


The β-Blocker Carvedilol Prevents Benzo(a)pyrene-Induced Lung Toxicity, Inflammation and Carcinogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Line and Cell Culture
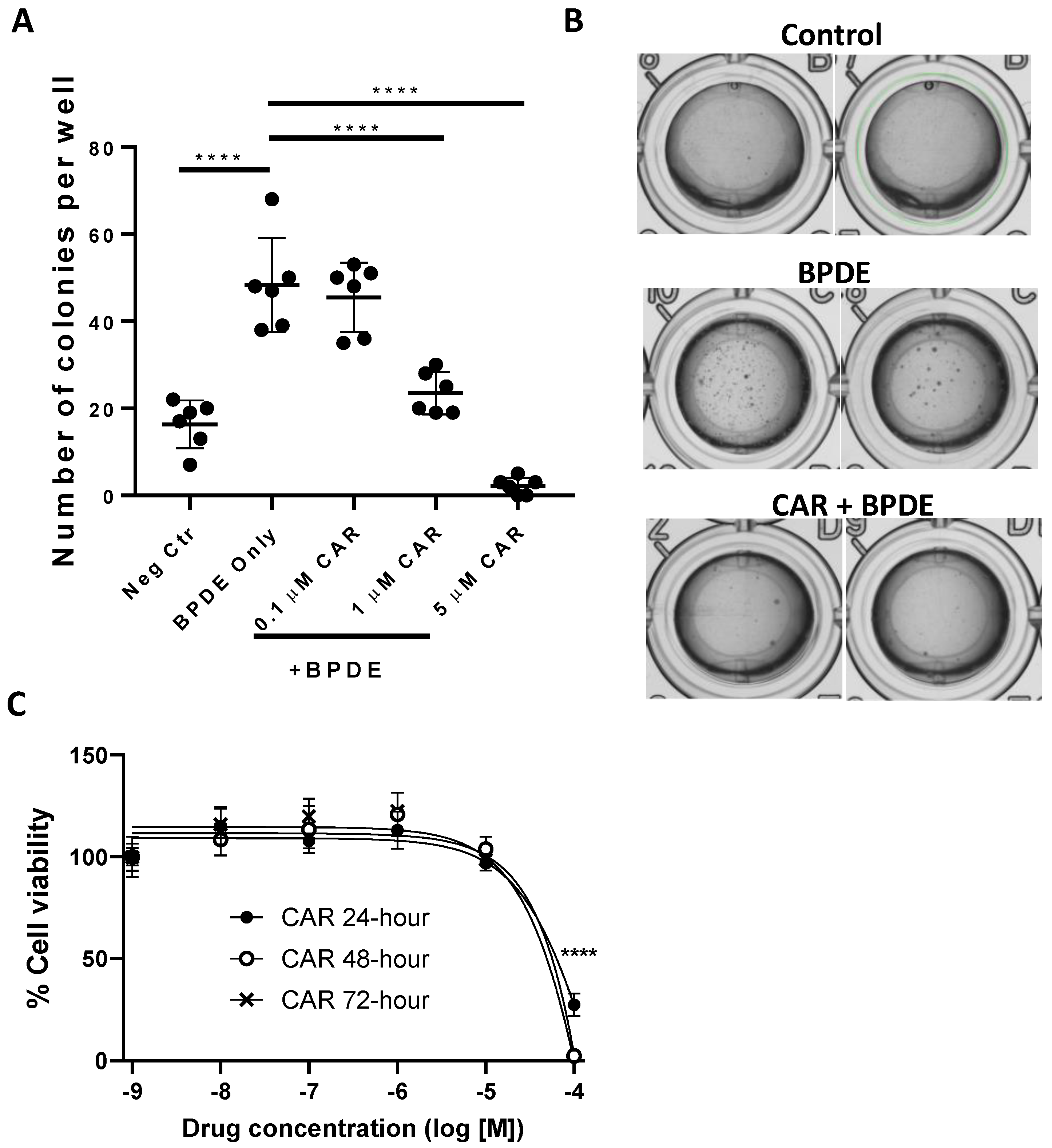
2.3. Anchorage-Independent Growth Assay in Soft Agar
2.4. MTT Cytotoxicity Assay
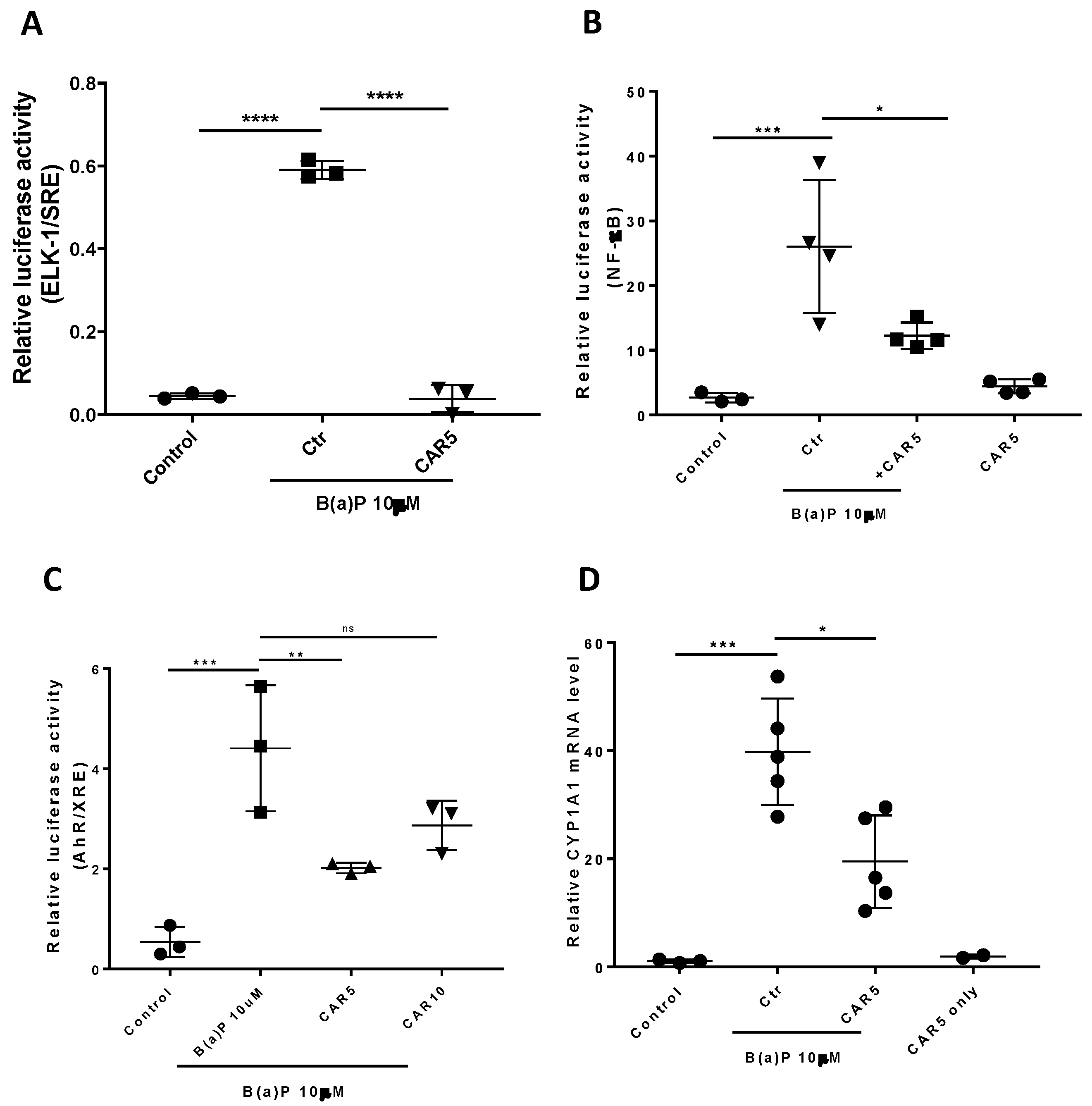
2.5. Dual Luciferase Reporter Assay
2.6. Real-Time RT-PCR Analysis
2.7. Animal Studies
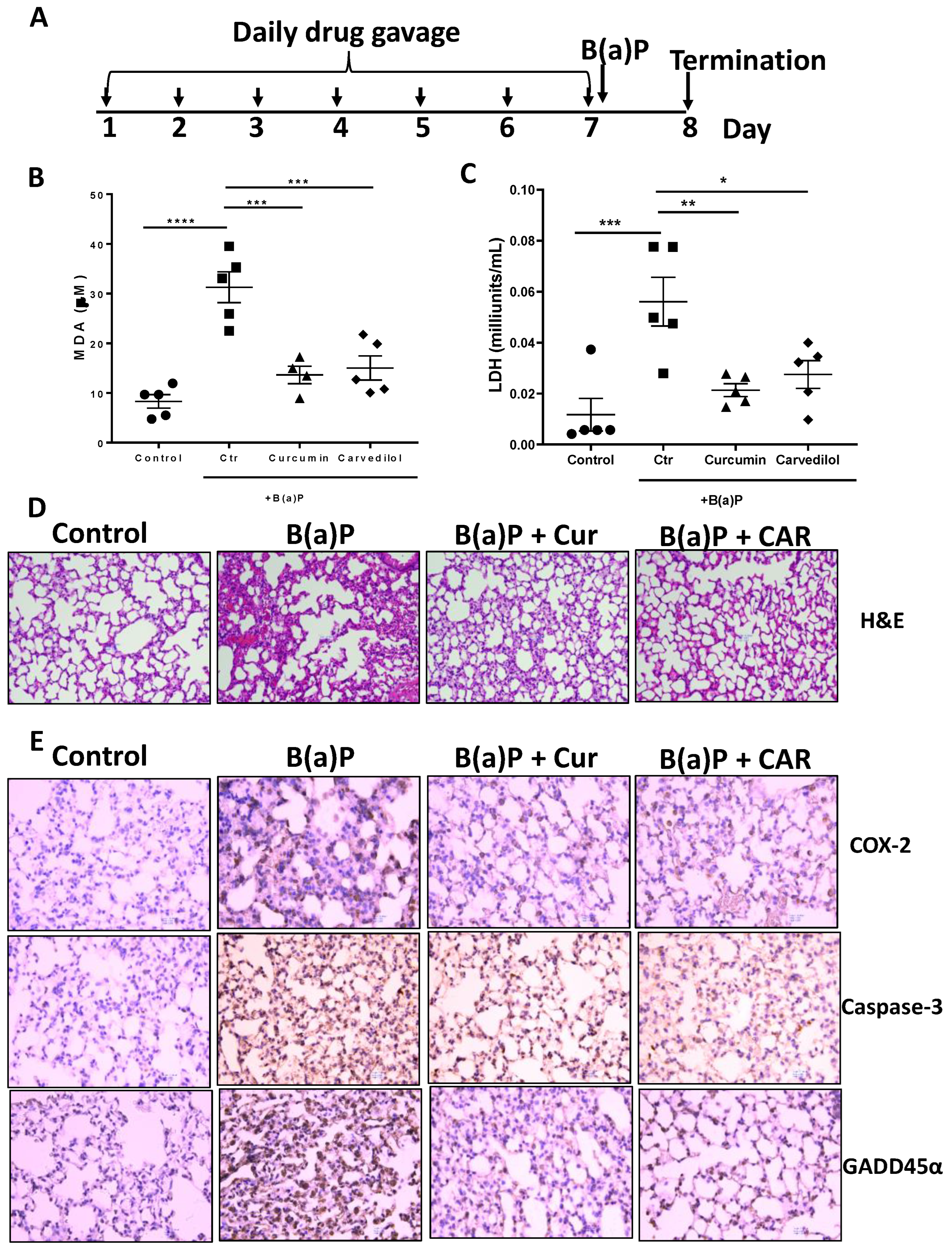
2.8. Acute Short-Term B(a)P Exposure Mouse Study
2.9. Lactate Dehydrogenase (LDH) Activity Assay
2.10. Lipid Hydroperoxide (LPO) Assay
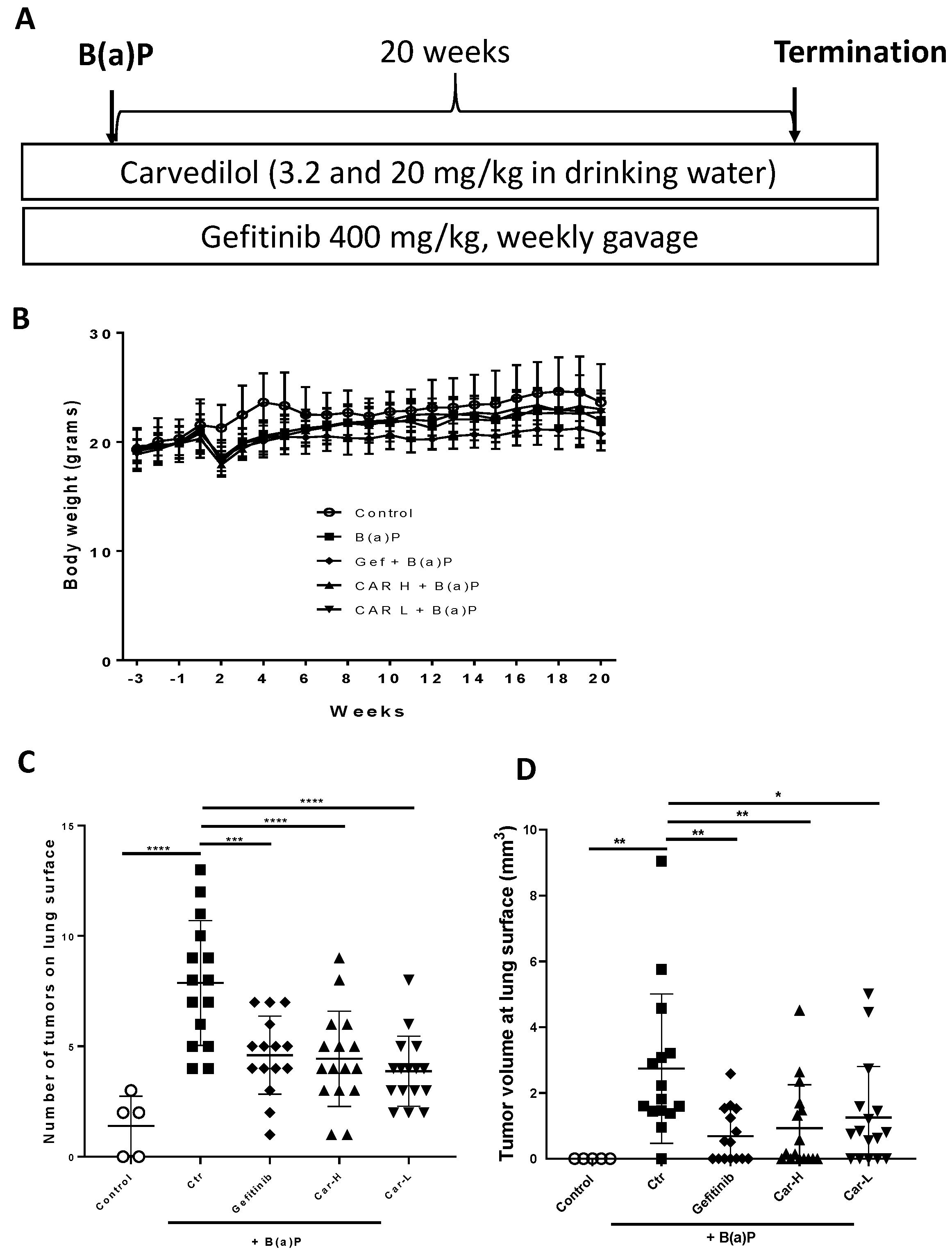
2.11. Long-Term Lung Tumorigenesis Study in Mice
2.12. Histology and Immunohistochemistry (IHC) Analysis
2.13. Statistical Analysis
3. Results
3.1. Effects of Carvedilol on BPDE-Induced Malignant Transformation of BEAS-2B Cells
3.2. Effects of Carvedilol on B(a)P-Induced ERK-1, AhR and NF-κB Signaling in BEAS-2B Cells
3.3. Effects of Carvedilol on Short Term B(a)P-Induced Lung Toxicity in Mice
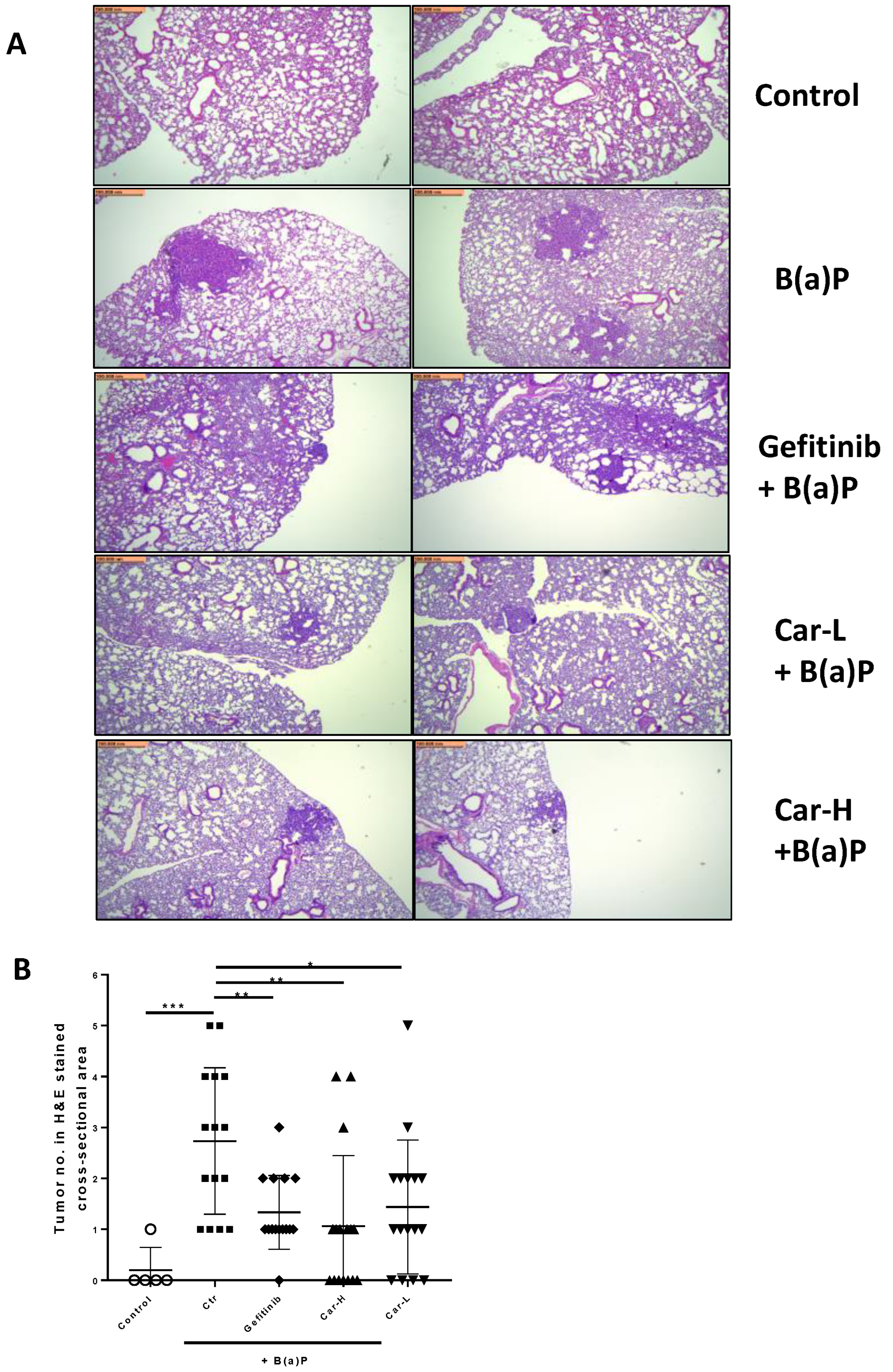
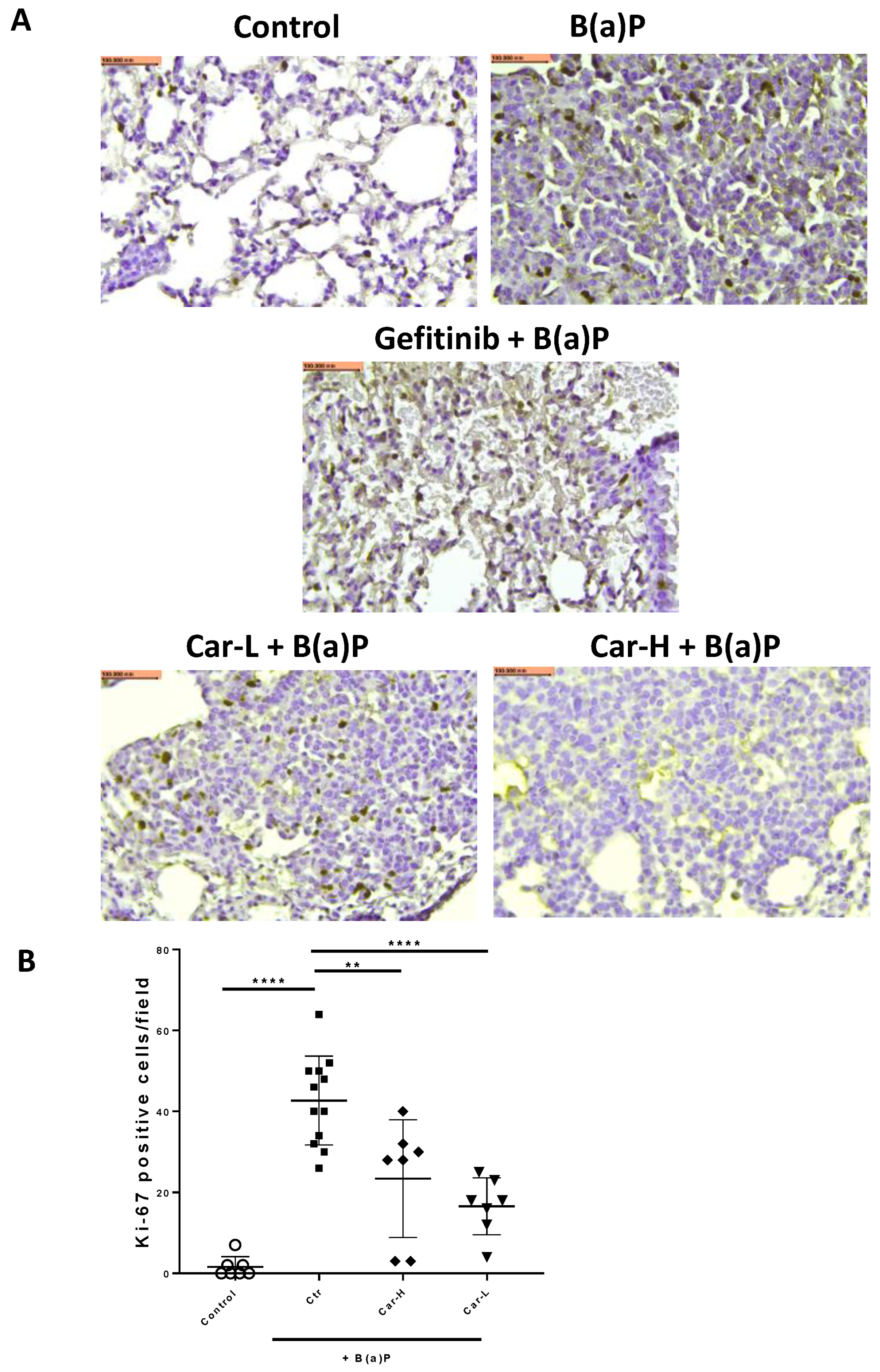
3.4. Effects of Carvedilol on B(a)P-Induced Lung Carcinogenesis in Mice
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef]
- Ashraf-Uz-Zaman, M.; Bhalerao, A.; Mikelis, C.M.; Cucullo, L.; German, N.A. Assessing the Current State of Lung Cancer Chemoprevention: A Comprehensive Overview. Cancers 2020, 12, 1265. [Google Scholar] [CrossRef]
- Patel, A.B.; Shaikh, S.; Jain, K.R.; Desai, C.; Madamwar, D. Polycyclic Aromatic Hydrocarbons: Sources, Toxicity, and Remediation Approaches. Front. Microbiol. 2020, 11, 562813. [Google Scholar] [CrossRef] [PubMed]
