
MicroRNA Expression Patterns Reveal a Role of the TGF-β Family Signaling in AML Chemo-Resistance

 , , and
, , and
Abstract
:Simple Summary
Abstract
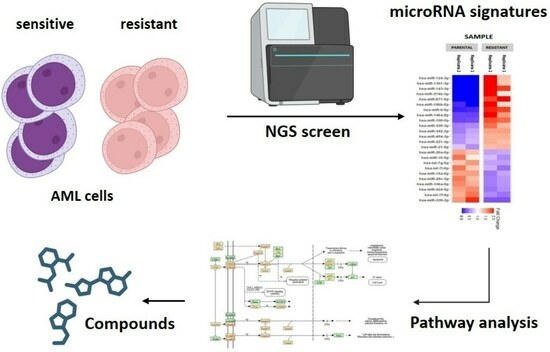
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. miRNA Expression Profiling
2.3. Bioinformatics
2.4. Cell Proliferation Assays
2.5. Western Blotting
2.6. Flow Cytometry Assays
2.7. Patient Samples and Quantitative Real-Time PCR
2.8. Statistical Analyses
3. Results
3.1. MiRNA Profiling Suggests the Involvement of TGF-β Family Signaling in Chemo-Resistance
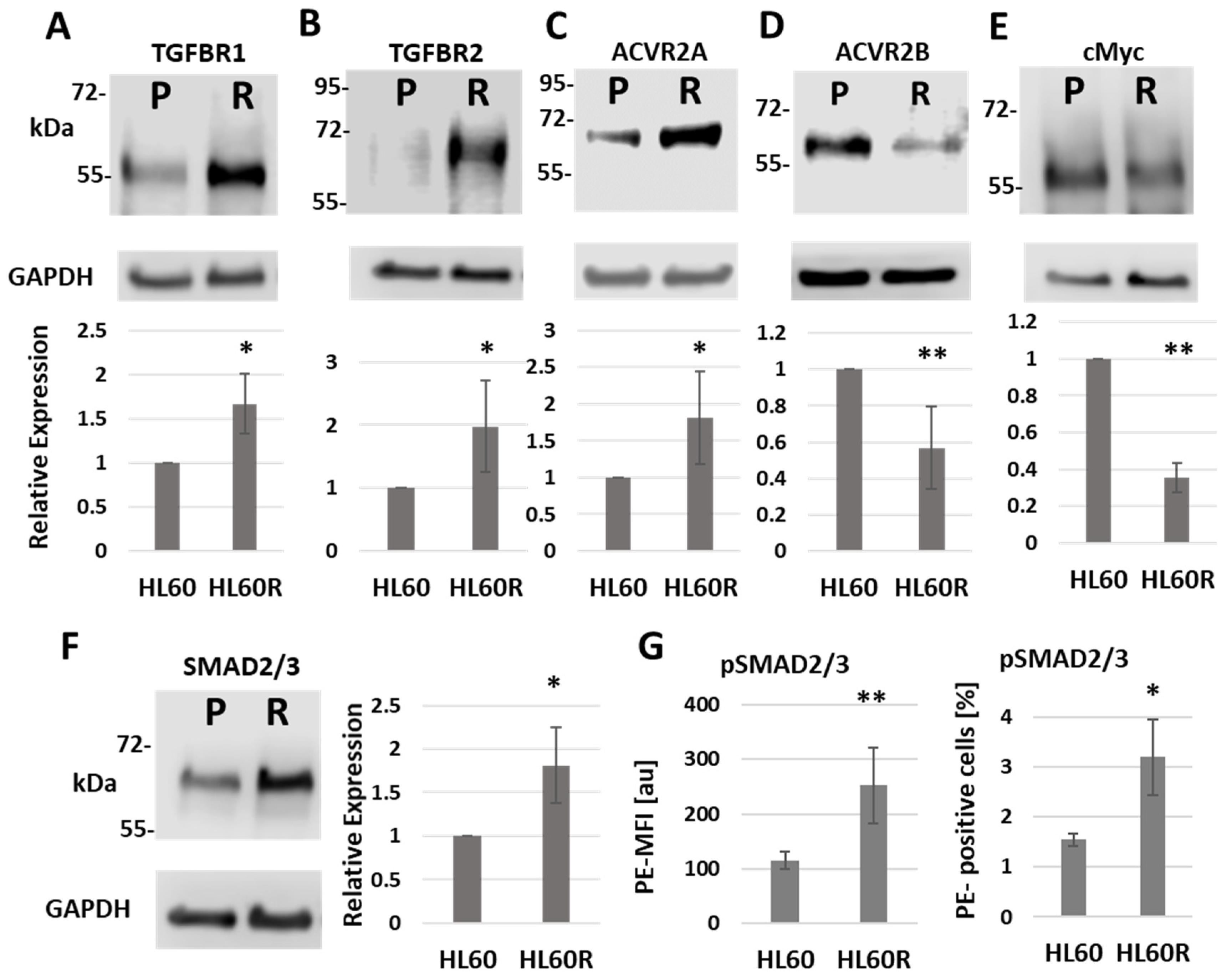
3.2. Chemo-Resistant Cells Have Altered Levels of TGF-β Signaling Proteins and Their Targets
3.3. TGF-β Receptor-Mediated Signaling Affects the Proliferation of Chemo-Resistant Cells, but Is Not a Major Determinant of Chemo-Resistance
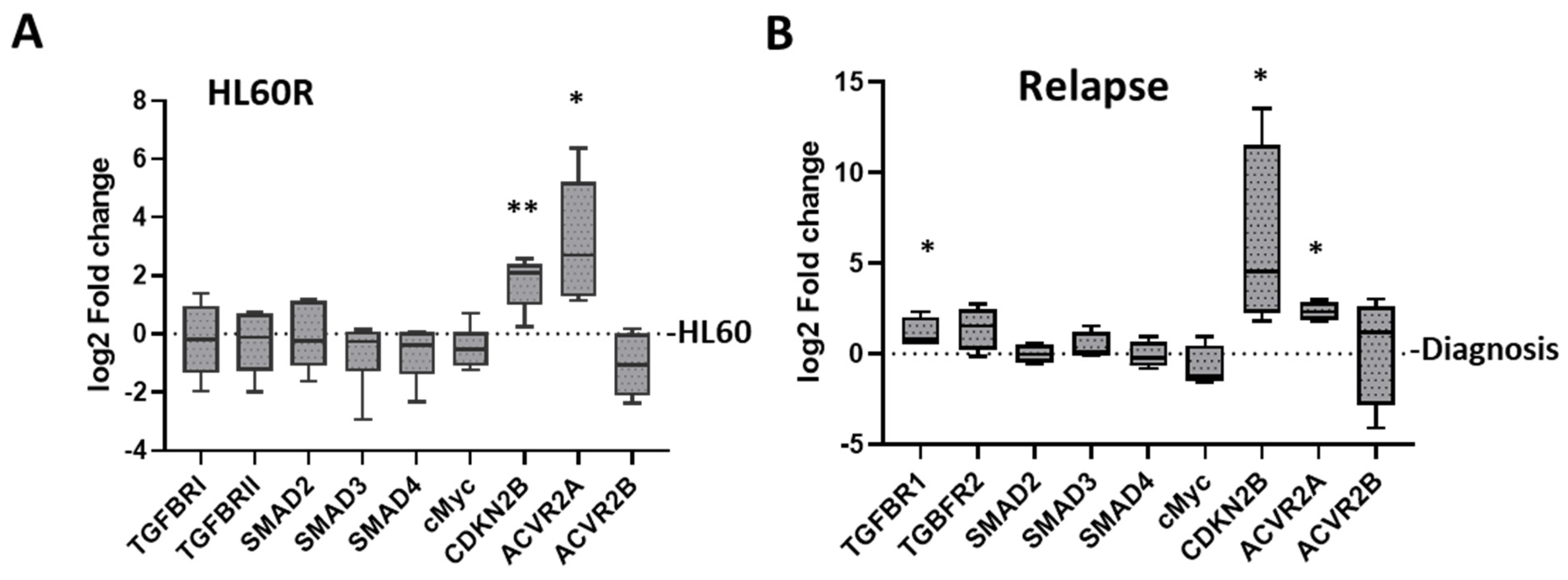
3.4. Changes in TGF-β Pathway Expression Associated with the Development of Resistance in Primary AML
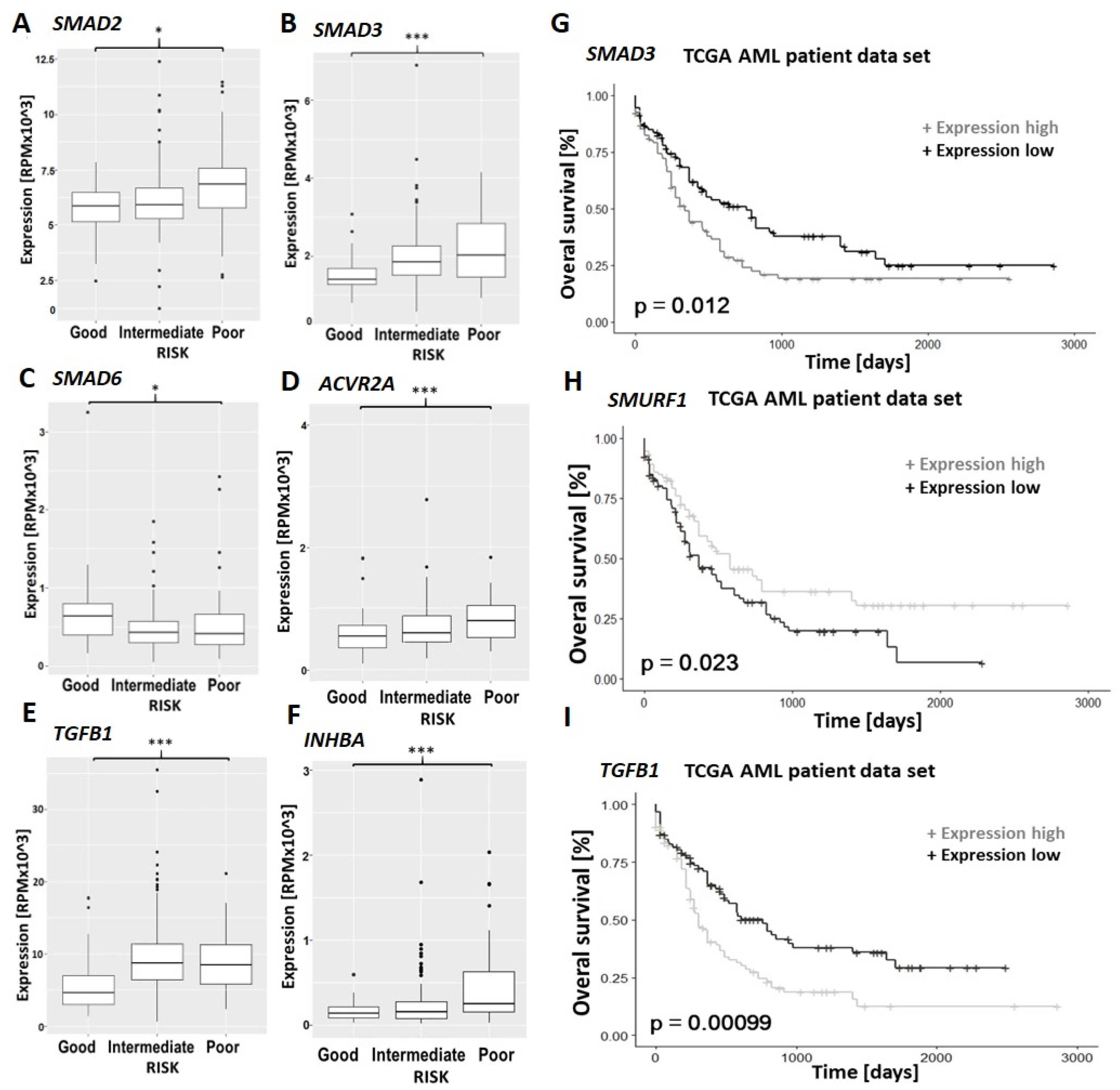
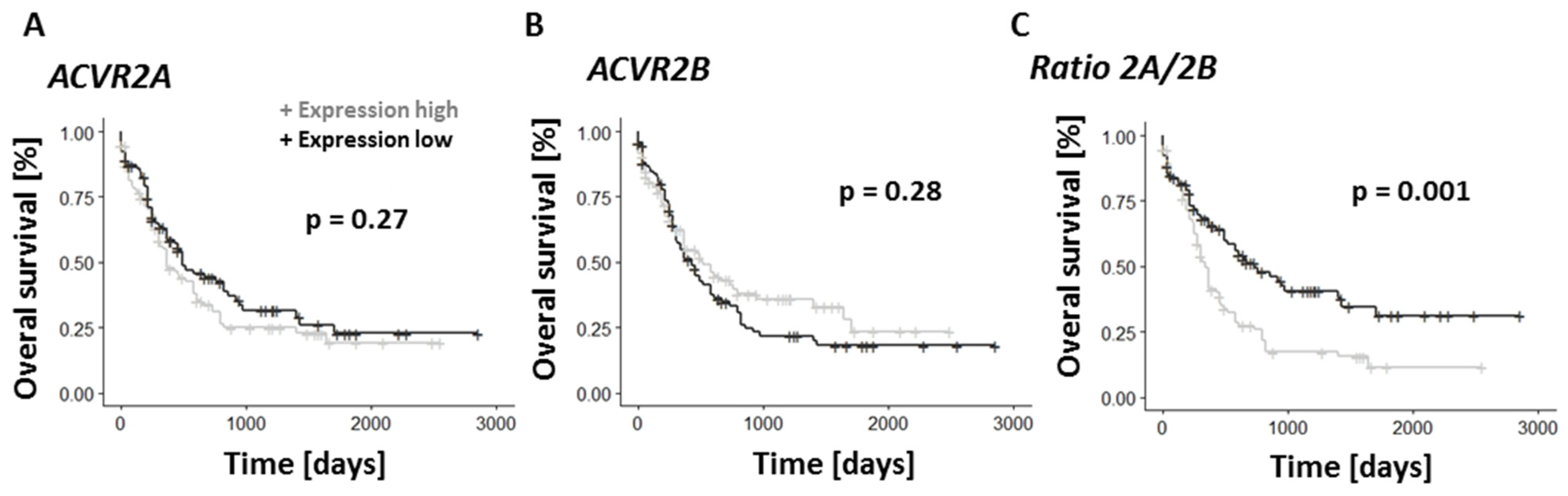
3.5. TGF-β Family Pathways Are Implicated in the Prognosis and Chemo-Resistance of Primary AML
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levin, M.; Stark, M.; Ofran, Y.; Assaraf, Y.G. Deciphering molecular mechanisms underlying chemoresistance in relapsed AML patients: Towards precision medicine overcoming drug resistance. Cancer Cell Int. 2021, 21, 53. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef]
- Tan, W.; Liu, B.; Qu, S.; Liang, G.; Luo, W.; Gong, C. MicroRNAs and cancer: Key paradigms in molecular therapy. Oncol. Lett. 2018, 15, 2735–2742. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, R.; Wang, J.; Qin, Z.; Xue, L. Circulating microRNAs as potential cancer biomarkers: The advantage and disadvantage. Clin. Epigenetics 2018, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, C.; Slack, F.J. MicroRNAs and the cancer phenotype: Profiling, signatures and clinical implications. Genome Med. 2013, 5, 111. [Google Scholar] [CrossRef]
