
Validation of a Proteomic Signature of Lung Cancer Risk from Bronchial Specimens of Risk-Stratified Individuals
 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
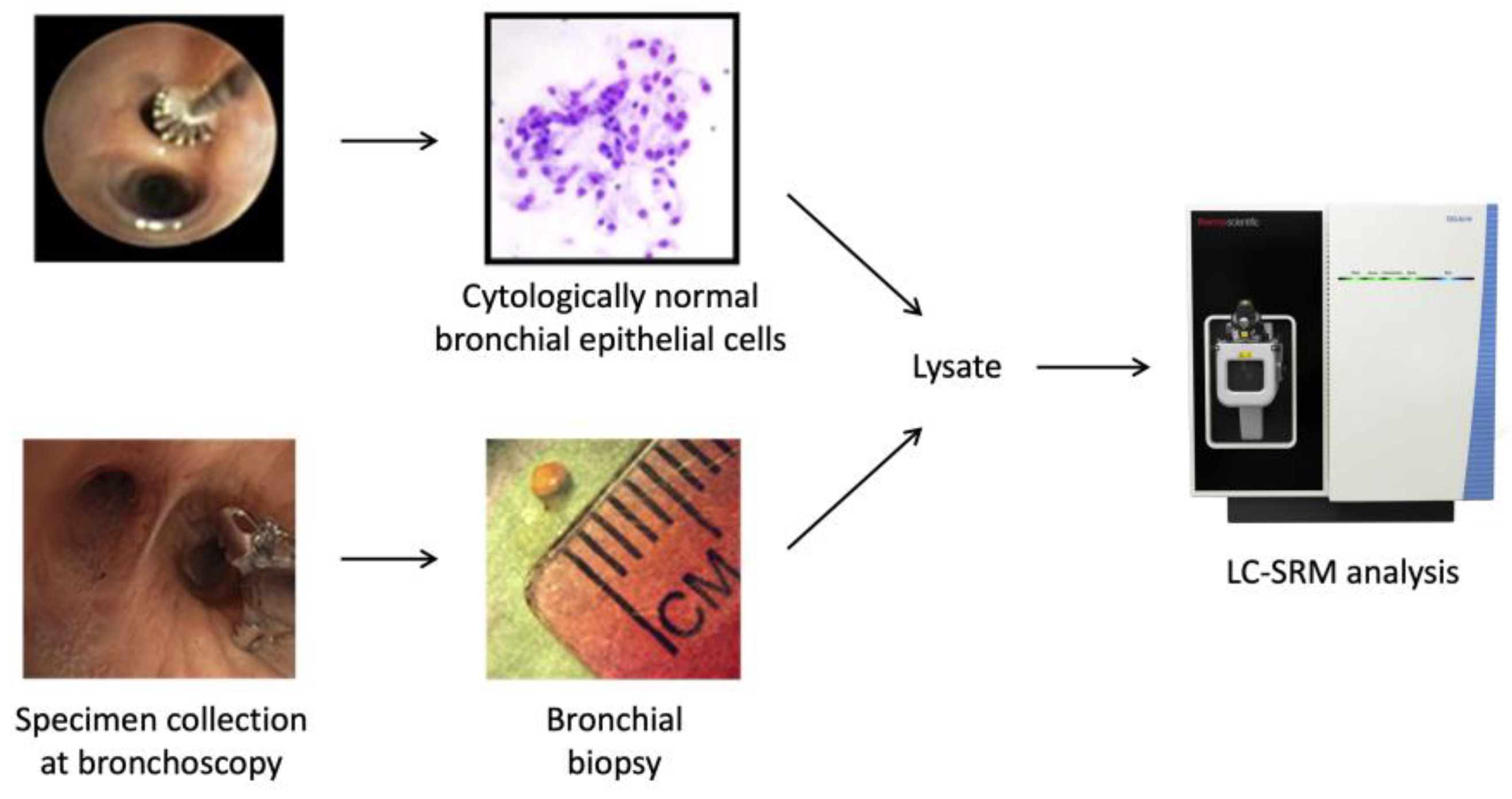
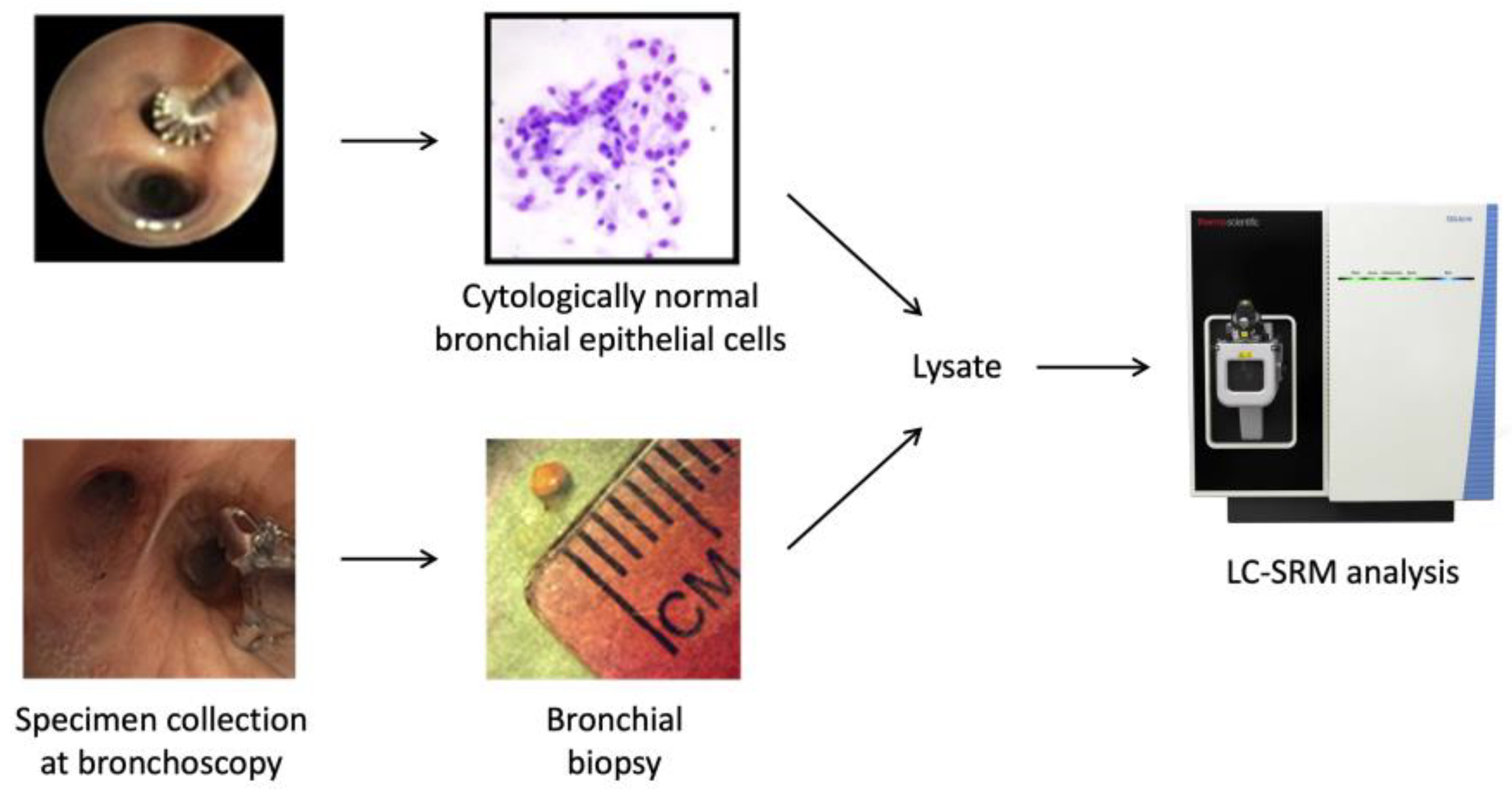
2.1. Patient Cohorts and Collection of Bronchial Specimens
2.2. Tryptic Digestion of Proteins
2.3. SRM Assay Development
2.4. LC-SRM Analysis
2.5. SRM Data Analysis
2.6. Statistical Analysis
3. Results
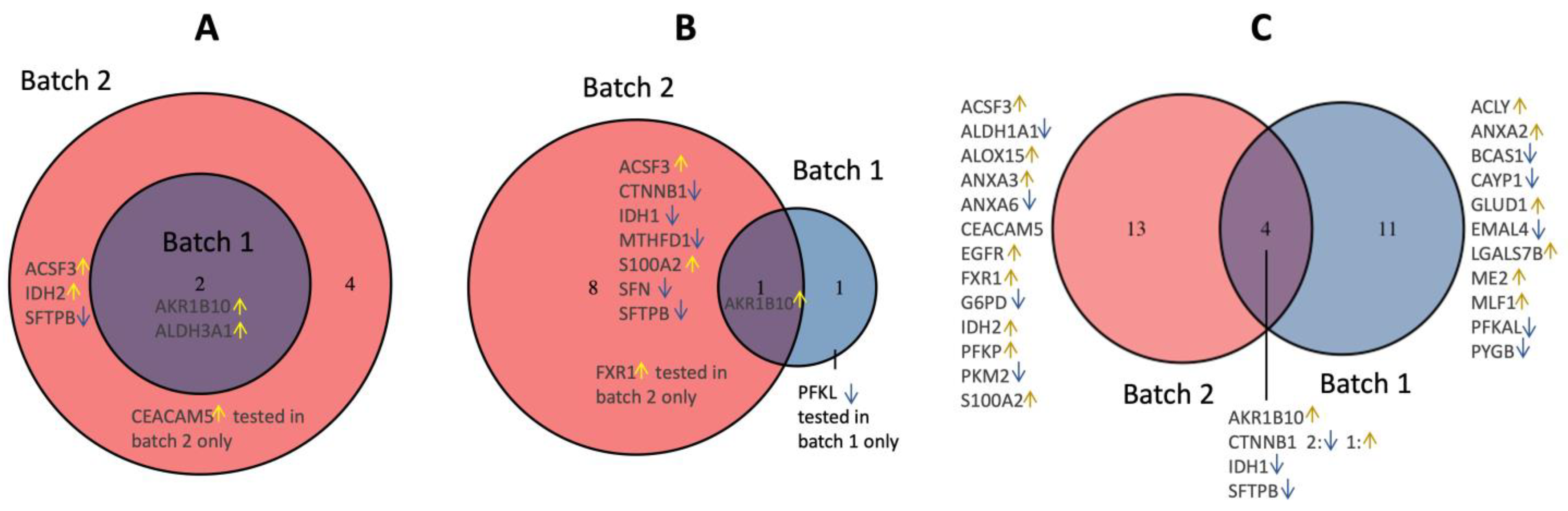
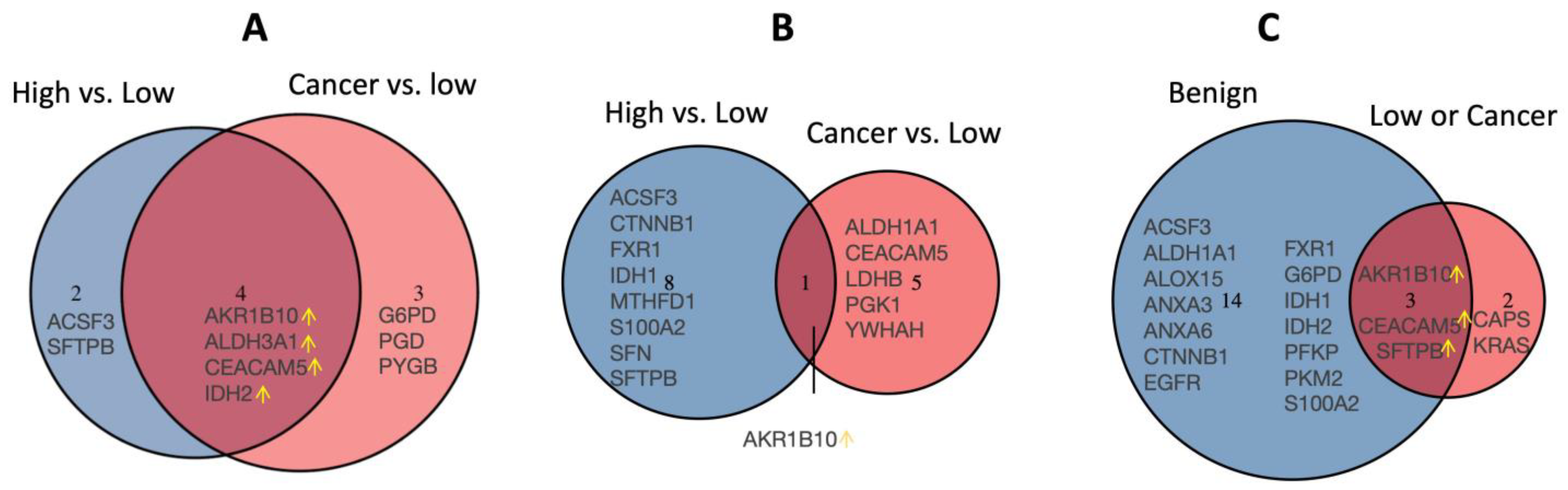
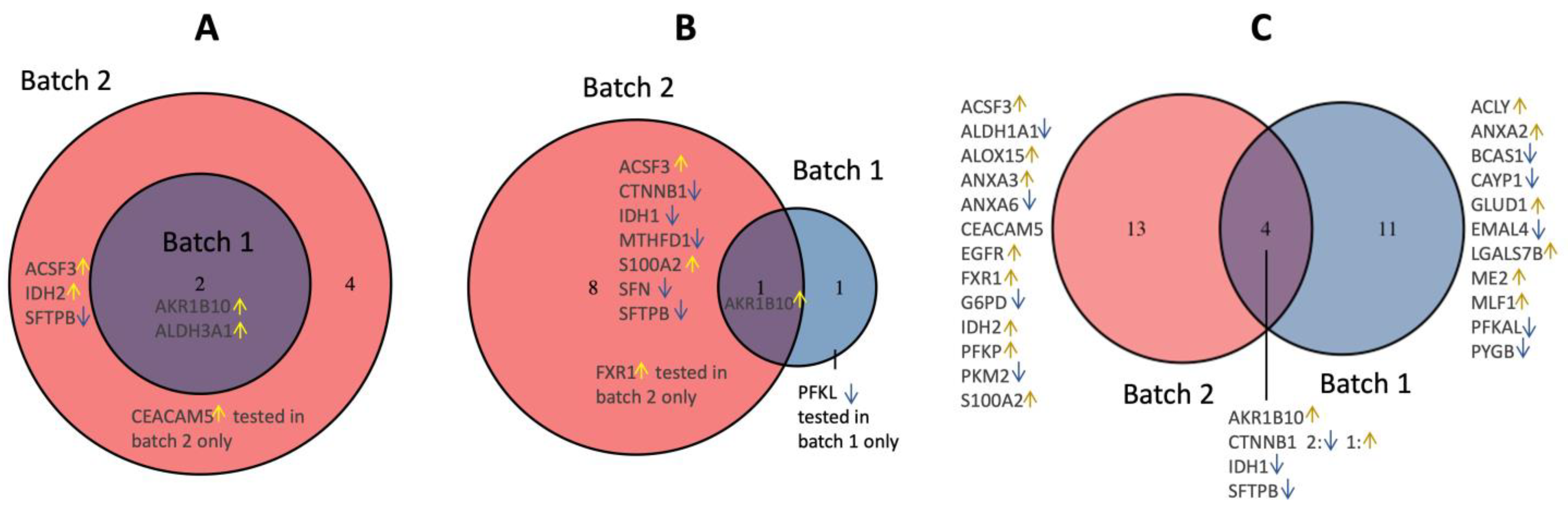
3.1. Candidate Protein Selection for Validation
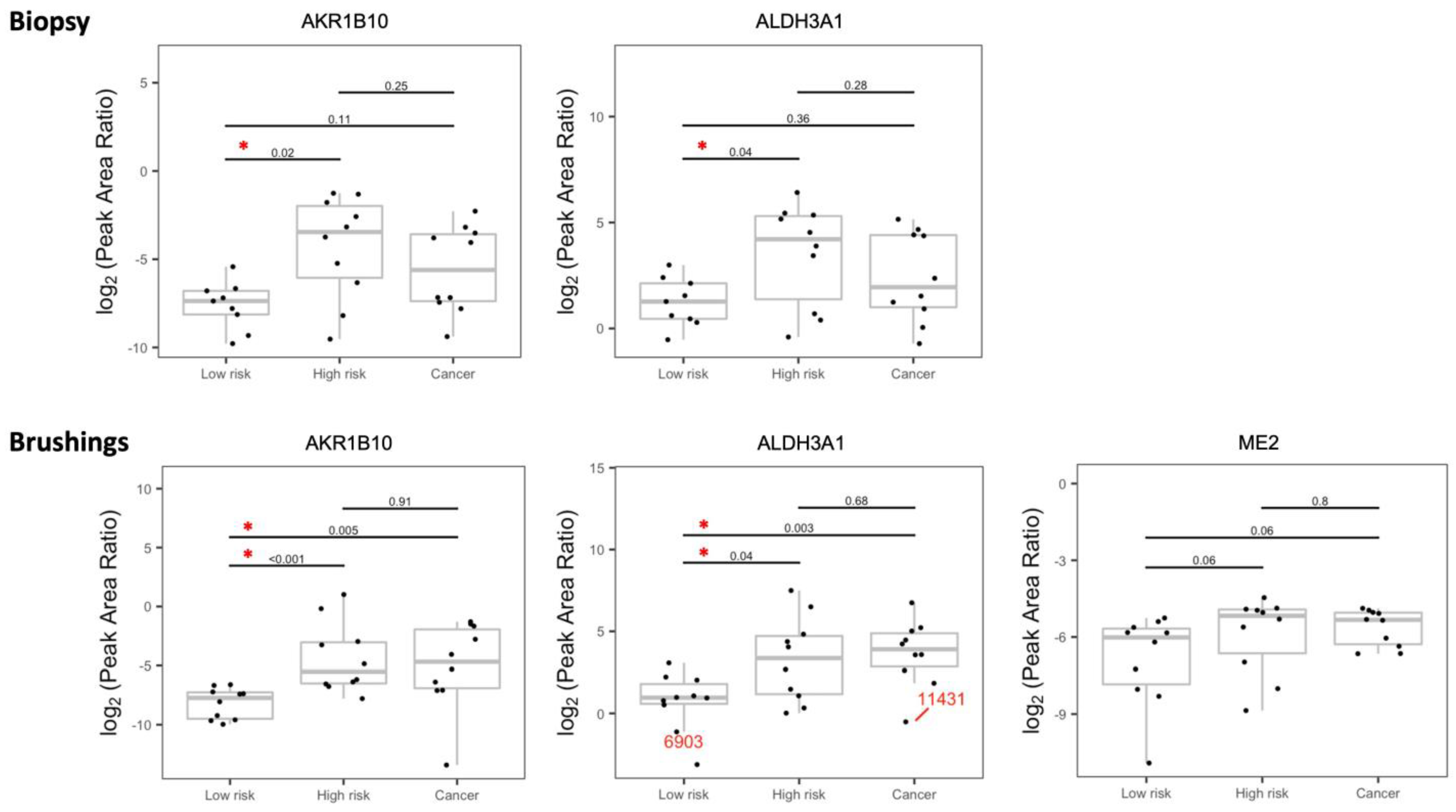
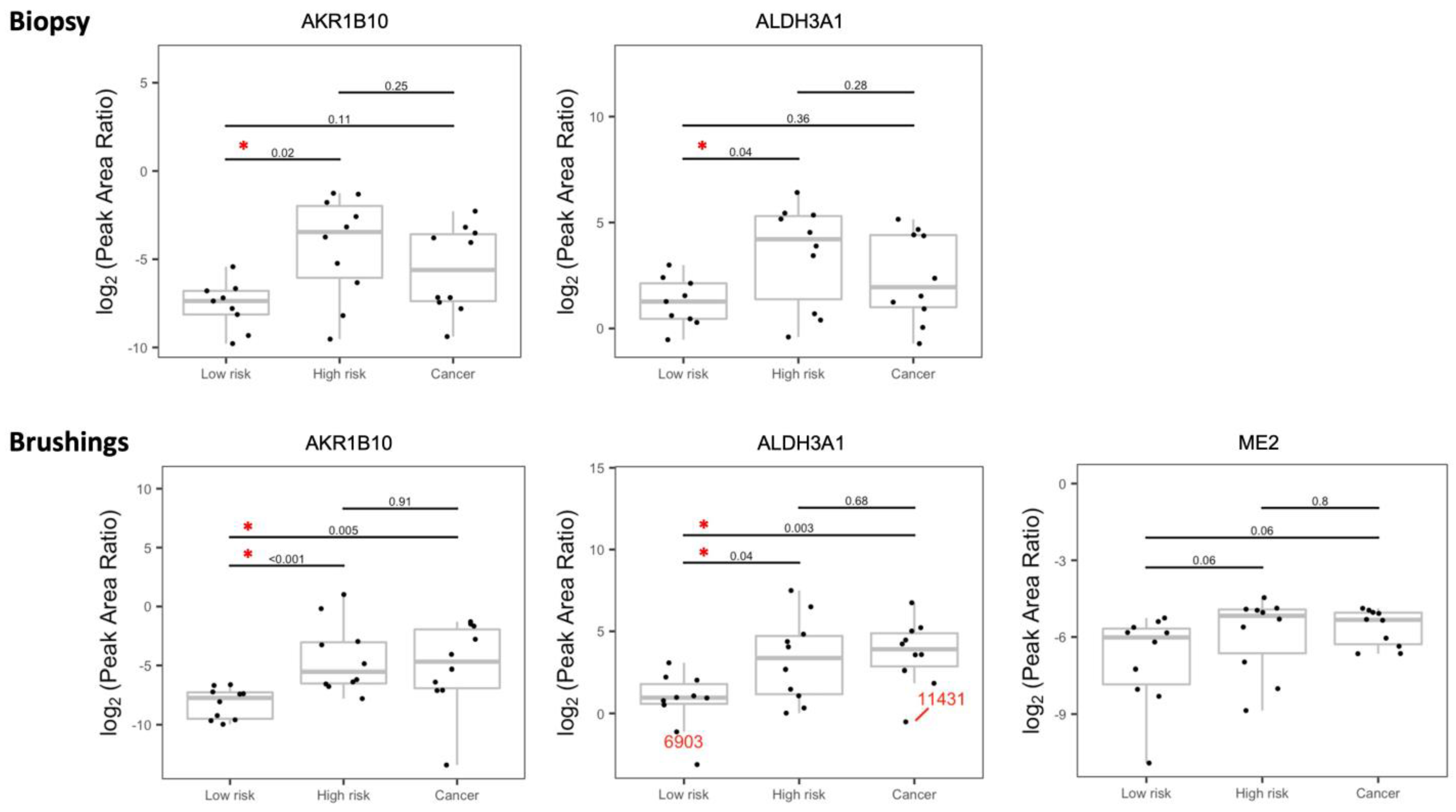
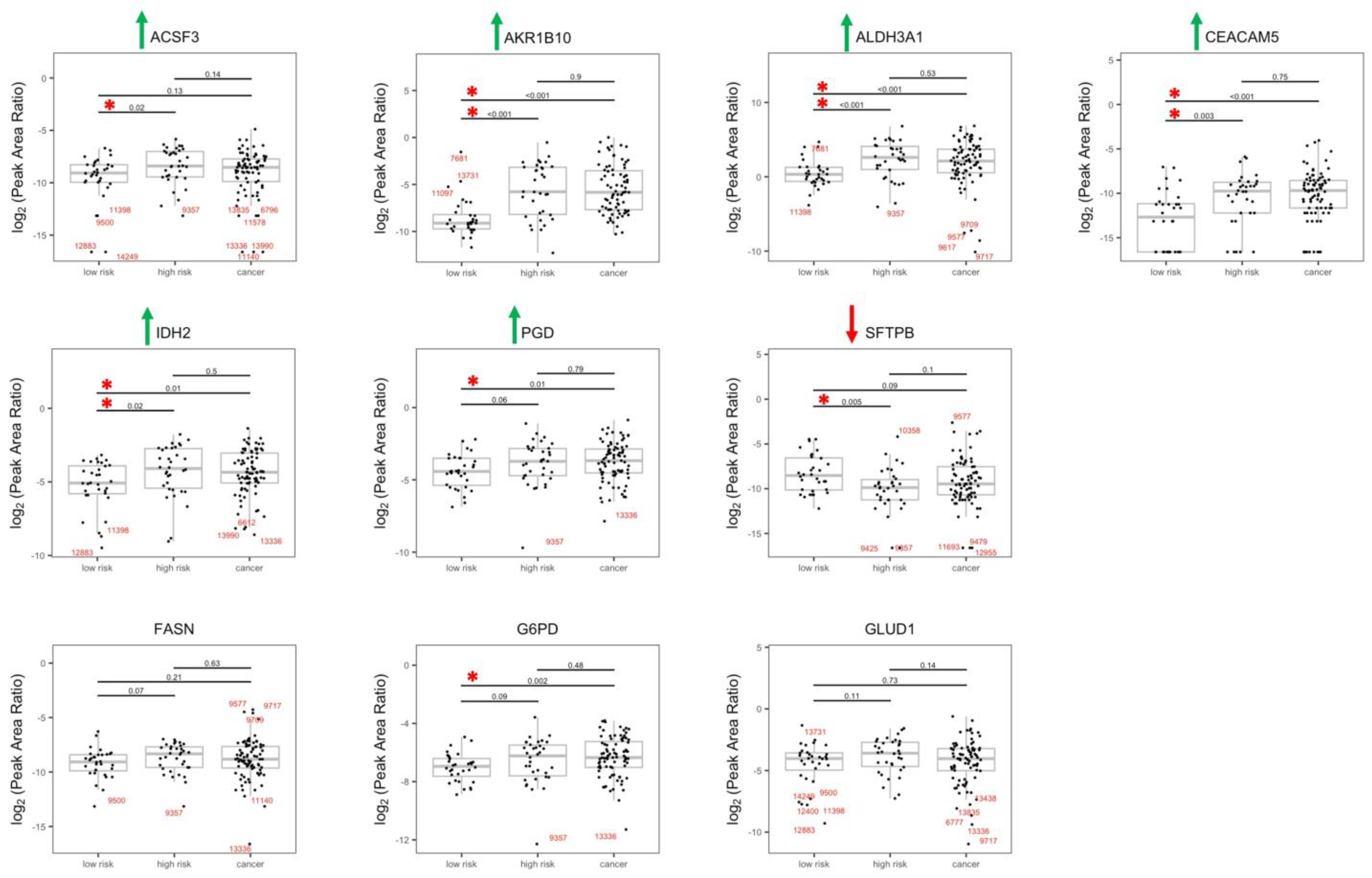
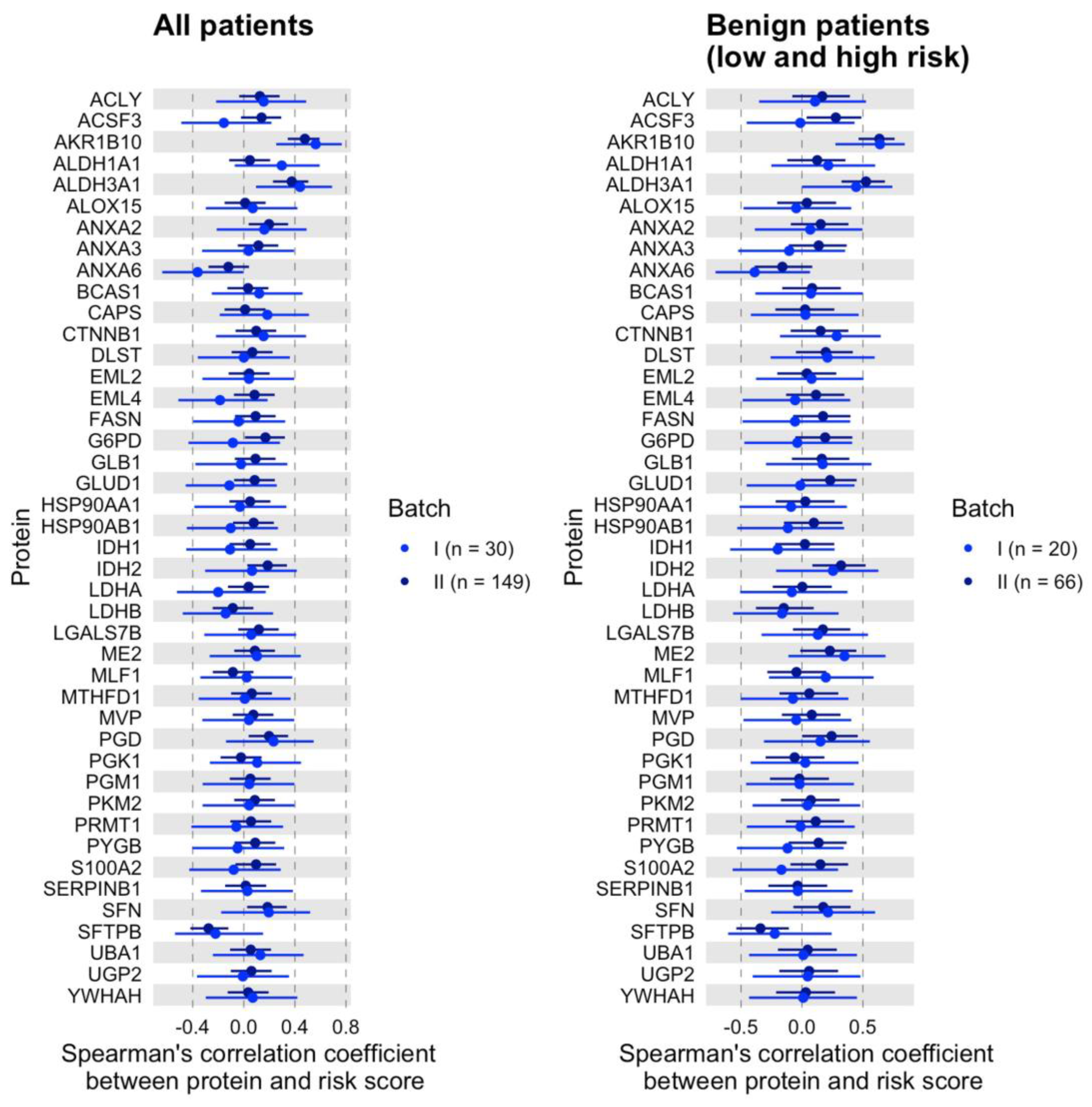
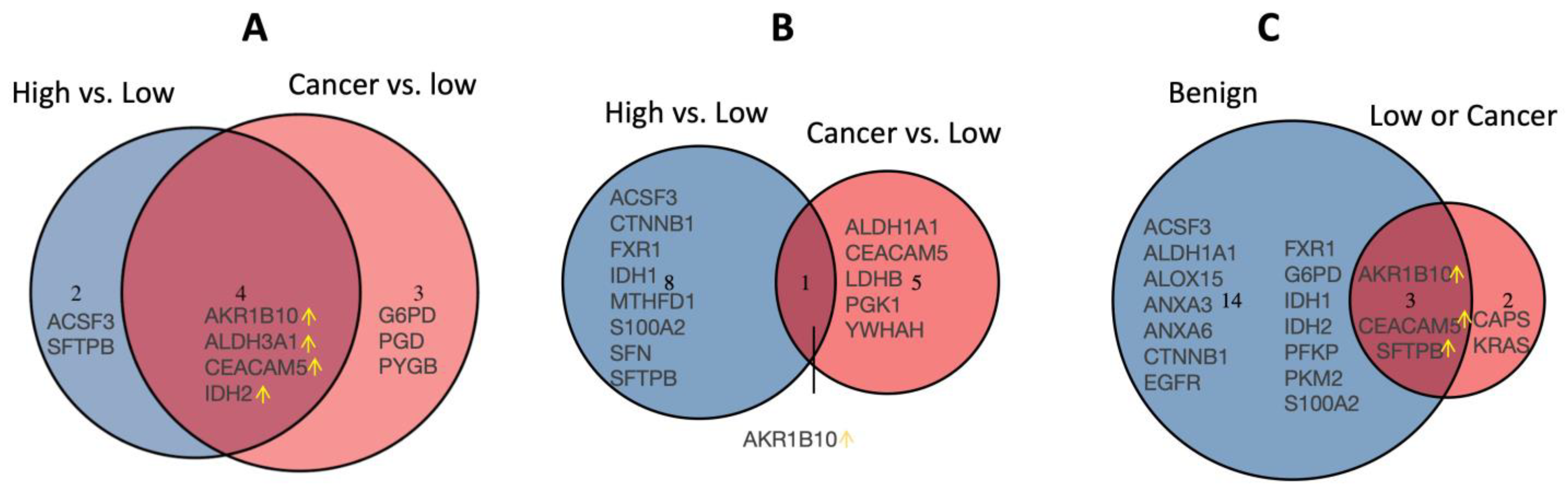
3.2. Validation of the Dysregulated Proteins
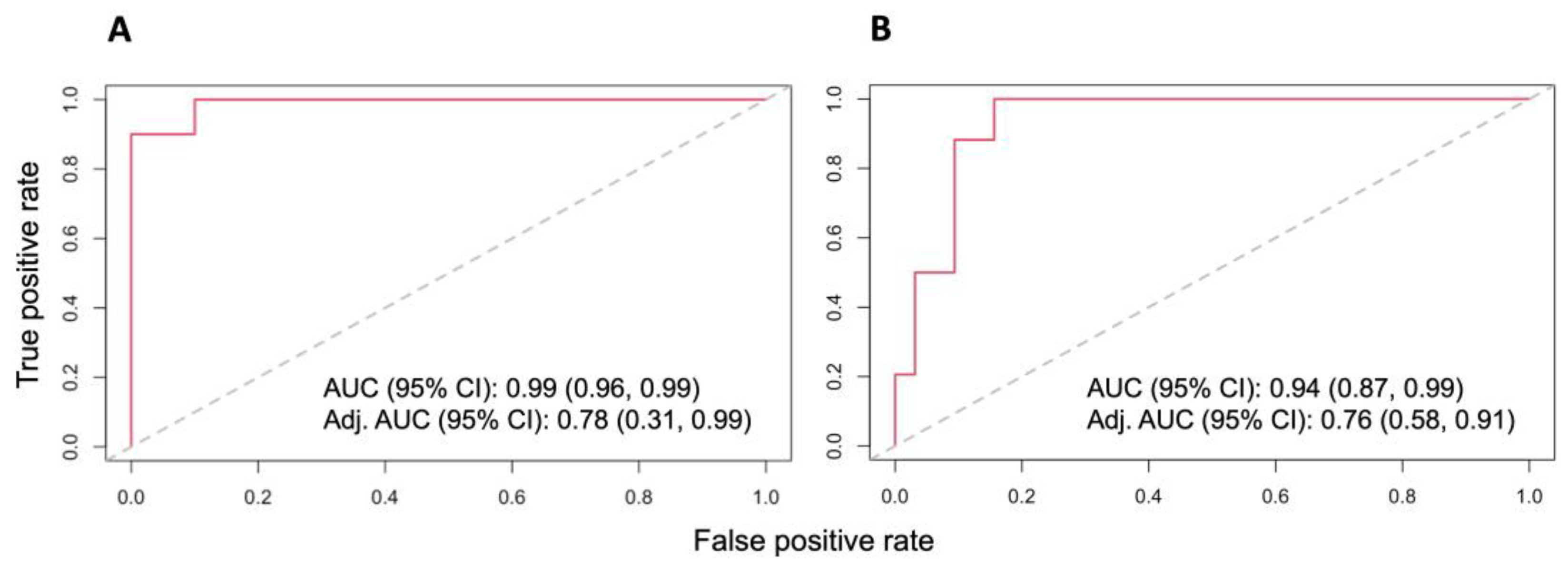
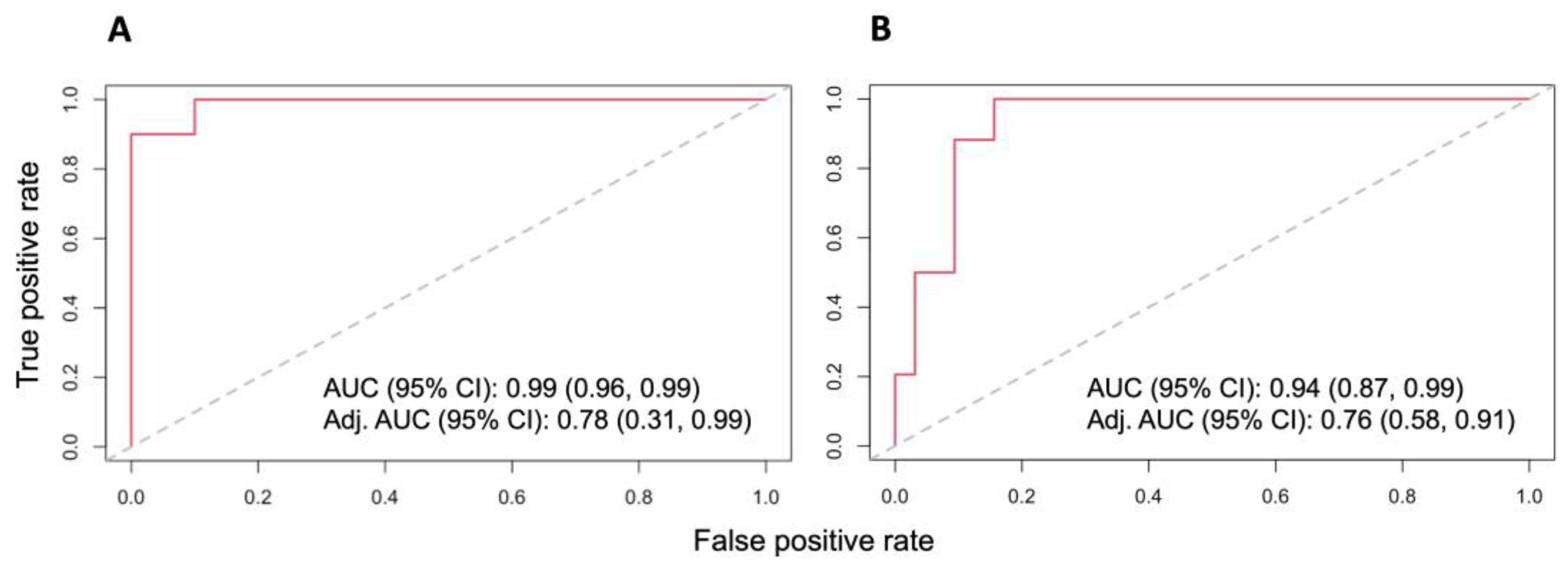
3.3. Dysregulated Proteins Discriminate High-Risk Groups from Low-Risk Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kadara, H.; Sivakumar, S.; Jakubek, Y.; San Lucas, F.A.; Lang, W.; McDowell, T.; Weber, Z.; Behrens, C.; Davies, G.E.; Kalhor, N.; et al. Driver Mutations in Normal Airway Epithelium Elucidate Spatiotemporal Resolution of Lung Cancer. Am. J. Respir. Crit. Care Med. 2019, 200, 742–750. [Google Scholar] [CrossRef]
- Spira, A.; Beane, J.E.; Shah, V.; Steiling, K.; Liu, G.; Schembri, F.; Gilman, S.; Dumas, Y.M.; Calner, P.; Sebastiani, P.; et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat. Med. 2007, 13, 361–366. [Google Scholar] [CrossRef]