- Kasala, E.R.; Bodduluru, L.N.; Barua, C.C.; Sriram, C.S.; Gogoi, R. Benzo(a)pyrene induced lung cancer: Role of dietary phytochemicals in chemoprevention. Pharmacol. Rep. 2015, 67, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Chang, X.; Puga, A.; Xia, Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons: Role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochem. Pharmacol. 2002, 64, 771–780. [Google Scholar] [CrossRef]
- Ali, R.; Shahid, A.; Ali, N.; Hasan, S.K.; Majed, F.; Sultana, S. Amelioration of Benzo[a]pyrene-induced oxidative stress and pulmonary toxicity by Naringenin in Wistar rats: A plausible role of COX-2 and NF-kappaB. Hum. Exp. Toxicol. 2017, 36, 349–364. [Google Scholar] [CrossRef]
- Wang, H.M.; Liao, Z.X.; Komaki, R.; Welsh, J.W.; O’Reilly, M.S.; Chang, J.Y.; Zhuang, Y.; Levy, L.B.; Lu, C.; Gomez, D.R. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann. Oncol. 2013, 24, 1312–1319. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Sun, H.; Diao, L.; Tong, P.; Liu, D.; Li, L.; Fan, Y.; Poteete, A.; Lim, S.O.; Howells, K.; et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with beta-blockers. Sci. Transl. Med. 2017, 9, eaao4307. [Google Scholar] [CrossRef]
- Sakamoto, A.; Yagi, K.; Okamura, T.; Harada, T.; Usuda, J. Perioperative Administration of an Intravenous Beta-Blocker Landiolol Hydrochloride in Patients with Lung Cancer: A Japanese Retrospective Exploratory Clinical Study. Sci. Rep. 2019, 9, 5217. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hamasaki, T.; Onda, K.; Nojiri, T.; Aragaki, M.; Horie, N.; Sato, N.; Hida, Y. Landiolol, an ultra-short acting beta-1 blocker, for preventing postoperative lung cancer recurrence: Study protocol for a phase III, multicenter randomized trial with two parallel groups of patients. Trials 2019, 20, 715. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.R.; Yan, S.X.; Heilbroner, S.P.; Sonett, J.R.; Stoopler, M.B.; Shu, C.; Halmos, B.; Wang, T.J.C.; Hei, T.K.; Cheng, S.K. Effects of beta-Adrenergic Antagonists on Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. J. Clin. Med. 2019, 8, 575. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.; Pereira, M.L.; Oliveira, M. Beta-Blockers and Cancer: Where Are We? Pharmaceuticals 2020, 13, 105. [Google Scholar] [CrossRef]