- De Leeuw, D.C.; van den Ancker, W.; Denkers, F.; de Menezes, R.X.; Westers, T.M.; Ossenkoppele, G.J.; van de Loosdrecht, A.A.; Smit, L. MicroRNA profiling can classify acute leukemias of ambiguous lineage as either acute myeloid leukemia or acute lymphoid leukemia. Clin. Cancer Res. 2013, 19, 2187–2196. [Google Scholar] [CrossRef]
- Wee, E.J.H.; Peters, K.; Nair, S.S.; Hulf, T.; Stein, S.; Wagner, S.; Bailey, P.; Lee, S.Y.; Qu, W.J.; Brewster, B.; et al. Mapping the regulatory sequences controlling 93 breast cancer-associated miRNA genes leads to the identification of two functional promoters of the Hsa-mir-200b cluster, methylation of which is associated with metastasis or hormone receptor status in advanced breast cancer. Oncogene 2012, 31, 4182–4195. [Google Scholar] [CrossRef]
- Bräuer-Hartmann, D.; Hartmann, J.-U.; Wurm, A.A.; Gerloff, D.; Katzerke, C.; Verga Falzacappa, M.V.; Pelicci, P.G.; Muller-Tidow, C.; Tenen, D.G.; Niederwieser, D. PML/RARα-Regulated miR-181a/b Cluster Targets the Tumor Suppressor RASSF1A in Acute Promyelocytic Leukemia. Cancer Res. 2015, 75, 3411–3424. [Google Scholar] [CrossRef]
- Hartmann, J.-U.; Bräuer-Hartmann, D.; Kardosova, M.; Wurm, A.A.; Wilke, F.; Schödel, C.; Gerloff, D.; Katzerke, C.; Krakowsky, R.; Yaeko Namasu, C.; et al. MicroRNA-143 targets ERK5 in granulopoiesis and predicts outcome of patients with acute myeloid leukemia. Cell Death Dis. 2018, 9, 814. [Google Scholar] [CrossRef]