- Spira, A.; Beane, J.; Shah, V.; Liu, G.; Schembri, F.; Yang, X.; Palma, J.; Brody, J.S. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc. Natl. Acad. Sci. USA 2004, 101, 10143–10148. [Google Scholar] [CrossRef]
- Ghosh, A.; Coakley, R.C.; Mascenik, T.; Rowell, T.R.; Davis, E.S.; Rogers, K.; Webster, M.J.; Dang, H.; Herring, L.E.; Sassano, M.F.; et al. Chronic E-Cigarette Exposure Alters the Human Bronchial Epithelial Proteome. Am. J. Respir. Crit. Care Med. 2018, 198, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.M.; Shyr, Y.; Yildiz, P.B.; Gonzalez, A.L.; Li, H.; Zhang, X.; Chaurand, P.; Yanagisawa, K.; Slovis, B.S.; Miller, R.F.; et al. Proteomic patterns of preinvasive bronchial lesions. Am. J. Respir. Crit. Care Med. 2005, 172, 1556–1562. [Google Scholar] [CrossRef]
- Kadara, H.; Wistuba, I.I. Field cancerization in non-small cell lung cancer: Implications in disease pathogenesis. Proc. Am. Thorac. Soc. 2012, 9, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.M.J.; Ji, X.; Zimmerman, L.J.; Li, M.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Zou, Y.; Qian, J.; Slebos, R.J.; et al. The airway epithelium undergoes metabolic reprogramming in individuals at high risk for lung cancer. JCI Insight 2016, 1, e88814. [Google Scholar] [CrossRef] [PubMed]
- Cerciello, F.; Choi, M.; Sinicropi-Yao, S.L.; Lomeo, K.; Amann, J.M.; Felley-Bosco, E.; Stahel, R.A.; Robinson, B.W.S.; Creaney, J.; Pass, H.I.; et al. Verification of a Blood-Based Targeted Proteomics Signature for Malignant Pleural Mesothelioma. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Tammemagi, M.C.; Katki, H.A.; Hocking, W.G.; Church, T.R.; Caporaso, N.; Kvale, P.A.; Chaturvedi, A.K.; Silvestri, G.A.; Riley, T.L.; Commins, J.; et al. Selection criteria for lung-cancer screening. N. Engl. J. Med. 2013, 368, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.M.J.; Gonzalez, A.L.; Li, M.; Seeley, E.H.; Zimmerman, L.J.; Zhang, X.J.; Manier, M.L.; Olson, S.J.; Shah, R.N.; Miller, A.N.; et al. Lung Cancer Diagnosis from Proteomic Analysis of Preinvasive Lesions. Cancer Res. 2011, 71, 3009–3017. [Google Scholar] [CrossRef]
- Petyuk, V.A.; Mayampurath, A.M.; Monroe, M.E.; Polpitiya, A.D.; Purvine, S.O.; Anderson, G.A.; Camp, D.G., 2nd; Smith, R.D. DtaRefinery, a software tool for elimination of systematic errors from parent ion mass measurements in tandem mass spectra data sets. Mol. Cell. Proteom. 2010, 9, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Gupta, N.; Pevzner, P.A. Spectral probabilities and generating functions of tandem mass spectra: A strike against decoy databases. J. Proteome Res. 2008, 7, 3354–3363. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Sharma, V.; Eckels, J.; Schilling, B.; Ludwig, C.; Jaffe, J.D.; MacCoss, M.J.; MacLean, B. Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Mol. Cell. Proteom. 2018, 17, 1239–1244. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Sdelci, S.; Rendeiro, A.F.; Rathert, P.; You, W.; Lin, J.G.; Ringler, A.; Hofstatter, G.; Moll, H.P.; Gurtl, B.; Farlik, M.; et al. MTHFD1 interaction with BRD4 links folate metabolism to transcriptional regulation. Nat. Genet. 2019, 51, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Li, W.; Song, Y.; Wang, Z.; Ju, R.; Ulman, A.; Hu, J.; Palomba, F.; Zhao, Y.; Le, J.P.; et al. UTX condensation underlies its tumour-suppressive activity. Nature 2021, 597, 726–731. [Google Scholar] [CrossRef]
- Alam, H.; Tang, M.; Maitituoheti, M.; Dhar, S.S.; Kumar, M.; Han, C.Y.; Ambati, C.R.; Amin, S.B.; Gu, B.; Chen, T.Y.; et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 2020, 37, 599–617.e597. [Google Scholar] [CrossRef]