- Jafri, S.H.; Ali, F.; Mollaeian, A.; Mojiz Hasan, S.; Hussain, R.; Akkanti, B.; Williams, J.; Shoukier, M.; El-Osta, H. Major Stressful Life Events and Risk of Developing Lung Cancer: A Case-Control Study. Clin. Med. Insights Oncol. 2019, 13, 1179554919835798. [Google Scholar] [CrossRef]
- Min, H.Y.; Boo, H.J.; Lee, H.J.; Jang, H.J.; Yun, H.J.; Hwang, S.J.; Smith, J.K.; Lee, H.J.; Lee, H.Y. Smoking-associated lung cancer prevention by blockade of the beta-adrenergic receptor-mediated insulin-like growth factor receptor activation. Oncotarget 2016, 7, 70936–70947. [Google Scholar] [CrossRef]
- Schuller, H.M.; Porter, B.; Riechert, A. Beta-adrenergic modulation of NNK-induced lung carcinogenesis in hamsters. J. Cancer Res. Clin. Oncol. 2000, 126, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Mayati, A.; Levoin, N.; Paris, H.; N’Diaye, M.; Courtois, A.; Uriac, P.; Lagadic-Gossmann, D.; Fardel, O.; Le Ferrec, E. Induction of intracellular calcium concentration by environmental benzo(a)pyrene involves a beta2-adrenergic receptor/adenylyl cyclase/Epac-1/inositol 1,4,5-trisphosphate pathway in endothelial cells. J. Biol. Chem. 2012, 287, 4041–4052. [Google Scholar] [CrossRef]
- Calo, L.A.; Semplicini, A.; Davis, P.A. Antioxidant and antiinflammatory effect of carvedilol in mononuclear cells of hypertensive patients. Am. J. Med. 2005, 118, 201–202. [Google Scholar] [CrossRef]
- Dandona, P.; Ghanim, H.; Brooks, D.P. Antioxidant activity of carvedilol in cardiovascular disease. J. Hypertens. 2007, 25, 731–741. [Google Scholar] [CrossRef]
