

Primary Cutaneous Anaplastic Large Cell Lymphoma—A Review of Clinical, Morphological, Immunohistochemical, and Molecular Features



Abstract
Simple Summary
Abstract
1. Introduction
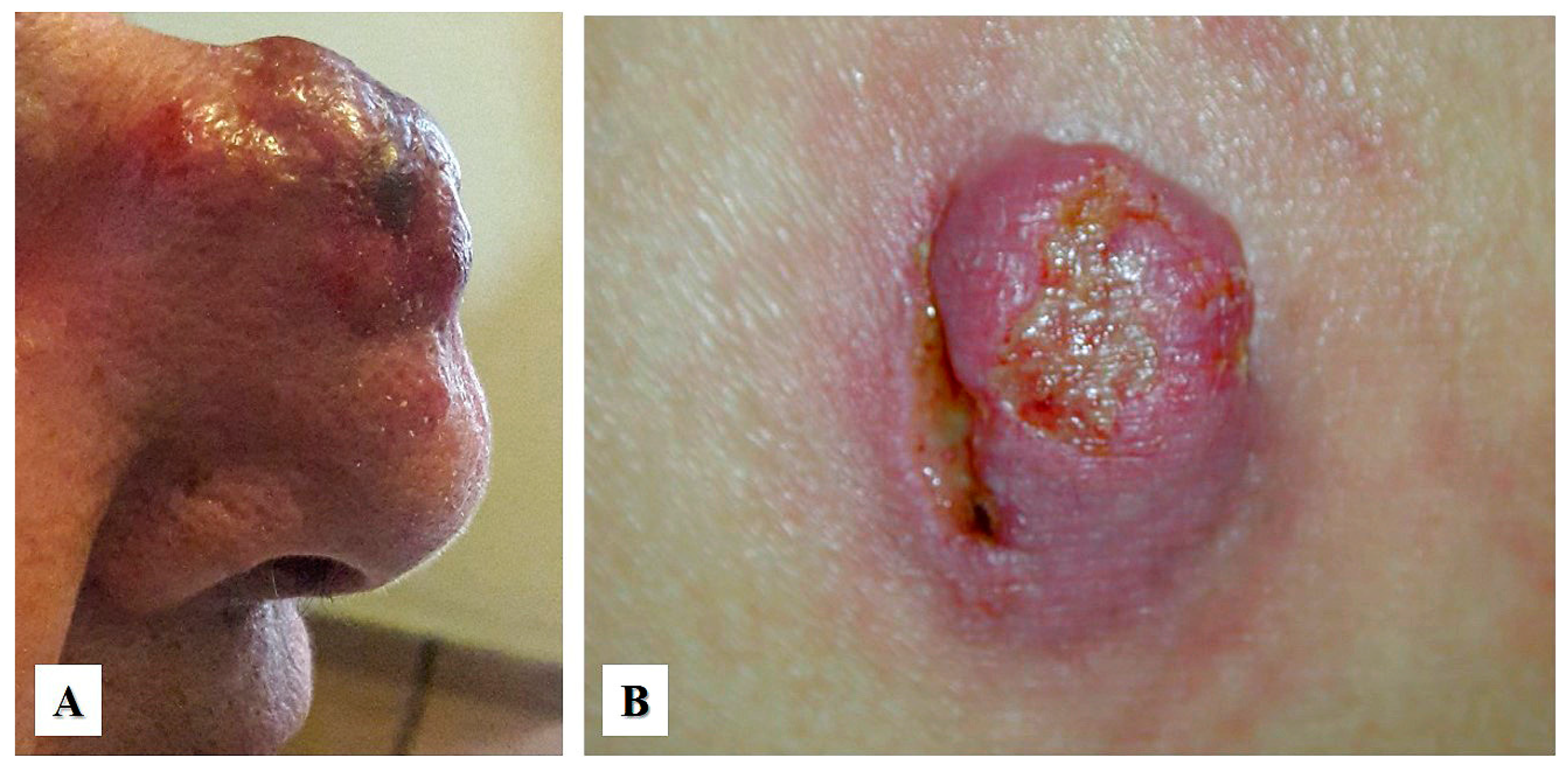
2. Clinical Presentation
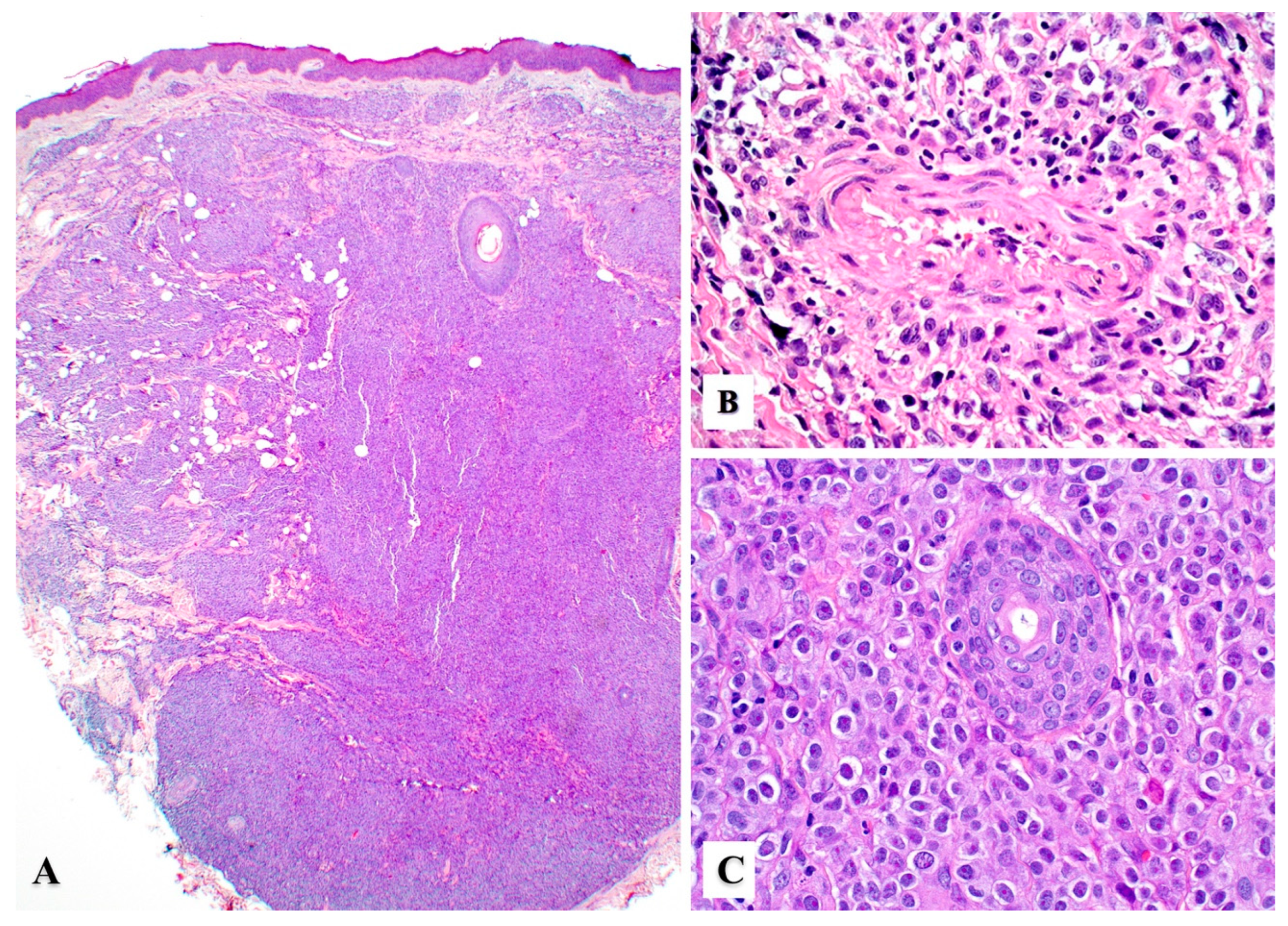
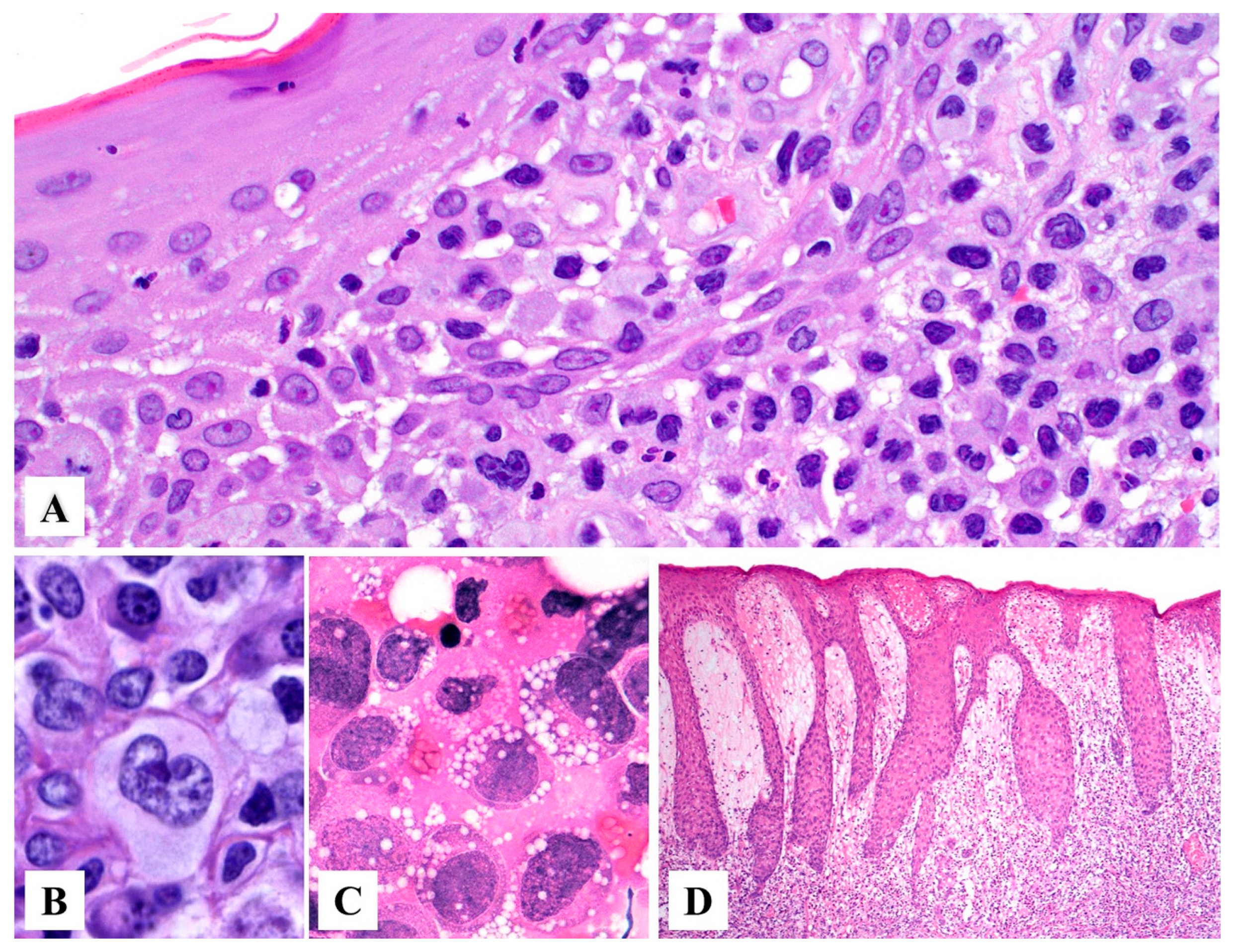
3. Histopathology
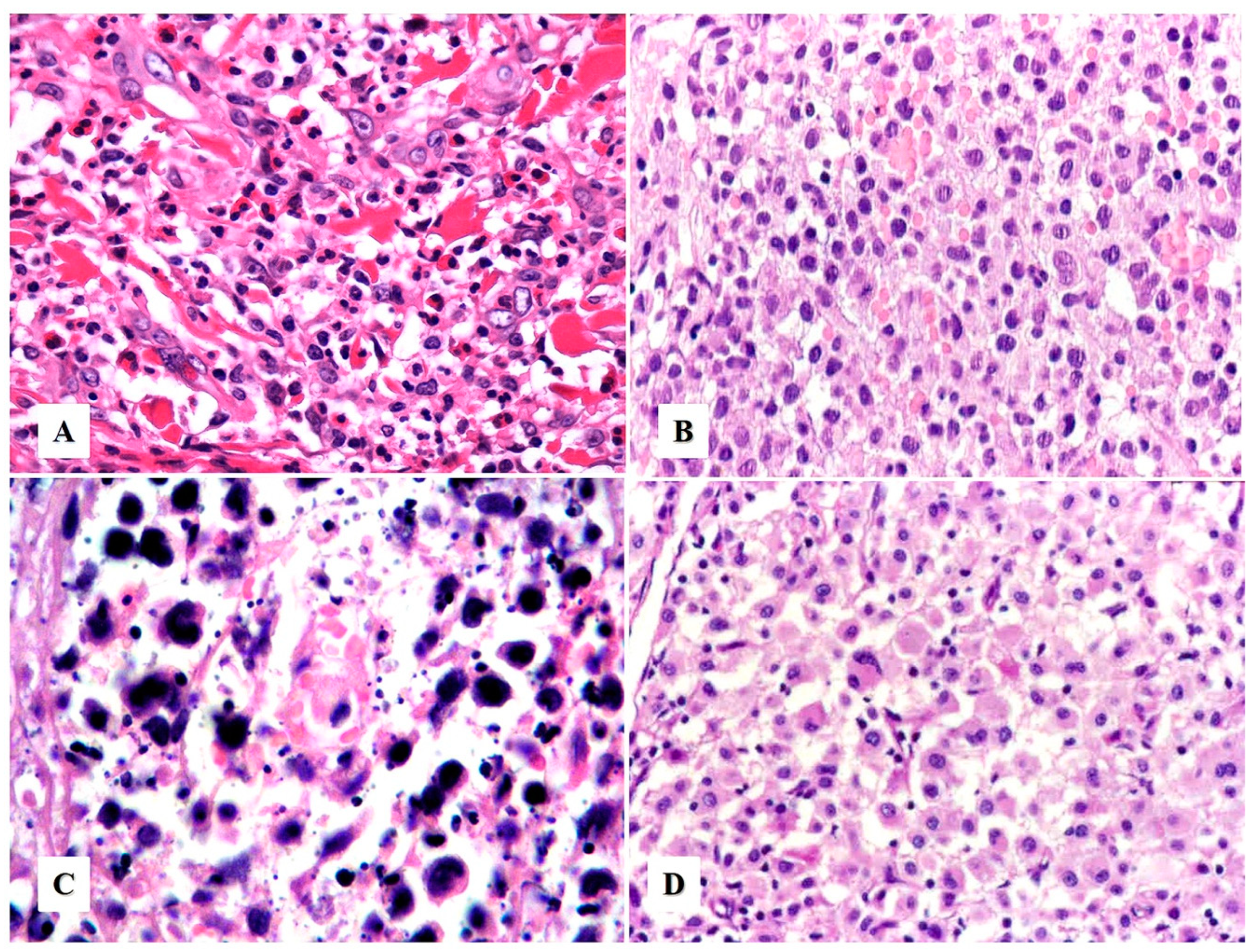
Morphologic Variants of Primary Cutaneous ALCL
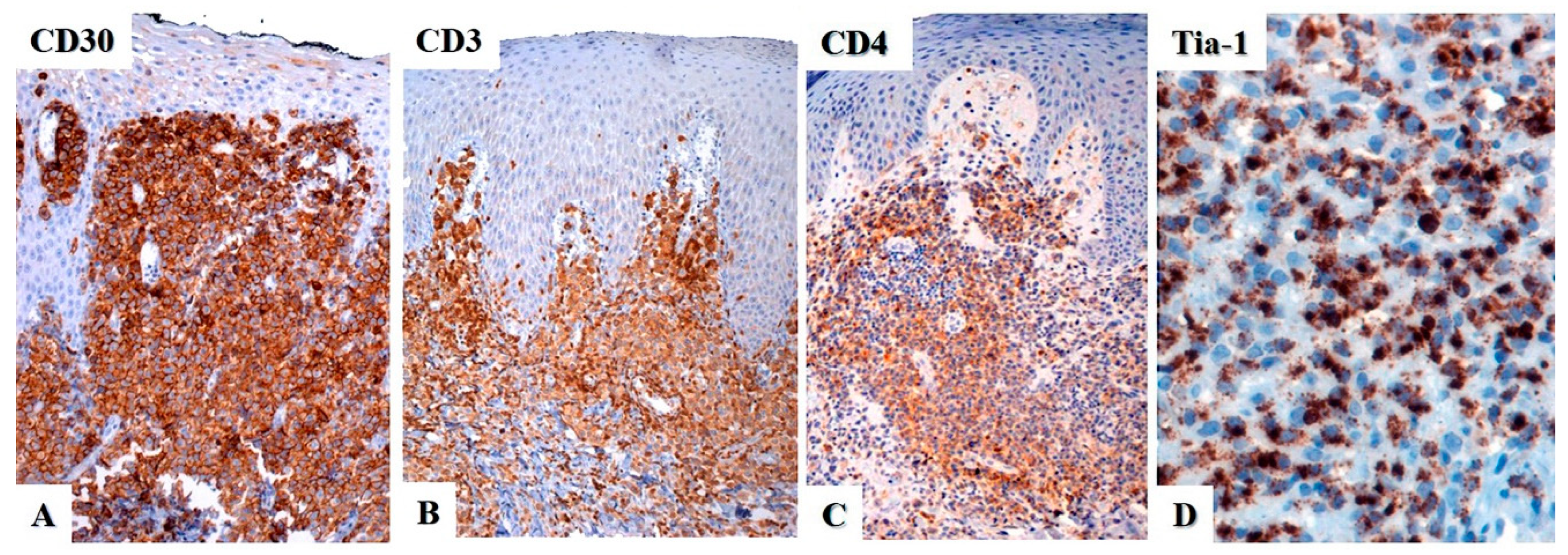
4. Immunohistochemistry
5. Differential Diagnosis
5.1. Differential Diagnosis with Other CD30+ T-Cell Lymphoproliferative Disorders
5.2. Differential Diagnosis with Entities Other Than CD30+ T-Cell Lymphoproliferative Disorders
6. Genetic and Molecular Features
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hague, C.; Farquharson, N.; Menasce, L.; Parry, E.; Cowan, R. Cutaneous T-cell lymphoma: Diagnosing subtypes and the challenges. Br. J. Hosp. Med. 2022, 83, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Irshaid, L.; Xu, M.L. ALCL by any other name: The many facets of anaplastic large cell lymphoma. Pathology 2020, 52, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Kempf, W.; Mitteldorf, C. Cutaneous T-cell lymphomas—An update 2021. Hematol. Oncol. 2021, 39 (Suppl. 1), 46–51. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Martin, J.M.; Wu, H.; Barta, S.K. CD30+ T-cell lymphoproliferative disorders. Chin. Clin. Oncol. 2019, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Paulli, M.; Kadin, M.E. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 392–396. [Google Scholar]
- Pina-Oviedo, S.; Ortiz-Hidalgo, C.; Carballo-Zarate, A.A.; Zarate-Osorno, A. ALK-Negative Anaplastic Large Cell Lymphoma: Current Concepts and Molecular Pathogenesis of a Heterogeneous Group of Large T-Cell Lymphomas. Cancers 2021, 13, 4667. [Google Scholar] [CrossRef]
- Kempf, W. Cutaneous CD30-Positive Lymphoproliferative Disorders. Surg. Pathol. Clin. 2014, 7, 203–228. [Google Scholar] [CrossRef]
- Berti, E.; Gianotti, R.; Alessi, E. Primary anaplastic large cell lymphoma of the skin. Dermatologica 1989, 178, 225–227. [Google Scholar] [CrossRef]