- Miao, T.W.; Du, L.Y.; Xiao, W.; Mao, B.; Wang, Y.; Fu, J.J. Identification of Survival-Associated Gene Signature in Lung Cancer Coexisting With COPD. Front. Oncol. 2021, 11, 600243. [Google Scholar] [CrossRef]
- Hu, R.; Huffman, K.E.; Chu, M.; Zhang, Y.; Minna, J.D.; Yu, Y. Quantitative Secretomic Analysis Identifies Extracellular Protein Factors That Modulate the Metastatic Phenotype of Non-Small Cell Lung Cancer. J. Proteome Res. 2016, 15, 477–486. [Google Scholar] [CrossRef]
- Wang, G.; Qiu, M.; Xing, X.; Zhou, J.; Yao, H.; Li, M.; Yin, R.; Hou, Y.; Li, Y.; Pan, S.; et al. Lung cancer scRNA-seq and lipidomics reveal aberrant lipid metabolism for early-stage diagnosis. Sci. Transl. Med. 2022, 14, eabk2756. [Google Scholar] [CrossRef] [PubMed]
- Kammer, M.N.; Massion, P.P. Noninvasive biomarkers for lung cancer diagnosis, where do we stand? J Thorac Dis 2020, 12, 3317–3330. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.; Parnell, A.; Pennington, S.R. Why have so few proteomic biomarkers “survived” validation? (Sample size and independent validation considerations). Proteomics 2014, 14, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem. Biol. Interact. 2015, 234, 236–246. [Google Scholar] [CrossRef]
- Van Weverwijk, A.; Koundouros, N.; Iravani, M.; Ashenden, M.; Gao, Q.; Poulogiannis, G.; Jungwirth, U.; Isacke, C.M. Metabolic adaptability in metastatic breast cancer by AKR1B10-dependent balancing of glycolysis and fatty acid oxidation. Nat. Commun. 2019, 10, 2698. [Google Scholar] [CrossRef]