- Gerloff, D.; Grundler, R.; Wurm, A.A.; Bräuer-Hartmann, D.; Katzerke, C.; Hartmann, J.-U.; Madan, V.; Muller-Tidow, C.; Duyster, J.; Tenen, D.G.; et al. NF-κB/STAT5/miR-155 network targets PU.1 in FLT3-ITD-driven acute myeloid leukemia. Leukemia 2015, 29, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Katzerke, C.; Madan, V.; Gerloff, D.; Bräuer-Hartmann, D.; Hartmann, J.-U.; Wurm, A.A.; Muller-Tidow, C.; Schnittger, S.; Tenen, D.G.; Niederwieser, D.; et al. Transcription factor C/EBPα-induced microRNA-30c inactivates Notch1 during granulopoiesis and is downregulated in acute myeloid leukemia. Blood 2013, 122, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Wurm, A.A.; Zjablovskaja, P.; Kardosova, M.; Gerloff, D.; Bräuer-Hartmann, D.; Katzerke, C.; Hartmann, J.-U.; Benoukraf, T.; Fricke, S.; Hilger, N.; et al. Disruption of the C/EBPα-miR-182 balance impairs granulocytic differentiation. Nat. Commun. 2017, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Pulikkan, J.A.; Dengler, V.; Peramangalam, P.S.; Peer Zada, A.A.; Müller-Tidow, C.; Bohlander, S.K.; Tenen, D.G.; Behre, G. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood 2010, 115, 1768–1778. [Google Scholar] [CrossRef]
- Krakowsky, R.H.E.; Wurm, A.A.; Gerloff, D.; Katzerke, C.; Bräuer-Hartmann, D.; Hartmann, J.-U.; Wilke, F.; Thiede, C.; Muller-Tidow, C.; Niederwieser, D.; et al. miR-451a abrogates treatment resistance in FLT3-ITD-positive acute myeloid leukemia. Blood Cancer J. 2018, 8, 36. [Google Scholar] [CrossRef]
- Ma, J.; Dong, C.; Ji, C. MicroRNA and drug resistance. Cancer Gene Ther. 2010, 17, 523–531. [Google Scholar] [CrossRef]
- Bhise, N.S.; Chauhan, L.; Shin, M.; Cao, X.; Pounds, S.; Lamba, V.; Lamba, J.K. MicroRNA-mRNA Pairs Associated with Outcome in AML: From In Vitro Cell-Based Studies to AML Patients. Front. Pharmacol. 2015, 6, 324. [Google Scholar] [CrossRef]
