
Awakening of Dormant Breast Cancer Cells in the Bone Marrow


Simple Summary
Abstract
1. Introduction
2. Breast Cancer Metastasis and Dormancy in the Bone Marrow
2.1. Hematogenous Transit of Cancer Cells to the BM HSC Niches
2.2. The Metastatic BM Niches and DTC Dormancy Signaling

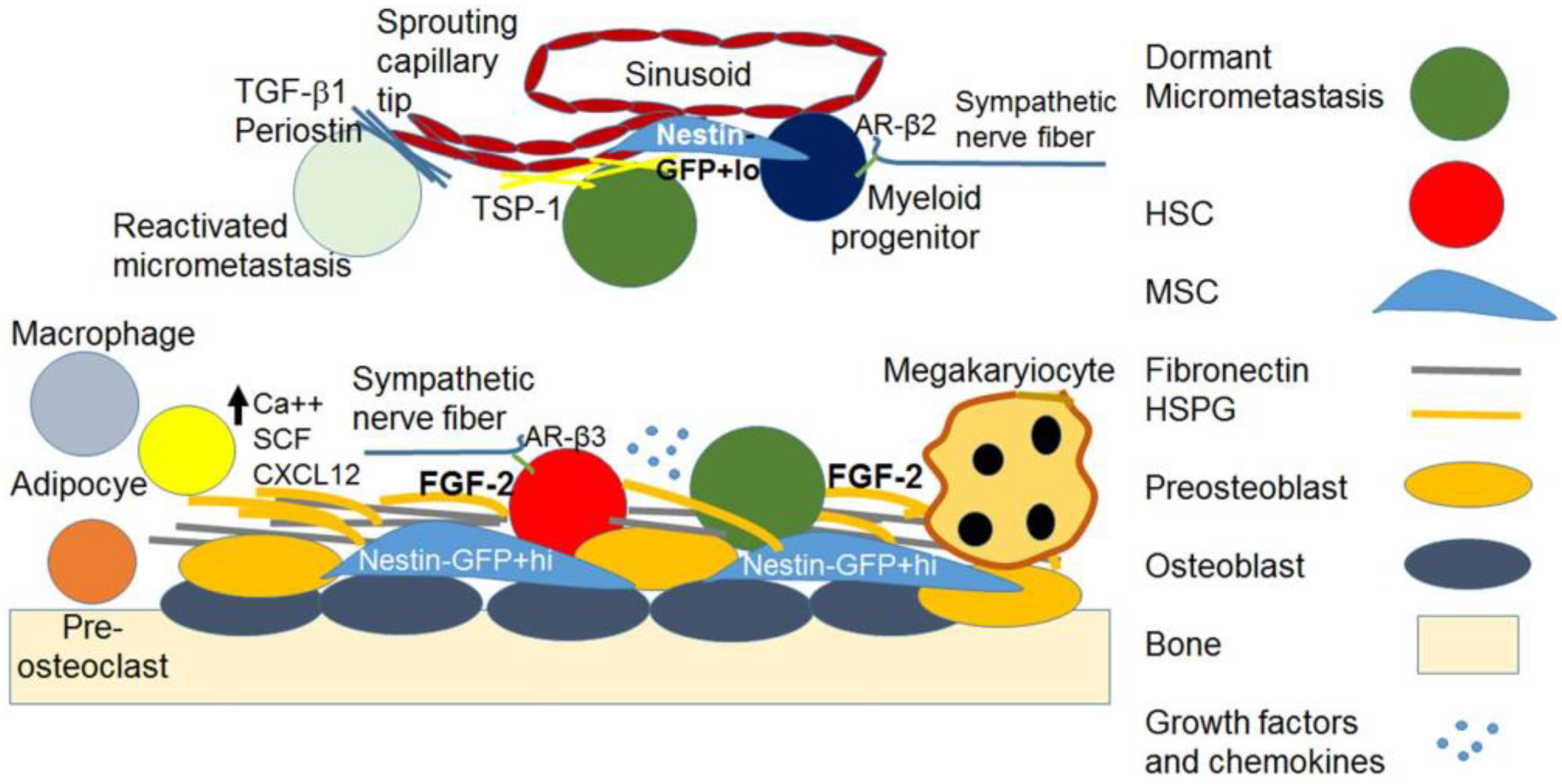{kind=link}
| Vehicle | Signaling | References | |
|---|---|---|---|
| Endosteal niche | MSCs | MERTK, AXL, TGFβR3, BMPR2, Alk5, NDRG1, ERK, p38, p21WAF1, p27Kip1, 15INK4b, PI3K, RhoA/GRAF, integrin α5β1, FGF-2, HSPG, fibronectin | [36,46,81,88,90,91,93,94,98,99,100,102,103,105,114,115,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136] |
| Inhibition of oncogene signaling | [92] | ||
| Non-canonical Wnt5a signaling, SIAH2, repression of β-catenin, LIF, RA | [94,142,143] | ||
| Hypoxia, acidic pH | LIF, STAT3, TGFβ, BMP signaling | [87,139,147] | |
| Redox signaling | [140] | ||
| Exosomes | miR-23b, -126, 127, -148a, -3p -197, -222, -223 | [94,95,125,126,127,141,153] | |
| Fusion with and cannibalizing MSCs | SDF-1a, decreased CXCL12 | [126,128,144,146] | |
| Microenvironmental remodeling | [129] | ||
| Preosteoblasts, SNO cells | Notch, Jagged1 | [48,149,150] | |
| Vascular niche | Endothelial cells | TSP1 | [46] |
| DARC, KAI1, p21Waf1, downregulated TBX2 | [154] | ||
| vWF, VCAM1, CXCL 1 and 2, BMP7, TGFβ-2, NFκB combined with ER in ER+ BC, NR2F1, ZFP281, PTEN | [8,91,92,93,155,156,157] | ||
| Prx1+ MSCs | CXCL12 | [158] | |
| Immune niche | CD4+ and CD8+ cells | INFγ | [46,166] |
| NK cells | DKK-1, inhibited canonical Wnt | [168] | |
| SDF-1/CXCR4 | Src, Akt, TRAIL, downregulated NK1R-Tr | [128,169] | |
| Cancer cell-intrinsic effects | TIE2 | p21Waf1, p27Kip1 | [170,171] |
| KAI1, MKK4/7, Nm23-H1 | [170] | ||
| IDO1 | mTOR | [172] | |
| Epigenetics | Repressive histones | altered p53 functions | [174,175,177] |
| downregulation of suppressor gene promoter methylation enzymes | |||
| downregulation of DNMT1 | silencing of a transcription network FOXM1, FOXD, FOXL EGR1/2/3, PPARγ, ELK1, Jun family upregulating p53, DEC2, NR2F1, RARβ | [78,176] | |
| NR2F1, RARβ | removal of acetyl groups from histone H3, HDACs | [20,93,178] | |
| NR2F1 | induced methylation of H3 residues H3K4, H3K9, H3K27, decreased expression of growth-promoting SOX9 | [176] | |
| processing alternative coding mRNA isoforms, non-coding RNAs, miRNAs, lnRNAs | [180,181] |