- Poturnajova, M.; Kozovska, Z.; Matuskova, M. Aldehyde dehydrogenase 1A1 and 1A3 isoforms—Mechanism of activation and regulation in cancer. Cell. Signal 2021, 87, 110120. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Symbol | Protein Name |
|---|---|
| ACLY | ATP citrate lyase |
| ACSF3 | Acyl-CoA synthetase family member 3 |
| AKR1B10 | Aldo-keto reductase family 1, member B10 (aldose reductase) |
| ALDH1A1 | Aldehyde dehydrogenase 1 family, member A1 |
| ALDH3A1 | Aldehyde dehydrogenase 3 family, memberA1 |
| ALOX15 | Arachidonate 15-lipoxygenase |
| ANXA2 | Annexin A2 |
| ANXA3 | Annexin A3 |
| ANXA6 | Annexin A6 |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3 related |
| BCAS1 | Breast carcinoma amplified sequence 1 |
| BRCA2 | Breast cancer type 2 susceptibility protein |
| CAPS | Calcyphosine |
| CEACAM5 | Carcinoembryonic antigen-related cell adhesion molecule 5 |
| CTNNB1 | Catenin (cadherin-associated protein), beta 1, 88kDa |
| DLG5 | Discs, large homolog 5 (Drosophila) |
| DLST | Dihydrolipoamide S-succinyltransferase |
| EGFR | Epidermal growth factor receptor |
| EML2 | Echinoderm microtubule associated protein like 2 |
| EML4 | Echinoderm microtubule associated protein like 4 |
| FASN | Fatty acid synthase |
| FXR1 | Fragile X mental retardation, autosomal homolog 1 |
| G6PD | Glucose-6-phosphate dehydrogenase |
| GLB1 | Galactosidase, beta 1 |
| GLUD1 | Glutamate dehydrogenase 1 |
| HSP90AA1 | Heat shock protein 90 kDa alpha (cytosolic), class A member 1 |
| HSP90AB1 | Heat shock protein 90 kDa alpha (cytosolic), class B member 1 |
| IDH1 | Isocitrate dehydrogenase 1 |
| IDH2 | Isocitrate dehydrogenase 2 |
| KMT2D | Histone-lysine N-methyltransferase 2D |
| KRAS | Kirsten rat sarcoma viral proto-oncogene |
| LDHA | Lactate dehydrogenase A |
| LDHB | Lactate dehydrogenase B |
