
Proteome-Wide Analysis Reveals TFEB Targets for Establishment of a Prognostic Signature to Predict Clinical Outcomes of Colorectal Cancer

 ,
,
Abstract
Simple Summary
Abstract
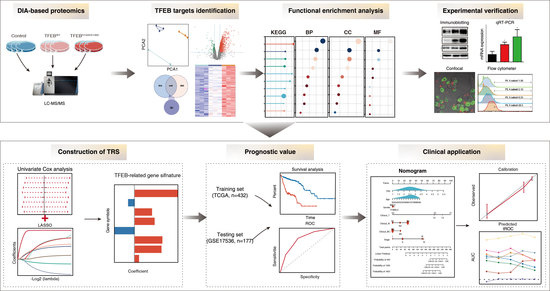
{kind=link}
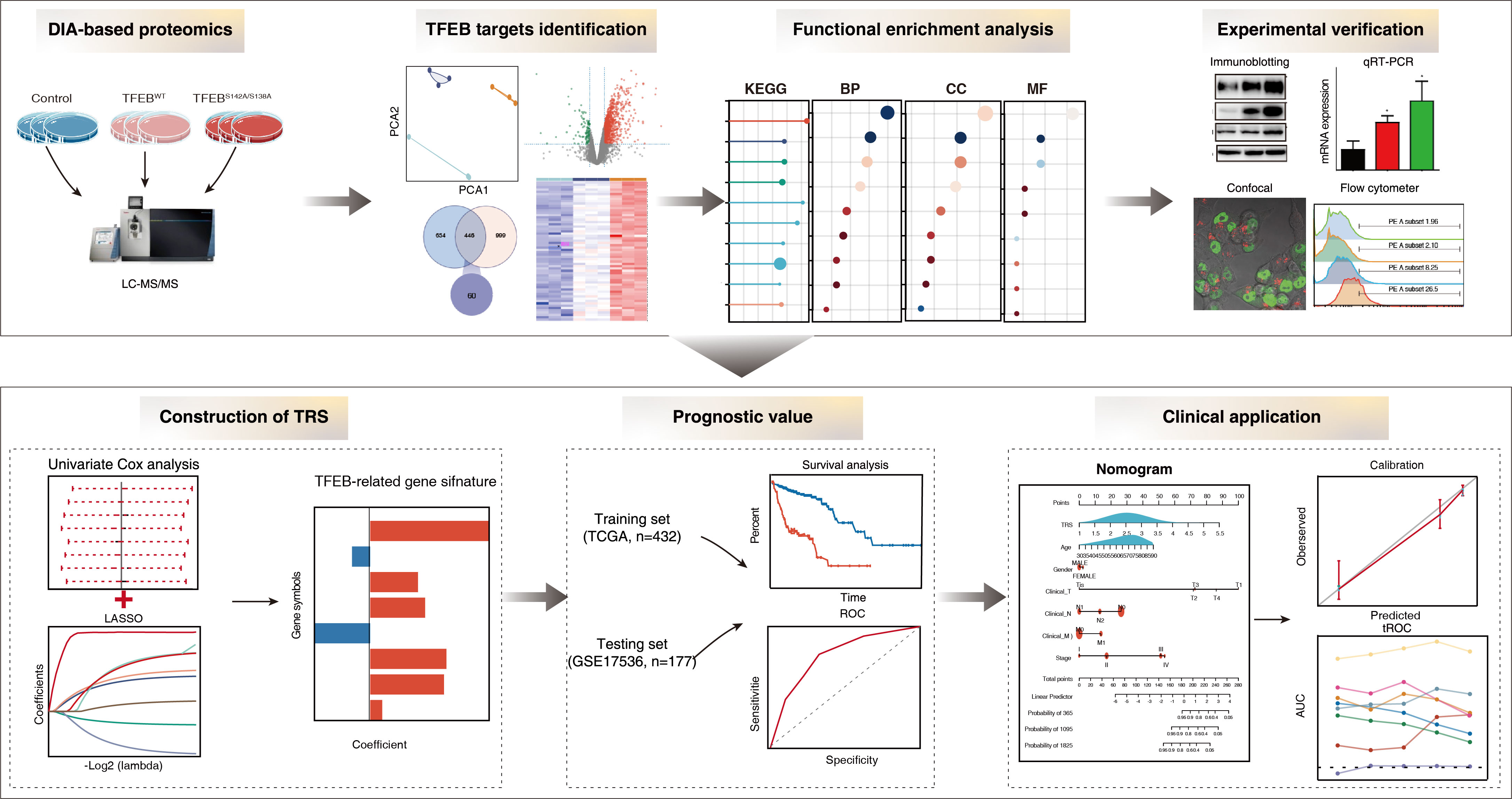
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Plasmid Construction, Retroviral Infection, and Transfection
2.3. Immunoblotting
2.4. Cell Proliferation Assays
2.5. Confocal Examination
2.6. Pinocytosis Assays
2.7. Mass Spectrometry (MS) Analysis
2.8. Analysis of Differentially Expressed TFEB Targets
2.9. Gene Ontology Analysis
2.10. Publicly Available mRNA Data
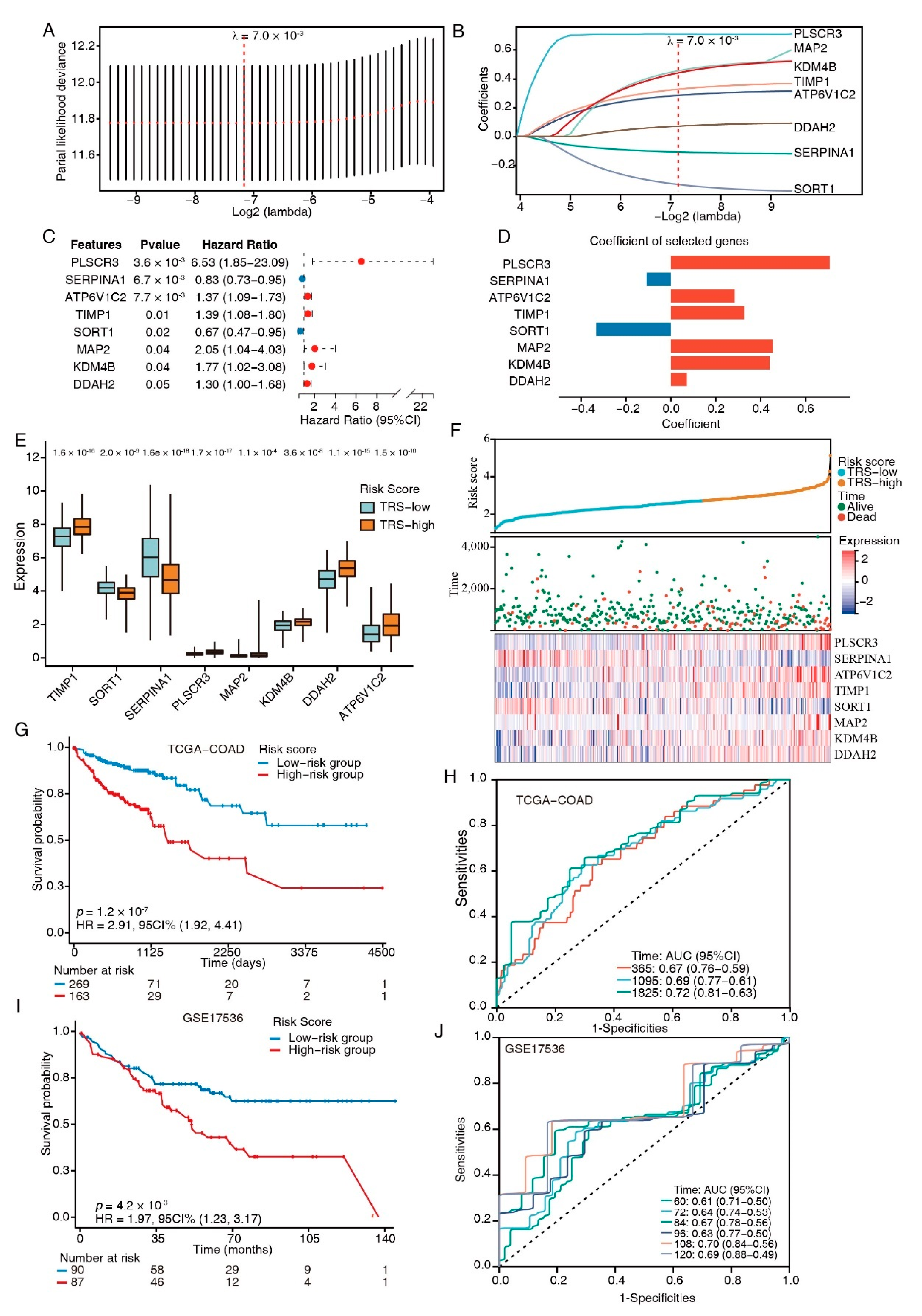
2.11. Signature Generation
2.12. Classification, Prediction, and Validation in GEO and TCGA
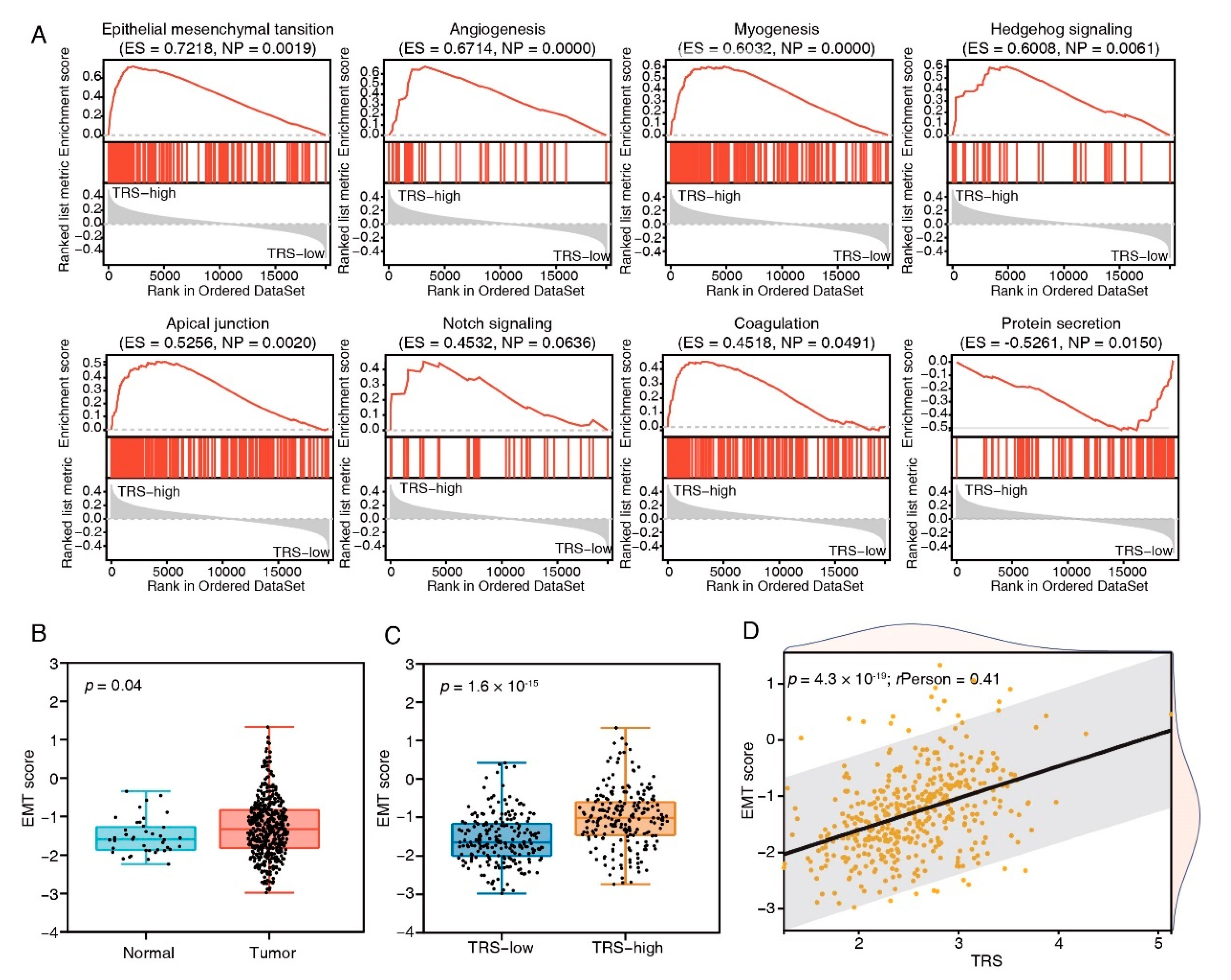
2.13. Gene Set Enrichment Analysis (GSEA)
2.14. Calculation of Epithelial–Mesenchymal Transition (EMT) Scores
3. Results
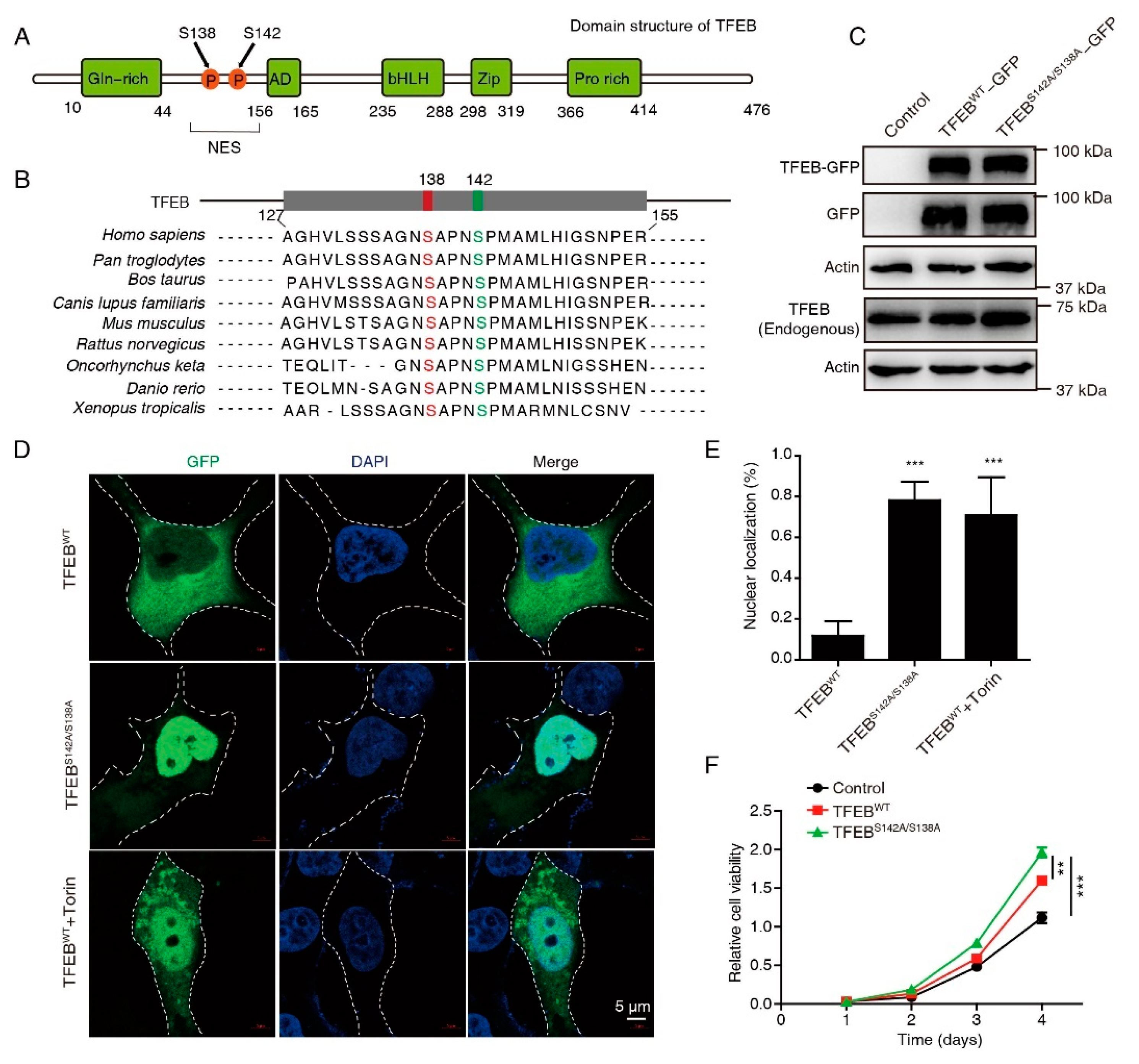
3.1. Constitutive Nuclear Import of TFEB Promotes CRC Proliferation