3. Reawakening of Dormant Cancer Cell
3.1. Clinical Cancer Variables Associated with Recurrence
3.2. Molecular Mechanisms Associated with Recurrence
3.2.1. Inflammation
| Mechanism | Vehicle and Function | Signaling | References | |
|---|---|---|---|---|
| Proliferative signaling | Dormant cell cycle activation | EGF, TGFβ1, integrins, adhesion molecules, periostin, stromal remodeling collagen I fibrotic niche ELEANORS | proliferative signaling chromatin regulation, upregulated CD44 | [42,46,121,129,222] [223] [195] |
| Inflammation | Type I inflammation | TNF-α, IFN-γ, IL-17, IL-6/sIL 6Rα CD4+ Th1 T-cells, M1-macrophages, ILC-1 | Dormancy if mIL-6 Rα signaling > sIL-6Rα signaling Dormancy if tumor inhibitory Th17 or ILC-3 cell signaling predominates IL-17 IL-22 promotes relapse | [209] |
| Type II anti-inflammatory classic signaling | IL-11, IL-22, IL-33, IL-6/mIL-6 Rα, CD4+ Th2 cells, IL-12, M2-macrophages, ILC-2 | Dormancy if mIL-6 Rα > sIL-6 Rα signaling Dormancy if tumor inhibitory Th17 or ILC-3 cell signaling predominates IL-17 IL-22 promotes relapse | [209] | |
| NETS | Panx1 ADRB3 VCAM-1, ICAM-1, E-selectin, IL-1β, IL-6, CXCL1 | Spermidine, immune escape, MMP9, cleaved laminin, activated integrins ROS Expanded MDSCs, cycle activates BC cells feed-forward inflammation α4β1-induced osteoclast activity | [210,212,213,214,215,216,219] | |
| Macrophages | VCAM-1 | NF-κB, TNFα, IL-1β, and IL-6 α4β1-induced osteoclast activity | [198,217,219] | |
| EGF | VCAM-1 | NF-κB α4β1-induced osteoclast activity | [218,219] | |
| COVID-19 infection | NETs, monocytes/macrophages | pro-inflammatory cytokines | [220] | |
| Stromal immune response | INFγ | activated stromal fibroblasts, blocked CXCL-12-NK cancer cells suppression | [221] |
3.2.2. Aging
3.2.3. Loss of FGF in Stroma
3.2.4. Increased Adipogenesis
3.2.5. Estrogen Deprivation
| Mechanism | Vehicle and Function | Signaling | References | |
|---|---|---|---|---|
| Aging | Estrogen deprivation | MSCs | angiopoietin-2, disrupted angiopoietin-1/Tie2 signaling, ER+ tumor cell survival via integrin β1 secreted IL-6, IL-8, activated TGFβ, TNFα signaling | [224,225,273] [121] |