- Koutova, L.; Sterbova, M.; Pazourkova, E.; Pospisilova, S.; Svobodova, I.; Horinek, A.; Lysak, D.; Korabecna, M. The impact of standard chemotherapy on miRNA signature in plasma in AML patients. Leuk. Res. 2015, 39, 1389–1395. [Google Scholar] [CrossRef]
- Inui, M.; Martello, G.; Piccolo, S. MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 2010, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Laurent, L.C.; Shamir, R. Towards computational prediction of microRNA function and activity. Nucleic Acids Res. 2010, 38, e160. [Google Scholar] [CrossRef]
- Artcibasova, A.V.; Korzinkin, M.B.; Sorokin, M.I.; Shegay, P.V.; Zhavoronkov, A.A.; Gaifullin, N.; Alekseev, B.Y.; Vorobyev, N.V.; Kuzmin, D.V.; Kaprin, A.D.; et al. MiRImpact, a new bioinformatic method using complete microRNA expression profiles to assess their overall influence on the activity of intracellular molecular pathways. Cell Cycle 2016, 15, 689–698. [Google Scholar] [CrossRef]
- Ben-Hamo, R.; Efroni, S. MicroRNA regulation of molecular pathways as a generic mechanism and as a core disease phenotype. Oncotarget 2015, 6, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Oellerich, T.; Baldauf, H.-M.; Schwarz, S.-M.; Thomas, D.; Flick, R.; Bohnenberger, H.; Kaderali, L.; Stegnmann, L.; Cremer, A.; et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat. Med. 2017, 23, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Hou, H.-A.; Tien, H.-F. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J. Biomed. Sci. 2020, 27, 81. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-D.; Lin, F.-M.; Wu, W.-Y.; Liang, C.; Huang, W.-C.; Chan, W.-L.; Tsai, W.-T.; Chen, G.-Z.; Lee, C.-J.; Chiu, C.-M.; et al. miRTarBase: A database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011, 39, D163–D169. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M.; Biecek, P. Drawing Survival Curves Using ’ggplot2’ [R Package Survminer Version 0.4.8]. 2020. Available online: https://rpkgs.datanovia.com/survminer/ (accessed on 25 September 2023).
- Qiu, Y.-Q. KEGG Pathway Database. In Encyclopedia of Systems Biology; Wolkenhauer, O., Cho, K.-H., Yokota, H., Dubitzky, W., Eds.; Springer: New York, NY, USA, 2013; pp. 1068–1069. [Google Scholar] [CrossRef]
- Dong, M.; Blobe, G.C. Role of transforming growth factor-beta in hematologic malignancies. Blood 2006, 107, 4589–4596. [Google Scholar] [CrossRef]
- Tabe, Y.; Shi, Y.X.; Zeng, Z.; Jin, L.; Shikami, M.; Hatanaka, Y.; Miida, T.; Hsu, F.J.; Andreeff, M.; Konopleva, M. TGF-β-Neutralizing Antibody 1D11 Enhances Cytarabine-Induced Apoptosis in AML Cells in the Bone Marrow Microenvironment. PLoS ONE 2013, 8, e62785. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J.; Beach, D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 1994, 371, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Alexandrow, M.G.; Moses, H.L. Transforming growth factor beta and cell cycle regulation. Cancer Res. 1995, 55, 1452–1457. [Google Scholar] [PubMed]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Aref, S.; Ebrahim, L.; El-Ashwah, S.; El Agdar, M.; Ayed, M. Relevance of plasma bone marrow Activin-A and CXCL-12 concentration levels as a biomarker in acute myeloid leukemia. Cancer Biomark. 2021, 32, 263–270. [Google Scholar] [CrossRef]
- Qiu, W.; Kuo, C.-Y.; Tian, Y.; Su, G.H. Dual Roles of the Activin Signaling Pathway in Pancreatic Cancer. Biomedicines 2021, 9, 821. [Google Scholar] [CrossRef]