- Sarfraz, H.; Gentille, C.; Ensor, J.; Wang, L.; Wong, S.; Ketcham, M.S.; Joshi, J.; Pingali, S.R.K. Primary cutaneous anaplastic large-cell lymphoma: A review of the SEER database from 2005 to 2016. Clin. Exp. Dermatol. 2021, 46, 1420–1426. [Google Scholar] [CrossRef]
- Kumar, S.; Pittaluga, S.; Raffeld, M.; Guerrera, M.; Seibel, N.L.; Jaffe, E.S. Primary cutaneous CD30-positive anaplastic large cell lymphoma in childhood: Report of 4 cases and review of the literature. Pediatr. Dev. Pathol. 2005, 8, 52–60. [Google Scholar] [CrossRef]
- Seo, A.N.; Lee, S.J.; Choi, Y.H.; Chung, H.Y.; Huh, J.; Yoon, G.S. Congenital primary cutaneous anaplastic large-cell lymphoma: A case report. Am. J. Dermatopathol. 2015, 37, 398–400. [Google Scholar] [CrossRef] [PubMed]
- King, R.L.; Musiek, A.C.; Feldman, A.L. Primary cutaneous CD30+ T-cell lymphoproliferative disorders. In Hematopathology of the Skin. Clinical & Pathological Approach; Gru, A.A., Schaffer, A., Eds.; Wolters Kluwer Health: Philadelphia, PA, USA, 2017; pp. 141–158. [Google Scholar]
- Philippe, E.; Creech, K.T.; Cook, N.; Segura, J. Recurrent Primary Cutaneous Anaplastic Large Cell Lymphoma With Systemic Involvement: A Case Report and Literature Review. Cureus 2021, 13, e14284. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Fernandez-Pol, S.; Kim, J. Primary cutaneous anaplastic large cell lymphoma. J. Cutan. Pathol. 2017, 44, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Poligone, B.; Wilson, L.D.; Subtil, A.; Heald, P. Primary cutaneous T-cell lymphoma localized to the lower leg: A distinct, locally aggressive cutaneous T-cell lymphoma. Arch. Dermatol. 2009, 145, 677–682. [Google Scholar] [CrossRef][Green Version]
- Prieto-Torres, L.; Rodriguez-Pinilla, S.M.; Onaindia, A.; Ara, M.; Requena, L.; Piris, M. CD30-positive primary cutaneous lymphoproliferative disorders: Molecular alterations and targeted therapies. Haematologica 2019, 104, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Hruska, C.J.; Bertoli, R.J.; Young, Y.D.; Burkhart, P.H.; Googe, P.B. Primary cutaneous anaplastic large cell lymphoma in a patient receiving adalimumab. JAAD Case Rep. 2015, 1, 56–59. [Google Scholar] [CrossRef]
