochemical Study of the Expression of Matrix Metalloproteinase-9 in Skin Lesions of Mycosis Fungoides. Am. J. Dermatopathol. 2010, 32, 162–169. [Google Scholar] [CrossRef]
- Sugaya, M.; Miyagaki, T.; Ohmatsu, H.; Suga, H.; Kai, H.; Kamata, M.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; et al. Association of the Numbers of CD163+ Cells in Lesional Skin and Serum Levels of Soluble CD163 with Disease Progression of Cutaneous T Cell Lymphoma. J. Dermatol. Sci. 2012, 68, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Furudate, S.; Fujimura, T.; Kakizaki, A.; Kambayashi, Y.; Asano, M.; Watabe, A.; Aiba, S. The Possible Interaction between Periostin Expressed by Cancer Stroma and Tumor-Associated Macrophages in Developing Mycosis Fungoides. Exp. Dermatol. 2016, 25, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Assaf, C.; Hwang, S.T. Mac Attack: Macrophages as Key Drivers of Cutaneous T-Cell Lymphoma Pathogenesis. Exp. Dermatol. 2016, 25, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Folkes, A.S.; Feng, M.; Zain, J.M.; Abdulla, F.; Rosen, S.T.; Querfeld, C. Targeting CD47 as a Cancer Therapeutic Strategy: The Cutaneous T-Cell Lymphoma Experience. Curr. Opin. Oncol. 2018, 30, 332–337. [Google Scholar] [CrossRef]
- Cao, X.; Wang, Y.; Zhang, W.; Zhong, X.; Gunes, E.G.; Dang, J.; Wang, J.; Epstein, A.L.; Querfeld, C.; Sun, Z.; et al. Targeting Macrophages for Enhancing CD47 Blockade–Elicited Lymphoma Clearance and Overcoming Tumor-Induced Immunosuppression. Blood 2022, 139, 3290–3302. [Google Scholar] [CrossRef]
- Rowden, G.; Phillips, T.M.; Lewis, M.G.; Wilkinson, R.D. Target Role of Langerhans Cells in Mycosis Fungoides: Transmission and Immuno-Electron Microscopic Studies. J. Cutan. Pathol. 1979, 6, 364–382. [Google Scholar] [CrossRef]
- Rabenhorst, A.; Schlaak, M.; Heukamp, L.C.; Förster, A.; Theurich, S.; von Bergwelt-Baildon, M.; Büttner, R.; Kurschat, P.; Mauch, C.; Roers, A.; et al. Mast Cells Play a Protumorigenic Role in Primary Cutaneous Lymphoma. Blood 2012, 120, 2042–2054. [Google Scholar] [CrossRef]
- Eder, J.; Rogojanu, R.; Jerney, W.; Erhart, F.; Dohnal, A.; Kitzwögerer, M.; Steiner, G.; Moser, J.; Trautinger, F. Mast Cells Are Abundant in Primary Cutaneous T-Cell Lymphomas: Results from a Computer-Aided Quantitative Immunohistological Study. PLoS ONE 2016, 11, e0163661. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobos, G.; Lazaridou, I.; de Masson, A. Mycosis Fungoides and Sézary Syndrome: Microenvironment and Cancer Progression. Cancers 2023, 15, 746. https://doi.org/10.3390/cancers15030746
Dobos G, Lazaridou I, de Masson A. Mycosis Fungoides and Sézary Syndrome: Microenvironment and Cancer Progression. Cancers. 2023; 15(3):746. https://doi.org/10.3390/cancers15030746
Chicago/Turabian StyleDobos, Gabor, Ingrid Lazaridou, and Adèle de Masson. 2023. "Mycosis Fungoides and Sézary Syndrome: Microenvironment and Cancer Progression" Cancers 15, no. 3: 746. https://doi.org/10.3390/cancers15030746
APA StyleDobos, G., Lazaridou, I., & de Masson, A. (2023). Mycosis Fungoides and Sézary Syndrome: Microenvironment and Cancer Progression. Cancers, 15(3), 746. https://doi.org/10.3390/cancers15030746