- Arab, H.H.; El-Sawalhi, M.M. Carvedilol alleviates adjuvant-induced arthritis and subcutaneous air pouch edema: Modulation of oxidative stress and inflammatory mediators. Toxicol. Appl. Pharmacol. 2013, 268, 241–248. [Google Scholar] [CrossRef]
- Chang, A.; Yeung, S.; Thakkar, A.; Huang, K.M.; Liu, M.M.; Kanassatega, R.S.; Parsa, C.; Orlando, R.; Jackson, E.K.; Andresen, B.T.; et al. Prevention of skin carcinogenesis by the beta-blocker carvedilol. Cancer Prev. Res. 2015, 8, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.M.; Liang, S.; Yeung, S.; Oiyemhonlan, E.; Cleveland, K.H.; Parsa, C.; Orlando, R.; Meyskens, F.L., Jr.; Andresen, B.T.; Huang, Y. Topically Applied Carvedilol Attenuates Solar Ultraviolet Radiation Induced Skin Carcinogenesis. Cancer Prev. Res. 2017, 10, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, K.H.; Liang, S.; Chang, A.; Huang, K.M.; Chen, S.; Guo, L.; Huang, Y.; Andresen, B.T. Carvedilol inhibits EGF-mediated JB6 P+ colony formation through a mechanism independent of adrenoceptors. PLoS ONE 2019, 14, e0217038. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Liu, X.; Zhang, Q.; Yu, Z.; Gao, D. Carvedilol suppresses malignant proliferation of mammary epithelial cells through inhibition of the ROSmediated PI3K/AKT signaling pathway. Oncol. Rep. 2019, 41, 811–818. [Google Scholar]
- Lin, C.S.; Lin, W.S.; Lin, C.L.; Kao, C.H. Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study. Int. J. Cardiol. 2015, 184, 9–13. [Google Scholar] [CrossRef]
- Chen, W.; Xu, X.; Bai, L.; Padilla, M.T.; Gott, K.M.; Leng, S.; Tellez, C.S.; Wilder, J.A.; Belinsky, S.A.; Scott, B.R.; et al. Low-dose gamma-irradiation inhibits IL-6 secretion from human lung fibroblasts that promotes bronchial epithelial cell transformation by cigarette-smoke carcinogen. Carcinogenesis 2012, 33, 1368–1374. [Google Scholar] [CrossRef]
