
Nivolumab after Induction Chemotherapy in Previously Treated Non-Small-Cell Lung Cancer Patients with Low PD-L1 Expression

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Participants
2.3. Assessment of Response
2.4. Immunohistochemistry of PD-L1 Expression
2.5. Blood Next-Generation Sequencing Analysis (NGS) by GuardantOMNI
2.6. Proteomics
2.6.1. Sample Preparation for Mass Spectrometry
2.6.2. LC-MS/MS Analysis
2.6.3. Protein Identification and Quantitation
2.6.4. Protein Expression Analysis
2.7. Flow Cytometry Analysis
2.8. Validation Cohort Selection and Enzyme-Linked Immunosorbent Assay (ELISA) for TFRC
2.9. Statistical Methods
3. Results
3.1. Patients Characteristics
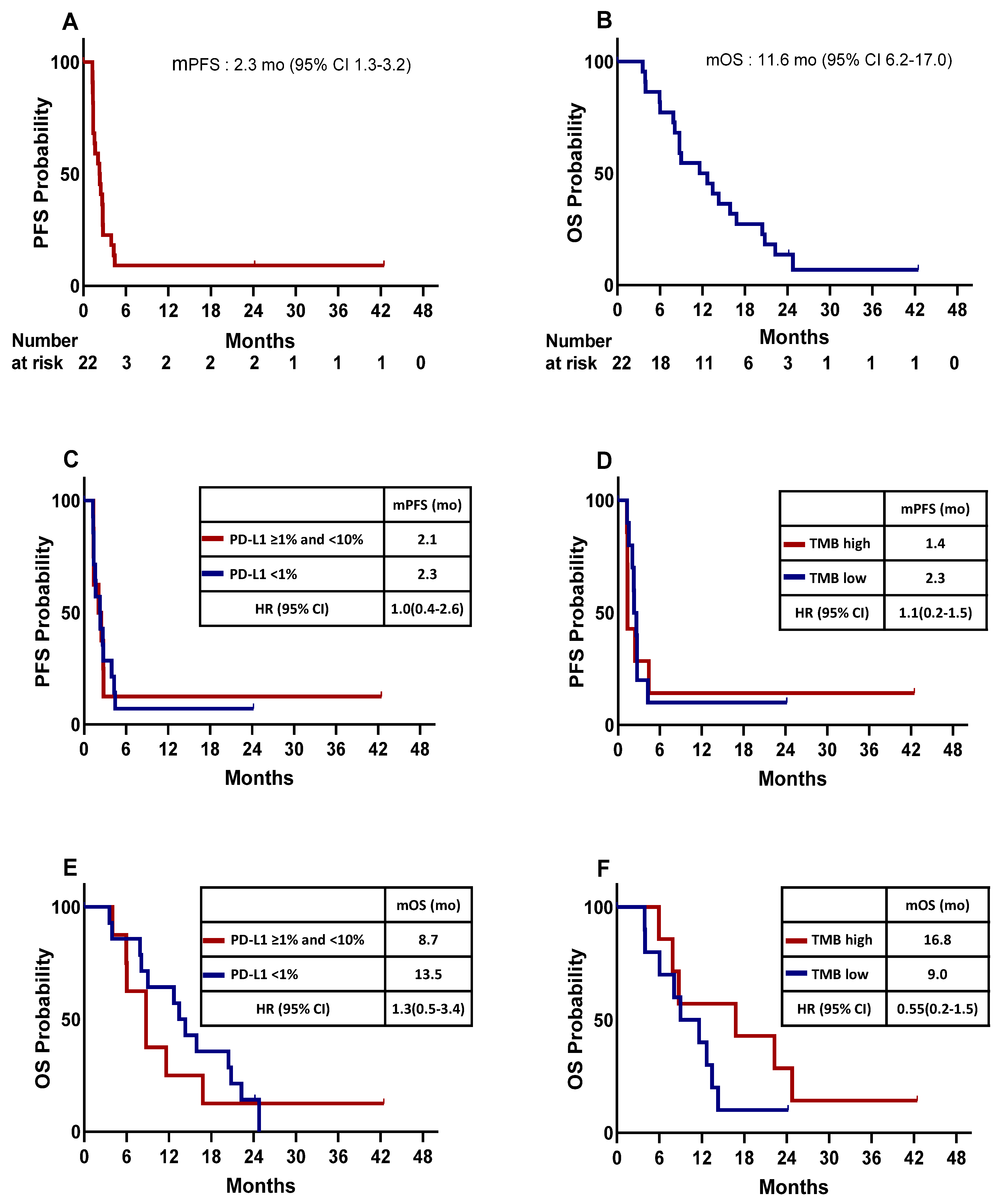
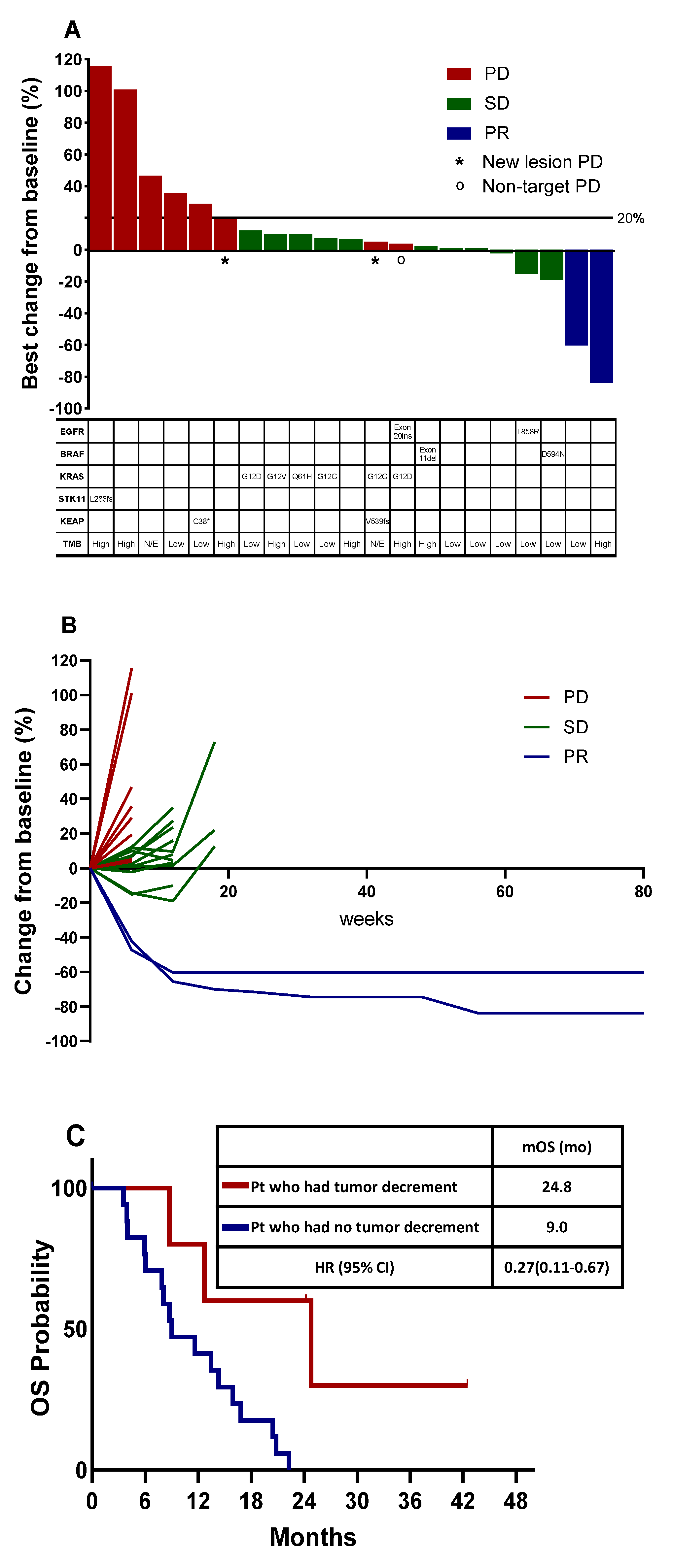
3.2. Treatment Efficacy
3.3. Tumor Burden Dynamics and Association with Survival
3.4. Blood NGS Analysis
3.5. MDSC Analysis in Peripheral Blood
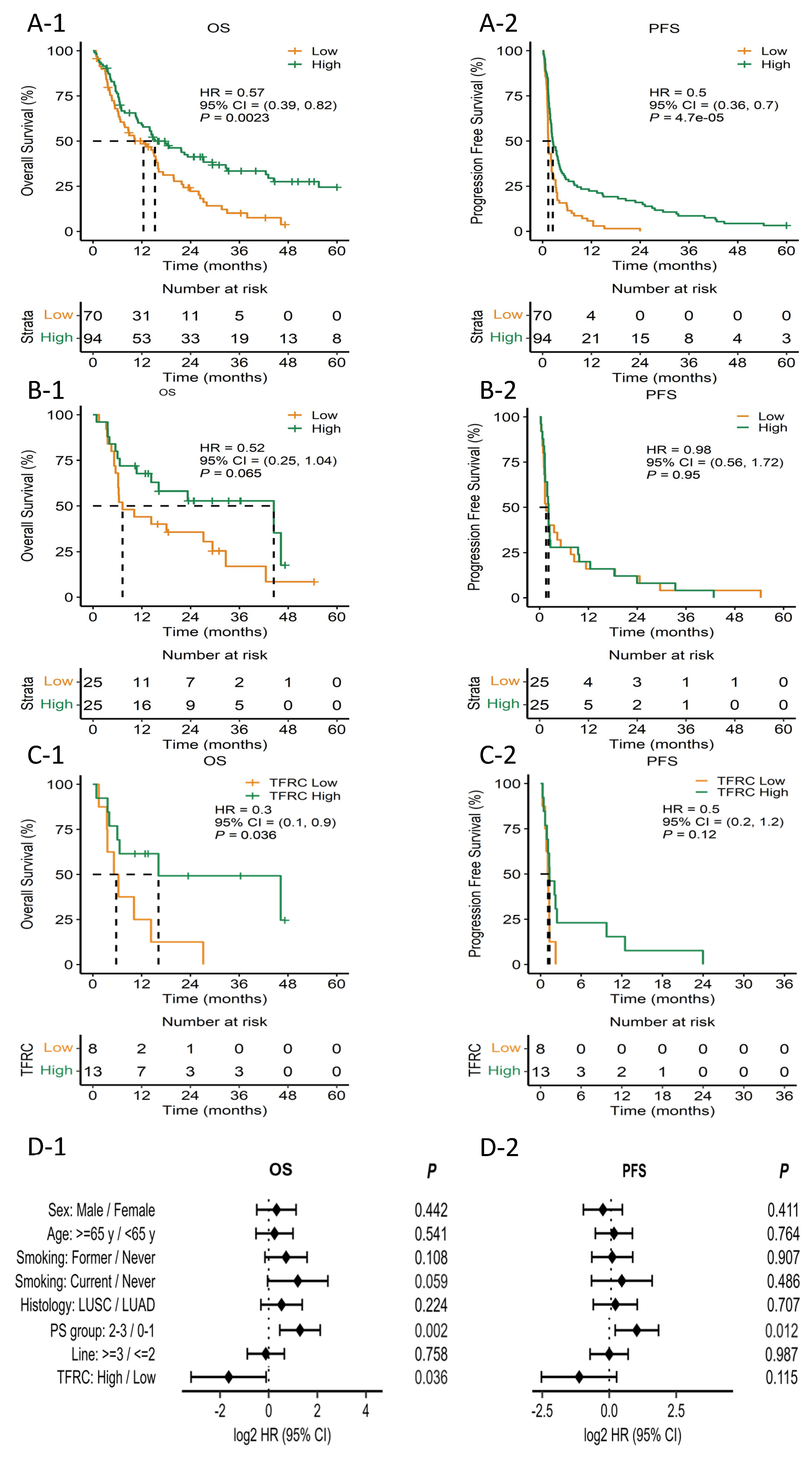
3.6. Protein Expression Analysis
3.7. Validation of Predictive Value of TFRC in an Independent Cohort
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Cappuzzo, F.; Horn, L.; Paz-Ares, L.; Borghaei, H.; Barlesi, F.; Steins, M.; Felip, E.; Spigel, D.; Dorange, C.; et al. OA03.05 analysis of early survival in patients with advanced non-squamous NSCLC treated with nivolumab vs docetaxel in CheckMate 057. J. Thorac. Oncol. 2017, 12, S253. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Hughes, E.; Scurr, M.; Campbell, E.; Jones, E.; Godkin, A.; Gallimore, A. T-cell modulation by cyclophosphamide for tumour therapy. Immunology 2018, 154, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Hu, Q.; Wang, X.; Feng, X.; He, Y.; Guo, Y.; Fu, D. Chemo-immunotherapy with doxorubicin prodrug and erythrocyte membrane-enveloped polymer nano-vaccine enhances antitumor activity. Biomed. Pharmacother. 