- Papathemeli, D.; Gräfe, R.; Hildebrandt, U.; Zettl, U.K.; Ulrich, J. Development of a primary cutaneous CD30(+) anaplastic large-cell T-cell lymphoma during treatment of multiple sclerosis with fingolimod. Mult. Scler. 2016, 22, 1888–1890. [Google Scholar] [CrossRef]
- Liu, H.L.; Hoppe, R.T.; Kohler, S.; Harvell, J.D.; Reddy, S.; Kim, Y.H. CD30+ cutaneous lymphoproliferative disorders: The Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J. Am. Acad. Dermatol. 2003, 49, 1049–1058. [Google Scholar] [CrossRef]
- Querfeld, C.; Khan, I.; Mahon, B.; Nelson, B.P.; Rosen, S.T.; Evens, A.M. Primary cutaneous and systemic anaplastic large cell lymphoma: Clinicopathologic aspects and therapeutic options. Oncology 2010, 24, 574–587. [Google Scholar]
- van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef]
- Zhang, X.R.; Chien, P.N.; Nam, S.Y.; Heo, C.Y. Anaplastic Large Cell Lymphoma: Molecular Pathogenesis and Treatment. Cancers 2022, 14, 1650. [Google Scholar] [CrossRef] [PubMed]
- Di Raimondo, C.; Parekh, V.; Song, J.Y.; Rosen, S.T.; Querfeld, C.; Zain, J.; Martinez, X.U.; Abdulla, F.R. Primary Cutaneous CD30+ Lymphoproliferative Disorders: A Comprehensive Review. Curr. Hematol. Malig. Rep. 2020, 15, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Benharroch, D.; Meguerian-Bedoyan, Z.; Lamant, L.; Amin, C.; Brugières, L.; Terrier-Lacombe, M.J.; Haralambieva, E.; Pulford, K.; Pileri, S.; Morris, S.W.; et al. ALK-positive lymphoma: A single disease with a broad spectrum of morphology. Blood 1998, 91, 2076–2084. [Google Scholar] [CrossRef]
- Pandiar, D.; Smitha, T. The “hallmark” cells. J. Oral. Maxillofac. Pathol. 2019, 23, 176–177. [Google Scholar] [CrossRef]