| Shift to adipose differentiation | increased BM adipocytes, RANKL, decreased bone forming osteoblasts - adipocyte leptin - increased β-oxidized lipid uptake - oxidative stress - adipocyte extracellular vesicles | - switch in MSCs differentiation potential from osteogenic to adipogenic, - TGFβ/BMP, PPARγ2 signaling, - Ob-R, FABP4, JAK/STAT3, PI3K/Akt, ERK, Rho/ROCK, Notch, TNF-α, ERK, NF-kB, IL-1β, CREB, IL-6, resistin - ligand-independent ER and Her2 receptor activation - CD36 cysteine oxidation - P450 epoxygenase- induced epoxyeicosatrienoic acid synthesis - Promotes ER+ and ER− BC cell proliferation, motility and metastasis - Hif1α | [59,199,226,227,228,255,263,264,265,266,267,268,269] | |
| Decreased osteoprogenitor cell proliferative capacity | [229] | |||
MSC senescence | loss FOXP1 expression, HOXB7 declines | miR-196 upregulation | [230,231] | |
Increased fibroblast metabolism, lower oxidation | Increased ALOX12 | Increased ERK signaling, radiation resistance | [232] | |
Niche fibroblasts | Increased periostin | [222] | ||
Increased MSC N-cadherin | MSCs steers to adipogenic differentiation | decrease in pro-osteogenic Wnt5a and Wnt10b signaling - decreased AXL dormancy signaling, - enhanced MERTK tumor promoting signaling. - Altered balance of sFRP2 canonical protumorigenic Wnt/β-catenin antagonist and sFRP1 dormancy sustainer | [142,233,234,235,236,244,245] | |
inflammation | MSCs | IL-6, IL-8, Il-1b, Il-6, Il-27, Il-1f9, CCl4, Ccl5, Tnfsf14, Ltb, TGF-β1 signaling, SMAD2 and 3, - CD133+ cancer cell renewal - IL-6/Notch induced endocrine resistance through gp130/gp80, STAT3, VEGF, PI3K/Akt signaling, EGFR and ERK | [97,121,159,196,212,238,239,240,241,242,243] | |
Loss of stem cell maintenance | Pericytes, decline with aging | Decrease in Bmp4, Bmp6, Bmp7, Kit ligand, TGF-β2, Dkk-1, Dkk3, Thbs2 | [97] | |
| FGF-2 synthetic loss by MSCs | - Lose damage repair capacity, proliferation, EGF, FGF-2, HGF, IGF signaling, FGF-2 expression, - increase oxidative stress - lose FGF-1- and FGF-2-mediated inhibition of adipogenesis | - MSC senescence through lnRNA-p2 β-catenin signaling suppression - decreased MMP-13, TIMP1, MMP-2-mediated fibrillary fibronectin degradation - induced collagen turnover - PPARγ2 adipogenic signaling | [103,231,248,249,250] |
3.3. Stromal Injury
3.4. Hypercoagulable State
| Mechanism | Vehicle and Function | Signaling | References | |
|---|---|---|---|---|
| Stromal injury | ||||
| Petrochemicals | Elevated IL-8, decreased DNA repair | [274] | ||
| Diesel exhaust | - increased inflammatory cytokines - decreased M1 and M2 macrophage chemotaxis | [275] | ||