| LGALS7B | Lectin, galactoside-binding, soluble, 7B |
| ME2 | Malic enzyme 2, NAD(+)-dependent, mitochondrial |
| MLF1 | Myeloid leukemia factor 1 |
| MTHFD1 | C-1-tetrahydrofolate synthase, cytoplasmic |
| MVP | Major vault protein |
| NFKB1 | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 |
| PFKL | Phosphofructokinase, liver |
| PFKP | Phosphofructokinase, platelet |
| PGD | Phosphogluconate dehydrogenase |
| PGK1 | Phosphoglycerate kinase 1 |
| PGM1 | Phosphoglucomutase 1 |
| PKM2 | Pyruvate kinase M2, muscle |
| PRMT1 | Protein arginine methyltransferase 1 |
| PYGB | Glycogen phosphorylase B |
| S100A2 | S100 calcium binding protein A2 |
| SERPINB1 | Serpin peptidase inhibitor, clade B (ovalbumin), member 1 |
| SFN | Stratifin |
| SFTPB | Surfactant protein B |
| UBA1 | Ubiquitin-like modifier activating enzyme 1 |
| UGP2 | UDP-glucose pyrophosphorylase 2 |
| YWHAH | 14-3-3 protein eta |
| Characteristics | Low Risk (n = 10) | High Risk (n = 10) | Lung Cancer (n = 10) | |
|---|---|---|---|---|
| Age | ||||
| Average + St.Dev. | 61.8 ± 5.7 | 69.5 ± 4 | 73.5 ± 7.8 | |
| Median (range) | 60.5 (55–74) | 69.5 (63–76) | 76 (57–82) | |
| Gender | ||||
| Male | 5 (50%) | 9 (90%) | 6 (60%) | |
| Female | 5 (50%) | 1 (10%) | 4 (40%) | |
| BMI | ||||
| Average + St.Dev. | 27.5 ± 6 | 27.8 ± 3.8 | 22.5 ± 1.9 | |
| Median (range) | 28 (19.7–37) | 27.5 (21.3–32.7) | 22.5 (19.3–25.8) | |
| Smoking history | ||||
| Never smoker | 4 (40%) | 0 (0%) | 1 (10%) | |
| Former smoker | 6 (60%) | 4 (40%) | 7 (70%) | |
| Current smoker | 0 (0%) | 6 (60%) | 2 (20%) | |
| Pack year (Average + St.Dev.) * | 30.9 ± 28.7 | 73.4 ± 30.1 | 51.2 ± 30.3 | |
| Pack year (median) | 23.5 (3.8–65) | 59 (47–129) | 45 (15–100) | |