- Guitart, J.; Martinez-Escala, M.E.; Deonizio, J.M.; Gerami, P.; Kadin, M.E. CD30(+) cutaneous lymphoproliferative disorders with pseudocarcinomatous hyperplasia are associated with a T-helper-17 cytokine profile and infiltrating granulocytes. J. Am. Acad. Dermatol. 2015, 72, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Burg, G.; Kempf, W.; Kazakov, D.V.; Dummer, R.; Frosch, P.J.; Lange-Ionescu, S.; Nishikawa, T.; Kadin, M.E. Pyogenic lymphoma of the skin: A peculiar variant of primary cutaneous neutrophil-rich CD30+ anaplastic large-cell lymphoma. Clinicopathological study of four cases and review of the literature. Br. J. Dermatol. 2003, 148, 580–586. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Dai, B.; Kong, J.C.; Lu, H.F.; Shi, D.R. Neutrophil/eosinophil-rich type of primary cutaneous anaplastic large cell lymphoma: A clinicopathological, immunophenotypic and molecular study of nine cases. Histopathology 2009, 55, 189–196. [Google Scholar] [CrossRef]
- Magro, C.M.; Momtahen, S.; Kiuru, M. Primary Cutaneous Small Cell Variant of Anaplastic Large Cell Lymphoma: A Case Series and Review of the Literature. Am. J. Dermatopathol. 2017, 39, 877–889. [Google Scholar] [CrossRef]
- Kempf, W.; Kazakov, D.V.; Paredes, B.E.; Laeng, H.R.; Palmedo, G.; Kutzner, H. Primary cutaneous anaplastic large cell lymphoma with angioinvasive features and cytotoxic phenotype: A rare lymphoma variant within the spectrum of CD30+ lymphoproliferative disorders. Dermatology 2013, 227, 346–352. [Google Scholar] [CrossRef]
- Onaindia, A.; de Villambrosía, S.G.; Prieto-Torres, L.; Rodríguez-Pinilla, S.M.; Montes-Moreno, S.; González-Vela, C.; Piris, M.A. DUSP22-rearranged anaplastic lymphomas are characterized by specific morphological features and a lack of cytotoxic and JAK/STAT surrogate markers. Haematologica 2019, 104, e158–e162. [Google Scholar] [CrossRef]
- Xue, Y.N.; Wang, Z.; Sun, J.F.; Chen, H. Primary cutaneous anaplastic large-cell lymphoma with 6p25.3 rearrangement exhibits a biphasic histopathologic pattern: Two case reports and literature review. J. Cutan. Pathol. 2021, 48, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Ena, L.; Cota, C.; Cerroni, L. Intralymphatic Spread Is a Common Finding in Cutaneous CD30+ Lymphoproliferative Disorders. Am. J. Surg. Pathol. 