| Chemo-, bio- and radiation-therapy | Stromal fibroblasts | injury and secretory senescence | [121,276,277,278,279] | |
| - HDAC inhibitor | - secretory senescence - chromatin remodeling rather than physical breaks in DNA | ATM, NF-κB, IL-6 and IL-8 osteopontin activation | [280] | |
| DNA damage response | NF-κB activation, TIFA, damaged chromatin, NF-κB, IL-6, IL-8 | [277] | ||
| Dietary fat, IL-1 | Secretory senescence | [281] | ||
| Osteoblast senescence | P27Kip1 secretory senescence | IL-6, osteoclastogenesis | [283,284] | |
| Oxidative and hypoxic stress | - TGF-β, TNF-α, IL-6 - lipid transport receptors, lipid metabolism | [121,140] | ||
| Colonization, feed-forward stromal injury | Secretory senescence | IL-6, IL-8, TNF-α, secretormes, nutrients, metabolites, inflammatory cells | [121,286] | |
| Hypercoagulable state | ||||
| Thrombin | PAR-1 on cancer cells | Enhanced binding to fibronectin, platelets, vWF, endothelial cells | [288,289,290] | |
Downregulated, inhibited p27KIP1, induced Skp2, cyclins D and A, and miRNA-222 | [291] | |||
| Mitogenic to fibroblasts, endothelial cells, and smooth muscle cells | [292,293] | |||
| angiogenesis through VEGF | [294] | |||
| increased thrombin potential, and thrombin generation peak in high-risk BC | High recurrence potential | [295,296] | ||
| FVIII, D-dimer levels | predictive of overall and disease-free survival | [297] |
3.5. Surgery, Associated Angiogenesis, Inflammation and Catecholamines
3.5.1. Tumors Produce Metastasis-Promoting Factors That Are Eliminated with Tumor Removal
3.5.2. Surgery Induces Catecholamines and Inflammatory Factors That May Promote the Growth of Dormant Micrometastases
3.6. Stress, Neuradrenergic Stimuli and Depression
| Mechanism | Vehicle and Function | Signaling | References | |
|---|---|---|---|---|
| Angiogenesis | ||||
| Endothelial cell stimulation | Endothelial cell tips | TGF-β1, periostin, Gli-1, Wnt | [159,160,162,311] | |
| Translocation of dormant BC cells from endosteal to the endothelial niche | L1CAM, YAP, MRTF, integrin β1, L1CAM | [307] | ||
| Intermittent hypoxia | Endothelial sprouting | Hif-1 and -2, angiogenic factors | [308] | |
| Surgery | Primary tumors (or local recurrences) secrete metastasis-stimulating factors | IL-6, IL-8, VEGF, EGF, PDGF-AA, MIF, SerpinE1, and M-CSF | Disseminated BC cell growth signals | [315] |