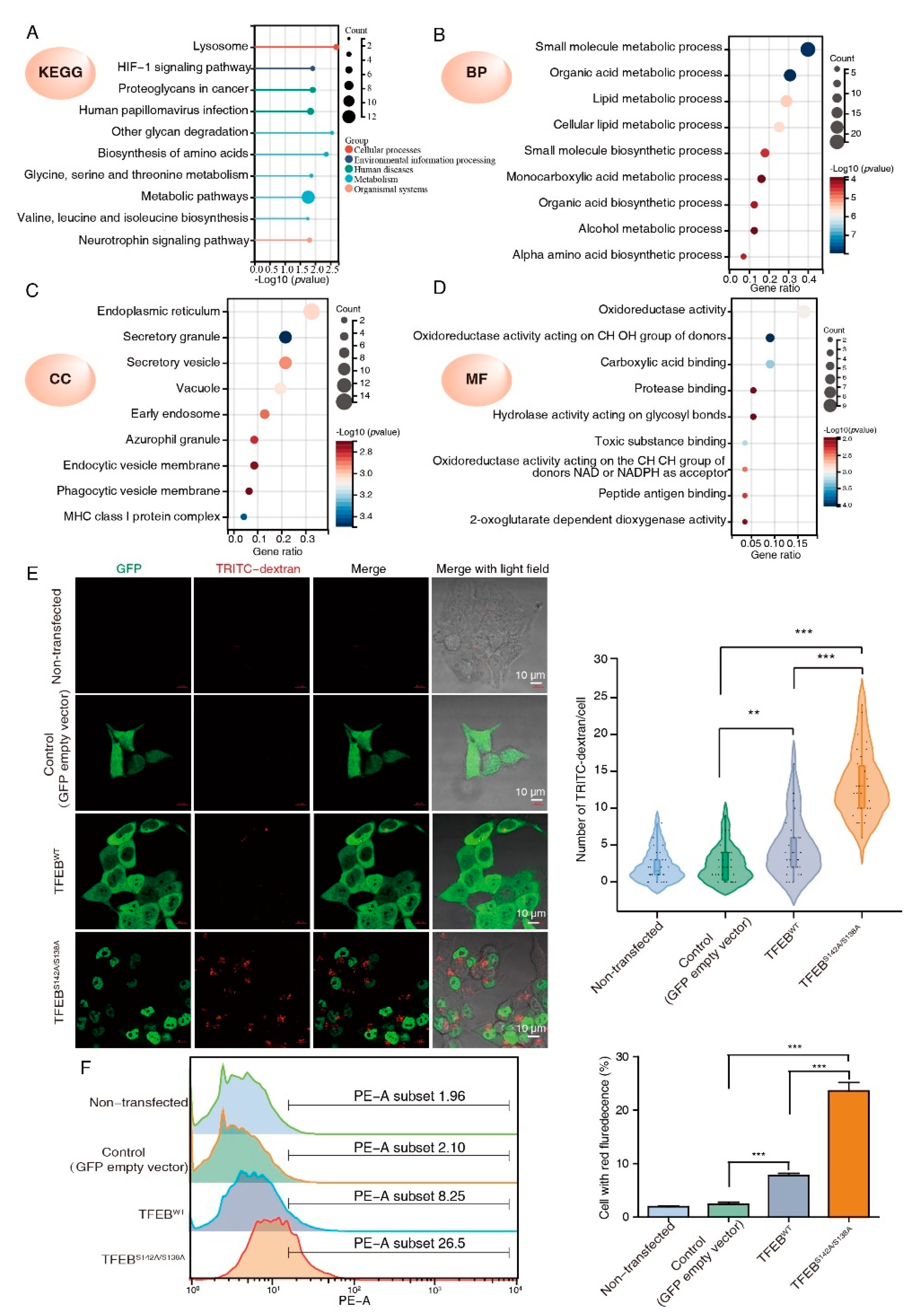
3.2. DIA-Based Proteomics Profiling Reveals Potential TFEB Targets
3.3. TFEB Increases Cellular Pinocytosis Rates
3.4. Establishment of a TFEB Target Signature for Prognosis
3.5. Tumor-Biology-Associated Potential Functions of the TFEB Signature
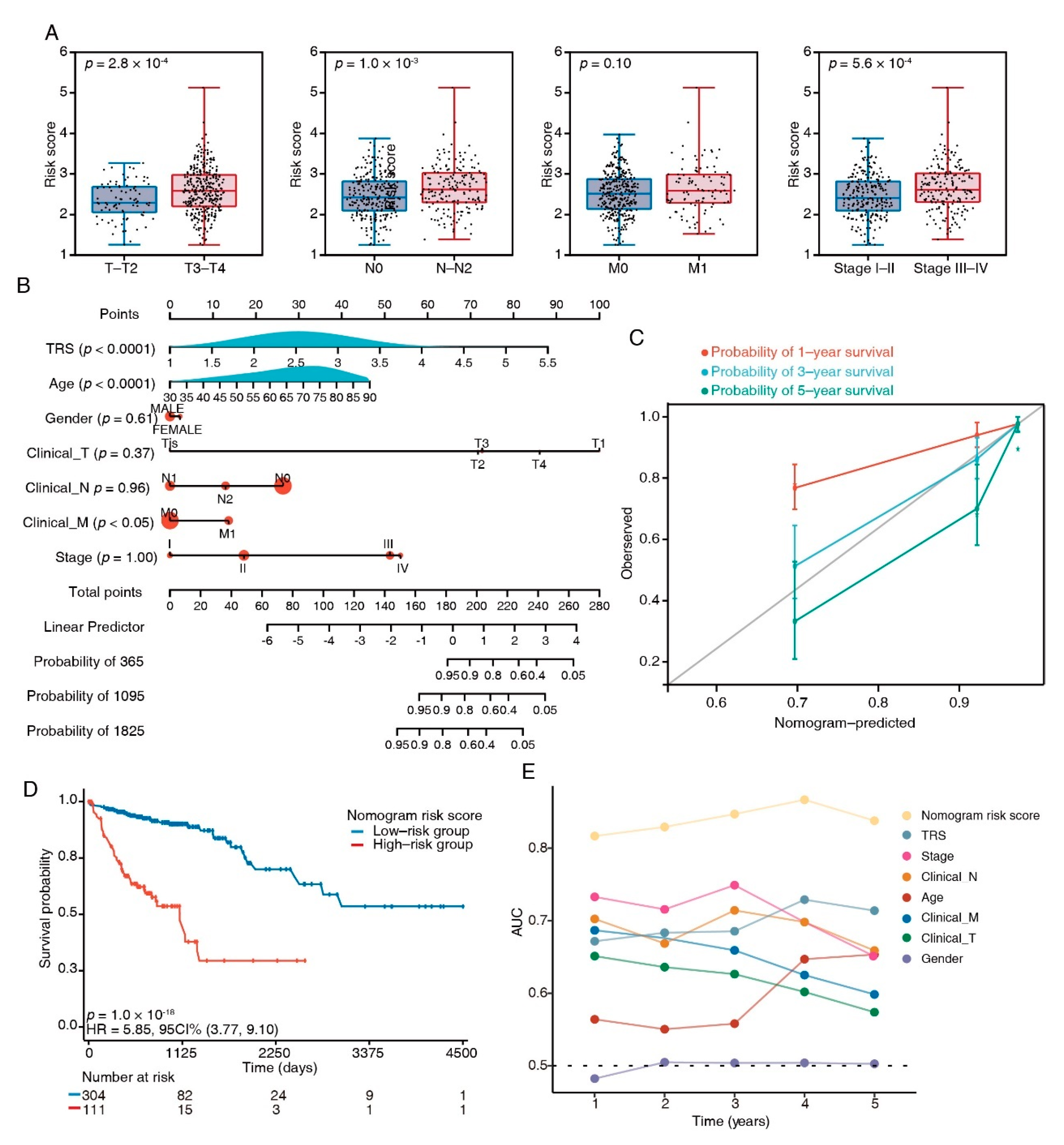
3.6. Combination of the TRS and Clinicopathological Characteristics Improves Survival Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Chen, B.; Scurrah, C.R.; McKinley, E.T.; Simmons, A.J.; Ramirez-Solano, M.A.; Zhu, X.; Markham, N.O.; Heiser, C.N.; Vega, P.N.; Rolong, A.; et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 2021, 184, 6262–6280.e6226. [Google Scholar] [CrossRef]
- Salvucci, M.; Crawford, N.; Stott, K.; Bullman, S.; Longley, D.B.; Prehn, J.H.M. Patients with mesenchymal tumours and high Fusobacteriales prevalence have worse prognosis in colorectal cancer (CRC). Gut 2022, 71, 1600–1612. [Google Scholar] [CrossRef]
- Bhagwat, A.S.; Vakoc, C.R. Targeting Transcription Factors in Cancer. Trends Cancer 2015, 1, 53–65. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef]