2015, 39, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, R.A.; Bashey, S.; Wysong, A.; Kim, J.; Kim, Y.H.; Gratzinger, D. Intravascular ALK-negative anaplastic large cell lymphoma with localized cutaneous involvement and an indolent clinical course: Toward recognition of a distinct clinicopathologic entity. Am. J. Surg. Pathol. 2013, 37, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Resnik, K.S.; Kutzner, H. Of lymphocytes and cutaneous epithelium: Keratoacanthomatous hyperplasia in CD30+ lymphoproliferative disorders and CD30+ cells associated with keratoacanthoma. Am. J. Dermatopathol. 2010, 32, 314–315. [Google Scholar] [CrossRef]
- Misra, M.; Raghuvanshi, S.; Goel, M.M.; Verma, S.P. ALK-negative primary cutaneous T-cell anaplastic large cell lymphoma, myxoid variant; masquerading as sarcoma: Unveiling the diagnostic dilemma. BMJ Case Rep. 2021, 14, e239350. [Google Scholar] [CrossRef]
- Wang, J.; Sun, N.C.; Nozawa, Y.; Arber, D.A.; Chu, P.; Chang, K.L.; Weiss, L.M. Histological and immunohistochemical characterization of extranodal diffuse large-cell lymphomas with prominent spindle cell features. Histopathology 2001, 39, 476–481. [Google Scholar] [CrossRef]
- Kempf, W. A new era for cutaneous CD30-positive T-cell lymphoproliferative disorders. Semin. Diagn. Pathol. 2017, 34, 22–35. [Google Scholar] [CrossRef]
- Wechsler, J.; Ingen-Housz-Oro, S.; Deschamps, L.; Brunet-Possenti, F.; Deschamps, J.; Delfau, M.H.; Calderaro, J.; Ortonne, N. Prevalence of T-cell antigen losses in mycosis fungoides and CD30-positive cutaneous T-cell lymphoproliferations in a series of 153 patients. Pathology 2022, 54, 729–737. [Google Scholar] [CrossRef]
- Xing, X.; Feldman, A.L. Anaplastic large cell lymphomas: ALK positive, ALK negative, and primary cutaneous. Adv. Anat. Pathol. 2015, 22, 29–49. [Google Scholar] [CrossRef]
- Yu, B.H.; Zhang, Y.; Xue, T.; Shui, R.H.; Lu, H.F.; Zhou, X.Y.; Zhu, X.Z.; Li, X.Q. The clinicopathological relevance of uniform CD56 expression in anaplastic large cell lymphoma: A retrospective analysis of 18 cases. Diagn. Pathol. 2021, 16, 1. [Google Scholar] [CrossRef]