- Fenaux, P.; Kiladjian, J.J.; Platzbecker, U. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood 2019, 133, 790–794. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Diez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Platzbecker, U.; Fenaux, P.; Zeidan, A.M.; Garcia-Manero, G.; Mufti, G.J.; Santini, V.; Diez-Campelo, M.; Finelli, C.; Jurcic, J.G.; et al. Luspatercept for myelodysplastic syndromes/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Leukemia 2022, 36, 1432–1435. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Count | % Coverage | FDR |
|---|---|---|---|
| Pathways in cancer | 186 | 4.1 | 5.89 × 10−5 |
| Proteoglycans in cancer | 81 | 1.78 | 0.00104 |
| Cell cycle | 54 | 1.19 | 0.0016 |
| ErbB signaling pathway | 40 | 0.88 | 0.0016 |
| Regulation of actin cytoskeleton | 83 | 1.83 | 0.0016 |
| Neurotrophin signaling pathway | 51 | 1.12 | 0.0019 |
| TGF-beta signaling pathway | 42 | 0.92 | 0.0029 |
| T cell receptor signaling pathway | 45 | 0.99 | 0.0034 |
| Salmonella infection | 89 | 1.96 | 0.0063 |
| MAPK signaling pathway | 102 | 2.25 | 0.0067 |
| Hippo signaling pathway | 60 | 1.32 | 0.0096 |
| Protein processing in endoplasmic reticulum | 64 | 1.41 | 0.0106 |
| Renal cell carcinoma | 31 | 0.68 | 0.0152 |
| Lipid and atherosclerosis | 76 | 1.67 | 0.0162 |
| EGFR tyrosine kinase inhibitor resistance | 34 | 0.75 | 0.0162 |
| miRs Upregulated in HL60R | Targets | miRs Downregulated in HL60R | Targets |
|---|---|---|---|
| hsa-miR-100-5p | CREBBP | hsa-let-7f-5p | TGFBR1 |
| CUL1 | THBS1 | ||
| ID1 | hsa-let-7g-5p | TGFBR1 | |
| BMP6 | THBS1 | ||
| BMPR1A | hsa-let-7i-5p | BMP4 | |
| E2F4 | THBS1 | ||
| FMOD | BMP2 | ||
| hsa-miR-124-3p | GREM1 | hsa-miR-106a-5p | BMP8B |
| ID1 | RGMB | ||
| ID2 | ACVR2A | ||
| ID4 | CDKN2B | ||
| RHOA | hsa-miR-15a-5p | IFNG | |
| ROCK1 | RPS6KB1 | ||
| ACVR2B | SMURF1 | ||