2020, 129, 110377. [Google Scholar] [CrossRef]
- Maccubbin, D.L.; Wing, K.R.; Mace, K.F.; Ho, R.L.; Ehrke, M.J.; Mihich, E. Adriamycin-induced modulation of host defenses in tumor-bearing mice. Cancer Res. 1992, 52, 3572–3576. [Google Scholar] [PubMed]
- Alizadeh, D.; Trad, M.; Hanke, N.T.; Larmonier, C.B.; Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014, 74, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Helman, E.; Artieri, C.; Vowles, J.V.; Yen, J.; Nance, T.; Sikora, M.; Gourneau, J.; Goel, M.; Mortimer, S.; Chudova, D.; et al. Abstract 5603: Analytical validation of a comprehensive 500-gene ctDNA panel designed for immuno-oncology and DNA damage research. Cancer Res. 2018, 78, 5603. [Google Scholar] [CrossRef]
- Si, H.; Kuziora, M.; Quinn, K.J.; Helman, E.; Ye, J.; Liu, F.; Scheuring, U.; Peters, S.; Rizvi, N.A.; Brohawn, P.Z.; et al. A blood-based assay for assessment of tumor mutational burden in first-line metastatic NSCLC treatment: Results from the MYSTIC study. Clin. Cancer Res. 2021, 27, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein KnowledgeBase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using David bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.I.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor Cells1. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Guo, J.; Weng, L.; Tang, W.; Jin, S.; Ma, W. Myeloid-derived suppressor cells—New and exciting players in lung cancer. J. Hematol. Oncol. 