- Dutta, S.; Saha, S.; Mahalanobish, S.; Sadhukhan, P.; Sil, P.C. Melatonin attenuates arsenic induced nephropathy via the regulation of oxidative stress and inflammatory signaling cascades in mice. Food Chem. Toxicol. 2018, 118, 303–316. [Google Scholar] [CrossRef]
- Kim, Y.S.; Chung, Y.H.; Seo, D.S.; Choi, H.S.; Lim, C.H. Twenty-Eight-Day Repeated Inhalation Toxicity Study of Aluminum Oxide Nanoparticles in Male Sprague-Dawley Rats. Toxicol Res. 2018, 34, 343–354. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, R.; Chen, X.; Lee, S.B.; Pan, J.; Xiong, D.; Hu, J.; Miller, M.S.; Szabo, E.; Lubet, R.A.; et al. Effect of weekly or daily dosing regimen of Gefitinib in mouse models of lung cancer. Oncotarget 2017, 8, 72447–72456. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Whipple, B.; Agar, J.; Zhao, J.; Pearce, D.A.; Kovacs, A.D. The acidified drinking water-induced changes in the behavior and gut microbiota of wild-type mice depend on the acidification mode. Sci. Rep. 2021, 11, 2877. [Google Scholar] [CrossRef] [PubMed]
- Hamed, R.; Awadallah, A.; Sunoqrot, S.; Tarawneh, O.; Nazzal, S.; AlBaraghthi, T.; Al Sayyad, J.; Abbas, A. pH-Dependent Solubility and Dissolution Behavior of Carvedilol--Case Example of a Weakly Basic BCS Class II Drug. AAPS PharmSciTech 2016, 17, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ono, K.; Dickstein, D.L.; Arrieta-Cruz, I.; Zhao, W.; Qian, X.; Lamparello, A.; Subnani, R.; Ferruzzi, M.; Pavlides, C.; et al. Carvedilol as a potential novel agent for the treatment of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 2321 e1-12. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, Z.; Lin, E.; Zhao, W.; Qian, X.; Freire, D.; Bilski, A.E.; Cheng, A.; Vempati, P.; Ho, L.; et al. Unintended effects of cardiovascular drugs on the pathogenesis of Alzheimer’s disease. PLoS ONE 2013, 8, e65232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiong, D.; Pan, J.; Wang, Y.; Hardy, M.; Kalyanaraman, B.; You, M. Chemoprevention of Lung Cancer with a Combination of Mitochondria-Targeted Compounds. Cancers 2022, 14, 2538. [Google Scholar] [CrossRef]