| hsa-miR-1301-3p | CREBBP | ACVR2A | |
| SKP1 | BAMBI | ||
| hsa-miR-143-3p | TNF | IFNG | |
| hsa-miR-146a-5p | RHOA | hsa-miR-16-5p | RPS6KB1 |
| ROCK1 | SMAD1 | ||
| hsa-miR-196b-5p | ACVR2B | SMURF1 | |
| FMOD | SMURF2 | ||
| GDF5 | hsa-miR-20a-5p | BAMBI | |
| hsa-miR-21-5p | TGFB2 | BMP2 | |
| TGIF1 | BMP8B | ||
| ZFYVE16 | RBL1 | ||
| hsa-miR-221-3p | ACVR2B | RGMB | |
| RHOA | SMAD6 | ||
| hsa-miR-335-3p | ID2 | TGFBR1 | |
| hsa-miR-342-3p | BMP7 | THBS1 | |
| ID4 | hsa-miR-29c-3p | FBN1 | |
| hsa-miR-374b-3p | TGIF1 | TGIF2 | |
| hsa-miR-454-3p | ACVR1 | hsa-miR-424-5p | ACVR2A |
| hsa-miR-671-5p | BMP8A | RPS6KB1 | |
| PPP2CA | SMURF1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reichelt, P.; Bernhart, S.; Wilke, F.; Schwind, S.; Cross, M.; Platzbecker, U.; Behre, G. MicroRNA Expression Patterns Reveal a Role of the TGF-β Family Signaling in AML Chemo-Resistance. Cancers 2023, 15, 5086. https://doi.org/10.3390/cancers15205086
Reichelt P, Bernhart S, Wilke F, Schwind S, Cross M, Platzbecker U, Behre G. MicroRNA Expression Patterns Reveal a Role of the TGF-β Family Signaling in AML Chemo-Resistance. Cancers. 2023; 15(20):5086. https://doi.org/10.3390/cancers15205086
Chicago/Turabian StyleReichelt, Paula, Stephan Bernhart, Franziska Wilke, Sebastian Schwind, Michael Cross, Uwe Platzbecker, and Gerhard Behre. 2023. "MicroRNA Expression Patterns Reveal a Role of the TGF-β Family Signaling in AML Chemo-Resistance" Cancers 15, no. 20: 5086. https://doi.org/10.3390/cancers15205086
APA StyleReichelt, P., Bernhart, S., Wilke, F., Schwind, S., Cross, M., Platzbecker, U., & Behre, G. (2023). MicroRNA Expression Patterns Reveal a Role of the TGF-β Family Signaling in AML Chemo-Resistance. Cancers, 15(20), 5086. https://doi.org/10.3390/cancers15205086