- Laissue, P. The forkhead-box family of transcription factors: Key molecular players in colorectal cancer pathogenesis. Mol. Cancer 2019, 18, 5. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Liang, J.; Jia, X.; Wang, K.; Zhao, N. High expression of TFEB is associated with aggressive clinical features in colorectal cancer. OncoTargets Ther. 2018, 11, 8089–8098. [Google Scholar] [CrossRef]
- Li, Y.; Hodge, J.; Liu, Q.; Wang, J.; Wang, Y.; Evans, T.D.; Altomare, D.; Yao, Y.; Murphy, E.A.; Razani, B.; et al. TFEB is a master regulator of tumor-associated macrophages in breast cancer. J. Immunother. Cancer 2020, 8, e000543. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018, 9, 3312. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, M.; Ding, X.; Yan, C.; Song, Z.; Chen, L.; Huang, X.; Wang, X.; Jian, Y.; Tang, G.; et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 2016, 18, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tang, B.; Wang, S.; Tang, L.; Wen, D.; Vlodavsky, I.; Yang, S.M. Non-enzymatic heparanase enhances gastric tumor proliferation via TFEB-dependent autophagy. Oncogenesis 2022, 11, 49. [Google Scholar] [CrossRef]
- Li, C.; Wang, L. TFEB-dependent autophagy is involved in scavenger receptor OLR1/LOX-1-mediated tumor progression. Autophagy 2022, 18, 462–464. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Zheng, C.C.; Huang, Z.J.; Zhang, W.X.; Long, Y.L.; Gao, G.B.; Sun, Y.; Xu, W.W.; Li, B.; et al. C20orf24 promotes colorectal cancer progression by recruiting Rin1 to activate Rab5-mediated mitogen-activated protein kinase/extracellular signal-regulated kinase signalling. Clin. Transl. Med. 2022, 12, e796. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Li, N.; Liu, W.T.; Liang, J.Z.; Sun, Y.; Zhang, W.X.; Fang, R.D.; Huang, S.L.; Sun, Z.H.; et al. Curcumol Overcomes TRAIL Resistance of Non-Small Cell Lung Cancer by Targeting NRH:Quinone Oxidoreductase 2 (NQO2). Adv. Sci. 2020, 7, 2002306. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Y.M.; Hu, Y.Y.; Ouyang, L.; Sun, Z.H.; Yin, X.F.; Li, N.; He, Q.Y.; Wang, Y. Inhibition of nuclear deacetylase Sirtuin-1 induces mitochondrial acetylation and calcium overload leading to cell death. Redox Biol. 2022, 53, 102334. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Song, Z.; Zhong, X.; Huang, M.; Shen, D.; Gao, P.; Qian, X.; Wang, M.; He, X.; Wang, T.; et al. Sangerbox: A comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta 2022, 1, e36. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef]
- Saftig, P.; Haas, A. Turn up the lysosome. Nat. Cell Biol. 2016, 18, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Irazoqui, J.E. Key Roles of MiT Transcription Factors in Innate Immunity and Inflammation. Trends Immunol. 2020, 41, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Steeg, P.S. Endocytosis: A pivotal pathway for regulating metastasis. Br. J. Cancer 2021, 124, 66–75. [Google Scholar] [CrossRef]
- Nnah, I.C.; Wang, B.; Saqcena, C.; Weber, G.F.; Bonder, E.M.; Bagley, D.; De Cegli, R.; Napolitano, G.; Medina, D.L.; Ballabio, A.; et al. TFEB-driven endocytosis coordinates MTORC1 signaling and autophagy. Autophagy 2019, 15, 151–164. [Google Scholar] [CrossRef]
- Li, G.; Huang, J.; Chen, S.; He, Y.; Wang, Z.; Peng, J. High Expression of ATP6V1C2 Predicts Unfavorable Overall Survival in Patients With Colon Adenocarcinoma. Front. Genet. 2022, 13, 930876. [Google Scholar] [CrossRef]
- Song, G.; Xu, S.; Zhang, H.; Wang, Y.; Xiao, C.; Jiang, T.; Wu, L.; Zhang, T.; Sun, X.; Zhong, L.; et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J. Exp. Clin. Cancer Res. 2016, 35, 148. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Feng, J.; Yang, S.; Huang, X.; Liao, Y.; Hu, Z.; Luo, M. miR-484/MAP2/c-Myc-positive regulatory loop in glioma promotes tumor-initiating properties through ERK1/2 signaling. J. Mol. Histol. 2018, 49, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lan, J.; Wang, G.; Guo, K.; Han, C.; Li, X.; Hu, J.; Cao, Z.; Luo, X. KDM4B facilitates colorectal cancer growth and glucose metabolism by stimulating TRAF6-mediated AKT activation. J. Exp. Clin. Cancer Res. 2020, 39, 12. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, T.; Iyama, S.; Toshima, S.; Sakata, A.; Usui, S.; Minami, Y.; Sato, Y.; Hizawa, N.; Noguchi, M. Dimethylarginine dimethylaminohydrolase 2 promotes tumor angiogenesis in lung adenocarcinoma. Virchows Arch. 2016, 468, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.; Rhost, S.; Rafnsdottir, S.; Hughes, E.; Magnusson, Y.; Ekholm, M.; Stal, O.; Ryden, L.; Landberg, G. Tumor co-expression of progranulin and sortilin as a prognostic biomarker in breast cancer. BMC Cancer 2021, 21, 185. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lu, Y.T.; Yeh, T.S.; Chan, Y.H.; Dash, S.; Yu, J.S. Identification of Fucosylated SERPINA1 as a Novel Plasma Marker for Pancreatic Cancer Using Lectin Affinity Capture Coupled with iTRAQ-Based Quantitative Glycoproteomics. Int. J. Mol. Sci. 2021, 22, 6079. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Zhu, S.; Han, Z.; Li, C.; Liang, J.; Wang, Y.; Zhang, S.; Zhang, J. Proteome-Wide Analysis Reveals TFEB Targets for Establishment of a Prognostic Signature to Predict Clinical Outcomes of Colorectal Cancer. Cancers 2023, 15, 744. https://doi.org/10.3390/cancers15030744
Huang Z, Zhu S, Han Z, Li C, Liang J, Wang Y, Zhang S, Zhang J. Proteome-Wide Analysis Reveals TFEB Targets for Establishment of a Prognostic Signature to Predict Clinical Outcomes of Colorectal Cancer. Cancers. 2023; 15(3):744. https://doi.org/10.3390/cancers15030744
Chicago/Turabian StyleHuang, Zijia, Sheng Zhu, Ziqin Han, Chen Li, Junze Liang, Yang Wang, Shuixing Zhang, and Jing Zhang. 2023. "Proteome-Wide Analysis Reveals TFEB Targets for Establishment of a Prognostic Signature to Predict Clinical Outcomes of Colorectal Cancer" Cancers 15, no. 3: 744. https://doi.org/10.3390/cancers15030744
APA StyleHuang, Z., Zhu, S., Han, Z., Li, C., Liang, J., Wang, Y., Zhang, S., & Zhang, J. (2023). Proteome-Wide Analysis Reveals TFEB Targets for Establishment of a Prognostic Signature to Predict Clinical Outcomes of Colorectal Cancer. Cancers, 15(3), 744. https://doi.org/10.3390/cancers15030744