- van Agen, B.; Maas, L.M.; Zwingmann, I.H.; Van Schooten, F.J.; Kleinjans, J.C. B[a]P-DNA adduct formation and induction of human epithelial lung cell transformation. Environ. Mol. Mutagen. 1997, 30, 287–292. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Yeung, S.; Huang, K.M.; Liang, S.; Andresen, B.T.; Huang, Y. Phosphoproteome profiling provides insight into the mechanism of action for carvedilol-mediated cancer prevention. Mol. Carcinog. 2018, 57, 997–1007. [Google Scholar] [CrossRef]
- Lerner-Marmarosh, N.; Miralem, T.; Gibbs, P.E.; Maines, M.D. Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 6870–6875. [Google Scholar] [CrossRef]
- Li, K.; Liu, Y.; Ding, Y.; Zhang, Z.; Feng, J.; Hu, J.; Chen, J.; Lian, Z.; Chen, Y.; Hu, K.; et al. BCL6 is regulated by the MAPK/ELK1 axis and promotes KRAS-driven lung cancer. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Yu, T.T.; Zhang, T.; Su, F.; Li, Y.L.; Shan, L.; Hou, X.M.; Wang, R.Z. ELK1 Promotes Epithelial-Mesenchymal Transition and the Progression of Lung Adenocarcinoma by Upregulating B7-H3. Oxid Med. Cell. Longev. 2021, 2021, 2805576. [Google Scholar] [CrossRef]
- Shimizu, Y.; Nakatsuru, Y.; Ichinose, M.; Takahashi, Y.; Kume, H.; Mimura, J.; Fujii-Kuriyama, Y.; Ishikawa, T. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Denissenko, M.F.; Pao, A.; Tang, M.; Pfeifer, G.P. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 1996, 274, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Pratheeshkumar, P.; Son, Y.O.; Divya, S.P.; Roy, R.V.; Hitron, J.A.; Wang, L.; Kim, D.; Dai, J.; Asha, P.; Zhang, Z.; et al. Luteolin inhibits Cr(VI)-induced malignant cell transformation of human lung epithelial cells by targeting ROS mediated multiple cell signaling pathways. Toxicol. Appl. Pharmacol. 