2020, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Yang, C.; Li, J.; Guo, Y.; Gan, D.; Zhang, C.; Wang, R.; Hua, L.; Zhu, L.; Ma, P.; Shi, J.; et al. Role of TFRC as a novel prognostic biomarker and in immunotherapy for pancreatic carcinoma. Front. Mol. Biosci. 2022, 9, 756895. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.L.; Zhang, N.P.; Xu, R.C.; Zhang, G.C.; Liu, Z.Y.; Abuduwaili, W.; Wang, F.; Yu, X.N.; Shi, X.; Song, G.Q.; et al. Tumor cell-imposed iron restriction drives immunosuppressive polarization of tumor-associated macrophages. J. Transl. Med. 2021, 19, 347. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Ferrara, N. Iron metabolism in the tumor microenvironment: Contributions of innate immune cells. Front. Immunol. 2020, 11, 626812. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Total (N = 22) |
|---|---|
| Age of diagnosis (years) | |
| Median (range) | 63 (37–72) |
| Sex, n (%) | |
| Male | 18 (82) |
| Female | 4 (18) |
| Tobacco use, n (%) | |
| Never | 4 (18) |
| Former | 11 (50) |
| Current | 7 (32) |
| ECOG PS, n (%) | |
| 0 | 3 (14) |
| 1 | 19 (86) |
| Previous line of therapy | |
| 1 | 16 (73) |
| 2 | 6 (27) |
| Tumor Histology, n (%) | |
| Adenocarcinoma | 16 (73) |
| Large cell neuroendocrine carcinoma | 5 (23) |
| Sarcomatoid carcinoma | 1 (4) |
| PD-L1 | |
| <1% | 14 (64) |
| ≥1% and <10% | 8 (36) |
| Duration since the last treatment to study enrollment | |
| <3 months | 17 (77) |
| ≥3 months | 5 (23) |
| Best response to prior treatment | |
| PR | 2 (9) |
| SD | 11 (50) |
| PD | 8 (36) |
| NE | 1 (4) |