| COPD | ||||
| Yes | 0 (0%) | 3 (30%) | 4 (40%) | |
| No | 10 (100%) | 7 (70%) | 6 (60%) | |
| Histology | ||||
| Adenocarcinoma | - | - | 2 (20%) | |
| Squamous cell carcinoma | - | - | 7 (70%) | |
| Small-cell lung cancer | - | - | 1 (10%) | |
| Path stage | ||||
| No surgery | 4 (40%) | |||
| Tx Nx Mx | - | - | 2 (20%) | |
| IA | - | - | 1 (10%) | |
| IB | - | - | 1 (10%) | |
| IIA | - | - | 1 (10%) | |
| IV | - | - | 1 (10%) |
| Characteristics | Low Risk (n = 32) | High Risk (n = 34) | Lung Cancer (n = 83) | |
|---|---|---|---|---|
| Age | ||||
| Average + St.Dev. | 63.1 ± 10.1 | 65.7 ± 7.4 | 66.4 ± 6.3 | |
| Median (range) | 61 (43–87) | 66 (48–87) | 66 (53–85) | |
| Gender | ||||
| Male | 14 (44%) | 22 (65%) | 56 (67%) | |
| Female | 18 (56%) | 12 (35%) | 27 (33%) | |
| BMI | ||||
| Average + St.Dev. | 27.9 ± 5.3 | 26.8 ± 5.2 | 27.1 ± 6.2 | |
| Median (range) | 27.6 (15.5–39.1) | 26.1 (19.1–39) | 26.6 (18.1–53) | |
| Smoking history | ||||
| Never smoker | 10 (31%) | 0 (0%) | 1 (1%) | |
| Former smoker | 19 (59%) | 14 (41%) | 36 (43%) | |
| Current smoker | 3 (9%) | 20 (59%) | 46 (54%) | |
| Pack year (Average + St.Dev.) | 14.9 ± 13.9 | 67.8 ± 25.7 | 53.2 ± 32.2 | |
| Pack year (median) | 18 (0–50) | 61.5 (33–135) | 50 (0–200) | |
| COPD | ||||
| Yes | 7 (22%) | 27 (79%) | 61 (73%) | |
| No | 25 (25%) | 7 (21%) | 22 (27%) | |
| Histology | ||||
| Adenocarcinoma | - | - | 37 (45%) | |
| Squamous cell carcinoma | - | - | 38 (46%) | |
| Adenosquamous cell ca. | - | - | 1 (1%) | |
| Non-small-cell lung cancer | - | - | 3 (3%) | |
| Large cell carcinoma | - | - | 1 (1%) | |
| Large cell neuroendocrine | - | - | 2 (2%) | |
| Small-cell lung cancer | - | - | 1 (10%) | |
| Path stage | ||||