2014, 281, 230–241. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, D.; Dai, J.; Zhang, Z. Human bronchial epithelial BEAS-2B cells, an appropriate in vitro model to study heavy metals induced carcinogenesis. Toxicol. Appl. Pharmacol. 2015, 287, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Gomez, G.; Rocha-Zavaleta, L.; Rodriguez-Sosa, M.; Petrosyan, P.; Rubio-Lightbourn, J. Benzo[a]pyrene activates an AhR/Src/ERK axis that contributes to CYP1A1 induction and stable DNA adducts formation in lung cells. Toxicol. Lett. 2018, 289, 54–62. [Google Scholar] [CrossRef]
- Ding, J.; Ning, B.; Gong, W.; Wen, W.; Wu, K.; Liang, J.; He, G.; Huang, S.; Sun, W.; Han, T.; et al. Cyclin D1 induction by benzo[a]pyrene-7,8-diol-9,10-epoxide via the phosphatidylinositol 3-kinase/Akt/MAPK- and p70s6k-dependent pathway promotes cell transformation and tumorigenesis. J. Biol. Chem. 2009, 284, 33311–33319. [Google Scholar] [CrossRef]
- Alzahrani, A.M.; Rajendran, P. Pinocembrin attenuates benzo(a)pyrene-induced CYP1A1 expression through multiple pathways: An in vitro and in vivo study. J. Biochem. Mol. Toxicol. 2021, 35, e22695. [Google Scholar] [CrossRef]
- Safe, S.; Lee, S.O.; Jin, U.H. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 2013, 135, 1–16. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Coelho, N.R.; Pimpao, A.B.; Correia, M.J.; Rodrigues, T.C.; Monteiro, E.C.; Morello, J.; Pereira, S.A. Pharmacological blockage of the AHR-CYP1A1 axis: A call for in vivo evidence. J. Mol. Med. 2022, 100, 215–243. [Google Scholar] [CrossRef]