| Surgically induced inflammatory responses | - eliminate dendritic cells - eliminate activation of cytotoxic T-cells tumor-directed responsible - macrophage, NK cell, monocytes dysfunction | eliminate immune suppression contributing to dormancy | [204,319,320] | |
| Surgically-induced stress | - Generation of catecholamines, β-adrenergic agonists, prostaglandins - EMT | - Elevated perioperative IL-6, IL-8, NF-kB, CRP - reduced IRF1 - promotes growth of micrometastases - increases GATA-1 GATA-2, EGR3, STAT3 activity - increased tumor-infiltrating monocytes - decreased tumor-infiltrating B cells - perioperative decline in stimulated IL-12 - perioperative decline in IFNγ, mobilization of CD16− monocytes - decreased expression of CD11a on circulating NK cells | [321,322,325,326] | |
| Stress, noradrenergic stimuli, depression | stressful events, sympathetic signaling, catecholamines | - systemic glucocorticoids and catecholamines - inflammatory response - direct activation of sympathetic nerve fiber signaling around osteoblasts and osteocytes the stem cell niche - suppression of anoikis and apoptosis - angiogenesis, stromal adhesion molecule expression - stromal protein remodeling | - downregulated inhibitory receptors on microglia - inflammation mediated by monocytes and macrophages - differentiation, maturation, proliferation of stromal cells, macrophages, Thy1/2+ cells - enhanced IL-33 expression by dendritic cells upon lipopolysaccharide stimulation mediated by - AR-β2, PKA, cAMP - promote dormant cell reactivation - activation of BCL-2, BCL-xL, MCL, pFAK, AR-β2 signaling - suppression of BAD - downregulates GAS6 in osteoblasts - ATF1, RAR, E2F | [206,333,334,335,336,339,345] |
| Chronic psychological stress | - immunosuppressive functions | accumulation of MDSCs - suppressed inflammation - accumulation of CD11b+Gr1+ Ly6G+Ly6Clow immature neutrophils - COX-2- PGE-2 - inhibits macrophages cytokine release Inhibits T-cell responsiveness - tumor promotion | [214,337,338] | |
| Depression and anxiety | circulating 5-HT bind BC cell receptors | - 5-HT Uptake by platelets and neurons - PTHRP production by BC via RUNX2 - inhibits osteoid maturation - activates osteoclasts through RANKL | [207,346] |
4. Potential Therapeutic Approaches
5. Conclusions
Funding
Conflicts of Interest
References
- American Cancer Society Facts and Figures. 2021. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2021.html (accessed on 6 June 2022).