| Stage 0 | 1 (1%) | |||
| IA | - | - | 15 (18%) | |
| IB | - | - | 11 (13%) | |
| IIA | - | - | 1 (1%) | |
| IIB | - | - | 9 (11%) | |
| IIIA | - | - | 15 (18%) | |
| IIIB | - | - | 6 (7%) | |
| IIIC | - | - | 2 (2%) | |
| IVA | - | - | 11 (13%) | |
| IVB | - | - | 2 (2%) | |
| Limited | - | - | 1 (1%) | |
| Unknown | - | - | 9 (11%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.M.J.; Chen, S.-C.; Wang, Y.-T.; Gao, Y.; Schepmoes, A.A.; Fillmore, T.L.; Shi, T.; Chen, H.; Rodland, K.D.; Massion, P.P.; et al. Validation of a Proteomic Signature of Lung Cancer Risk from Bronchial Specimens of Risk-Stratified Individuals. Cancers 2023, 15, 4504. https://doi.org/10.3390/cancers15184504
Rahman SMJ, Chen S-C, Wang Y-T, Gao Y, Schepmoes AA, Fillmore TL, Shi T, Chen H, Rodland KD, Massion PP, et al. Validation of a Proteomic Signature of Lung Cancer Risk from Bronchial Specimens of Risk-Stratified Individuals. Cancers. 2023; 15(18):4504. https://doi.org/10.3390/cancers15184504
Chicago/Turabian StyleRahman, S.M. Jamshedur, Sheau-Chiann Chen, Yi-Ting Wang, Yuqian Gao, Athena A. Schepmoes, Thomas L. Fillmore, Tujin Shi, Heidi Chen, Karin D. Rodland, Pierre P. Massion, and et al. 2023. "Validation of a Proteomic Signature of Lung Cancer Risk from Bronchial Specimens of Risk-Stratified Individuals" Cancers 15, no. 18: 4504. https://doi.org/10.3390/cancers15184504
APA StyleRahman, S. M. J., Chen, S.-C., Wang, Y.-T., Gao, Y., Schepmoes, A. A., Fillmore, T. L., Shi, T., Chen, H., Rodland, K. D., Massion, P. P., Grogan, E. L., & Liu, T. (2023). Validation of a Proteomic Signature of Lung Cancer Risk from Bronchial Specimens of Risk-Stratified Individuals. Cancers, 15(18), 4504. https://doi.org/10.3390/cancers15184504