- Sehgal, A.; Kumar, M.; Jain, M.; Dhawan, D.K. Synergistic effects of piperine and curcumin in modulating benzo(a)pyrene induced redox imbalance in mice lungs. Toxicol. Mech. Methods 2012, 22, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G. The role of gefitinib in lung cancer treatment. Clin. Cancer Res. 2004, 12 Pt 2, 4233s–4237s. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.L.; Cheng, H.Y.; Lysko, P.G.; McKenna, P.J.; Feuerstein, R.; Gu, J.L.; Lysko, K.A.; Davis, L.L.; Feuerstein, G. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J. Pharmacol. Exp. Ther. 1992, 263, 92–98. [Google Scholar] [PubMed]
- Chen, M.; Liang, S.; Shahid, A.; Andresen, B.T.; Huang, Y. The beta-Blocker Carvedilol Prevented Ultraviolet-Mediated Damage of Murine Epidermal Cells and 3D Human Reconstructed Skin. Int. J. Mol. Sci. 2020, 21, 798. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahid, A.; Chen, M.; Lin, C.; Andresen, B.T.; Parsa, C.; Orlando, R.; Huang, Y. The β-Blocker Carvedilol Prevents Benzo(a)pyrene-Induced Lung Toxicity, Inflammation and Carcinogenesis. Cancers 2023, 15, 583. https://doi.org/10.3390/cancers15030583
Shahid A, Chen M, Lin C, Andresen BT, Parsa C, Orlando R, Huang Y. The β-Blocker Carvedilol Prevents Benzo(a)pyrene-Induced Lung Toxicity, Inflammation and Carcinogenesis. Cancers. 2023; 15(3):583. https://doi.org/10.3390/cancers15030583
Chicago/Turabian StyleShahid, Ayaz, Mengbing Chen, Carol Lin, Bradley T. Andresen, Cyrus Parsa, Robert Orlando, and Ying Huang. 2023. "The β-Blocker Carvedilol Prevents Benzo(a)pyrene-Induced Lung Toxicity, Inflammation and Carcinogenesis" Cancers 15, no. 3: 583. https://doi.org/10.3390/cancers15030583
APA StyleShahid, A., Chen, M., Lin, C., Andresen, B. T., Parsa, C., Orlando, R., & Huang, Y. (2023). The β-Blocker Carvedilol Prevents Benzo(a)pyrene-Induced Lung Toxicity, Inflammation and Carcinogenesis. Cancers, 15(3), 583. https://doi.org/10.3390/cancers15030583