- Geller, S.; Canavan, T.N.; Pulitzer, M.; Moskowitz, A.J.; Myskowski, P.L. ALK-positive primary cutaneous anaplastic large cell lymphoma: A case report and review of the literature. Int. J. Dermatol. 2018, 57, 515–520. [Google Scholar] [CrossRef]
- Oschlies, I.; Lisfeld, J.; Lamant, L.; Nakazawa, A.; d’Amore, E.S.; Hansson, U.; Hebeda, K.; Simonitsch-Klupp, I.; Maldyk, J.; Müllauer, L.; et al. ALK-positive anaplastic large cell lymphoma limited to the skin: Clinical, histopathological and molecular analysis of 6 pediatric cases. A report from the ALCL99 study. Haematologica 2013, 98, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Pulitzer, M.; Ogunrinade, O.; Lin, O.; Steinherz, P. ALK-positive (2p23 rearranged) anaplastic large cell lymphoma with localization to the skin in a pediatric patient. J. Cutan. Pathol. 2015, 42, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.; Gu, J.; Aung, P.P.; Nagarajan, P.; Curry, J.L.; Huen, A.; Ivan, D.; Prieto, V.G.; Tetzlaff, M.T.; Duvic, M.; et al. Is immunohistochemical expression of GATA3 helpful in the differential diagnosis of transformed mycosis fungoides and primary cutaneous CD30-positive T cell lymphoproliferative disorders? Virchows. Arch. 2021, 479, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Mitteldorf, C.; Robson, A.; Tronnier, M.; Pfaltz, M.C.; Kempf, W. Galectin-3 Expression in Primary Cutaneous CD30-Positive Lymphoproliferative Disorders and Transformed Mycosis Fungoides. Dermatology 2015, 231, 164–170. [Google Scholar] [CrossRef]
- Olsen, S.H.; Ma, L.; Schnitzer, B.; Fullen, D.R. Clusterin expression in cutaneous CD30-positive lymphoproliferative disorders and their histologic simulants. J. Cutan. Pathol. 2009, 36, 302–307. [Google Scholar] [CrossRef]
- Magro, C.M.; Dyrsen, M.E. Cutaneous lymphocyte antigen expression in benign and neoplastic cutaneous B- and T-cell lymphoid infiltrates. J. Cutan. Pathol. 