| Response | Total (N = 22) n (%) [95% CI] |
|---|---|
| ORR, n (%) [95% CI] | 2 (9) [1.9–26.1] |
| Complete response (CR) | 0 (0) |
| Partial response (PR) | 2 (9) |
| Stable disease (SD) | 11 (50) |
| Progressive disease (PD) | 9 (41) |
| DCR, n (%) [95% CI] | 13 (59) [38.5–77.5] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, B.-C.; Park, C.; Lee, S.-J.; Hong, S.; Hwang, J.-E.; Kwon, K.; Kim, J.Y.; Kim, K.-H.; Kim, H.Y.; Lee, G.K.; et al. Nivolumab after Induction Chemotherapy in Previously Treated Non-Small-Cell Lung Cancer Patients with Low PD-L1 Expression. Cancers 2023, 15, 4460. https://doi.org/10.3390/cancers15184460
Ahn B-C, Park C, Lee S-J, Hong S, Hwang J-E, Kwon K, Kim JY, Kim K-H, Kim HY, Lee GK, et al. Nivolumab after Induction Chemotherapy in Previously Treated Non-Small-Cell Lung Cancer Patients with Low PD-L1 Expression. Cancers. 2023; 15(18):4460. https://doi.org/10.3390/cancers15184460
Chicago/Turabian StyleAhn, Beung-Chul, Charny Park, Sang-Jin Lee, Sehwa Hong, Ji-Eun Hwang, Kyoungsuk Kwon, Jin Young Kim, Kyung-Hee Kim, Hyae Young Kim, Geon Kook Lee, and et al. 2023. "Nivolumab after Induction Chemotherapy in Previously Treated Non-Small-Cell Lung Cancer Patients with Low PD-L1 Expression" Cancers 15, no. 18: 4460. https://doi.org/10.3390/cancers15184460
APA StyleAhn, B.-C., Park, C., Lee, S.-J., Hong, S., Hwang, J.-E., Kwon, K., Kim, J. Y., Kim, K.-H., Kim, H. Y., Lee, G. K., Lee, Y., & Han, J.-Y. (2023). Nivolumab after Induction Chemotherapy in Previously Treated Non-Small-Cell Lung Cancer Patients with Low PD-L1 Expression. Cancers, 15(18), 4460. https://doi.org/10.3390/cancers15184460