2008, 35, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Tomita, A.; Takata, K.; Shimoyama, Y.; Yoshino, T.; Tomita, Y.; Nakamura, S. Primary cutaneous CD30 positive T-cell lymphoproliferative disorders with aberrant expression of PAX5: Report of three cases. Pathol. Int. 2012, 62, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Karai, L.J.; Kadin, M.E.; Hsi, E.D.; Sluzevich, J.C.; Ketterling, R.P.; Knudson, R.A.; Feldman, A.L. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am. J. Surg. Pathol. 2013, 37, 1173–1181. [Google Scholar] [CrossRef]
- Mélard, P.; Idrissi, Y.; Andrique, L.; Poglio, S.; Prochazkova-Carlotti, M.; Berhouet, S.; Boucher, C.; Laharanne, E.; Chevret, E.; Pham-Ledard, A.; et al. Molecular alterations and tumor suppressive function of the DUSP22 (Dual Specificity Phosphatase 22) gene in peripheral T-cell lymphoma subtypes. Oncotarget 2016, 7, 68734–68748. [Google Scholar] [CrossRef]
- Feldman, A.L.; Dogan, A.; Smith, D.I.; Law, M.E.; Ansell, S.M.; Johnson, S.H.; Porcher, J.C.; Ozsan, N.; Wieben, E.D.; Eckloff, B.W.; et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 2011, 117, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Chen, Y.M.; Hung, W.T.; Li, J.P.; Chen, D.Y.; Lan, J.L.; Tan, T.H. Downregulation of the phosphatase JKAP/DUSP22 in T cells as a potential new biomarker of systemic lupus erythematosus nephritis. Oncotarget 2016, 7, 57593–57605. [Google Scholar] [CrossRef] [PubMed]
- Kao, E.Y.; Mukkamalla, S.K.R.; Lynch, D.T. ALK Negative Anaplastic Large Cell Lymphoma. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S. Genetic alterations in primary cutaneous CD30+ anaplastic large cell lymphoma. Genes Chromosomes Cancer 2003, 37, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Vitiello, P.; Ronchi, A.; Casale, B.; Calogero, A.; Sagnelli, E.; Costa Nachtigal, G.; Troiani, T.; Franco, R.; Argenziano, G.; et al. Primary Cutaneous Anaplastic Large Cell Lymphoma (pcALCL) in the Elderly and the Importance of Sport Activity Training. Int. J. Environ. Res. Public Health 2020, 17, 839. [Google Scholar] [CrossRef]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood 2014, 124, 3768–3771. [Google Scholar] [CrossRef]
- Macgrogan, G.; Vergier, B.; Dubus, P.; Beylot-Barry, M.; Belleannee, G.; Delaunay, M.M.; Eghbali, H.; Beylot, C.; Rivel, J.; Trojani, M.; et al. CD30-positive cutaneous large cell lymphomas. A comparative study of clinicopathologic and molecular features of 16 cases. Am. J. Clin. Pathol. 1996, 105, 440–450. [Google Scholar] [CrossRef]
- Schwarting, R.; Behling, E.; Allen, A.; Arguello-Guerra, V.; Budak-Alpdogan, T. CD30+ Lymphoproliferative Disorders as Potential Candidates for CD30-Targeted Therapies. Arch. Pathol. Lab. Med. 2022, 146, 415–432. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lymphomatoid papulosis, type C (diffuse large cell type)
|
ALK+ systemic anaplastic large cell lymphoma with skin involvement
|
ALK-negative systemic anaplastic large cell lymphoma with skin involvement
|
Mycosis fungoides with CD30+ large cell transformation
|
Classic Hodgkin lymphoma involving skin
|
Peripheral T lymphoma, not otherwise specified, involving skin
|
Subcutaneous panniculitis-like T-cell lymphoma
|
Primary cutaneous gamma-delta T-cell lymphoma
|
Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma
|
Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Hidalgo, C.; Pina-Oviedo, S. Primary Cutaneous Anaplastic Large Cell Lymphoma—A Review of Clinical, Morphological, Immunohistochemical, and Molecular Features. Cancers 2023, 15, 4098. https://doi.org/10.3390/cancers15164098
Ortiz-Hidalgo C, Pina-Oviedo S. Primary Cutaneous Anaplastic Large Cell Lymphoma—A Review of Clinical, Morphological, Immunohistochemical, and Molecular Features. Cancers. 2023; 15(16):4098. https://doi.org/10.3390/cancers15164098
Chicago/Turabian StyleOrtiz-Hidalgo, Carlos, and Sergio Pina-Oviedo. 2023. "Primary Cutaneous Anaplastic Large Cell Lymphoma—A Review of Clinical, Morphological, Immunohistochemical, and Molecular Features" Cancers 15, no. 16: 4098. https://doi.org/10.3390/cancers15164098
APA StyleOrtiz-Hidalgo, C., & Pina-Oviedo, S. (2023). Primary Cutaneous Anaplastic Large Cell Lymphoma—A Review of Clinical, Morphological, Immunohistochemical, and Molecular Features. Cancers, 15(16), 4098. https://doi.org/10.3390/cancers15164098