- Braun, S.; Pantel, K.; Muller, P.; Janni, W.; Hepp, F.; Kentenich, C.R.; Gastroph, S.; Wischnik, A.; Dimpfl, T.; Kindermann, G.; et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N. Engl. J. Med. 2000, 342, 525–533. [Google Scholar] [CrossRef]
- Hartkopf, A.D.; Brucker, S.Y.; Taran, F.A.; Harbeck, N.; von Au, A.; Naume, B.; Pierga, J.Y.; Hoffmann, O.; Beckmann, M.W.; Rydén, L.; et al. Disseminated tumour cells from the bone marrow of early breast cancer patients: Results from an international pooled analysis. Eur. J. Cancer 2021, 154, 128–137. [Google Scholar] [CrossRef]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, T.; Liu, A.Y.; Ouyang, G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget 2015, 6, 39550–39563. [Google Scholar] [CrossRef]
- Braun, S.; Kentenich, C.; Janni, W.; Hepp, F.; de Waal, J.; Willegroth, F.; Sommer, H.; Pantel, K. Lack of an effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high risk breast cancer patients. J. Clin. Onc. 2000, 18, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Naumov, G.N.; Townson, J.L.; MacDonald, I.C.; Wilson, S.M.; Bramwell, V.H.; Groom, A.C.; Chambers, A.F. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res. Treat. 2003, 82, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Chen, X.; Cowley, N.; Ottewell, P.D.; Hawkins, R.J.; Hunter, K.D.; Hobbs, J.K.; Brown, N.J.; Holen, I. Osteoblast-derived paracrine and juxtacrine signals protect disseminated breast cancer cells from stress. Cancers 2021, 13, 1366. [Google Scholar] [CrossRef]
- Coombes, R.C.; Powles, T.J.; Abbott, M.; De Rivas, L.; Ford, H.T.; McCready, V.R.; Neville, A.M.; Gazet, J.C. Physical test for distant metastases in patients with breast cancer. J. R. Soc. Med. 1980, 73, 617–623. [Google Scholar] [CrossRef]
- Dearnaley, D.P.; Sloane, J.P.; Ormerod, M.G.; Steele, K.; Coombes, R.C.; Clink, H.M.; Powles, T.J.; Ford, H.T.; Gazet, J.C.; Neville, A.M. Increased detection of mammary carcinoma cells in marrow smears using antisera to epithelial membrane antigen. Br. J. Cancer 1981, 44, 85–90. [Google Scholar] [CrossRef]
- Redding, W.H.; Coombes, R.C.; Monaghan, P.; Clink, H.M.; Imrie, S.F.; Dearnaley, D.P.; Ormerod, M.G.; Sloane, J.P.; Gazet, J.C.; Powles, T.J.; et al. Detection of micrometastases in patients with primary breast cancer. Lancet 1983, 2, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Mansi, J.L.; Berger, U.; Easton, D.; McDonnell, T.; Redding, W.H.; Gazet, J.C.; McKinna, A.; Powles, T.J.; Coombes, R.C. Micrometastases in bone marrow in patients with primary breast cancer: Evaluation as an early predictor of bone metastases. Br. Med. J. Clin. Res. Ed. 1987, 295, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Mansi, J.L.; Easton, D.; Berger, U.; Gazet, J.C.; Ford, H.T.; Dearnaley, D.; Coombes, R.C. Bone marrow micrometastases in primary breast cancer: Prognostic significance after 6 years’ follow-up. Eur. J. Cancer 1991, 27, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Mansi, J.L.; Gogas, H.; Bliss, J.M.; Gazet, J.C.; Berger, U.; Coombes, R.C. Outcome of primary-breast-cancer patients with micrometastases: A long-term follow-up study. Lancet 1999, 354, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Jatoi, I.; Anderson, W.F.; Jeong, J.H.; Redmond, C.K. Breast cancer adjuvant therapy: Time to consider its time-dependent effects. J. Clin. Oncol. 2011, 29, 2301–2304. [Google Scholar] [CrossRef]
- Naume, B.; Synnestvedt, M.; Falk, R.S.; Wiedswang, G.; Weyde, K.; Risberg, T.; Kersten, C.; Mjaaland, I.; Vindi, L.; Sommer, H.H.; et al. Clinical outcome with correlation to disseminated tumor cell (DTC) status after DTC-guided secondary adjuvant treatment with docetaxel in early breast cancer. J. Clin. Oncol. 2014, 32, 3848–3857. [Google Scholar] [CrossRef]
- Borgen, E.; Rypdal, M.C.; Sosa, M.S.; Renolen, A.; Schlichting, E.; Lonning, P.E.; Synnestvedt, M.; Aguirre-Ghiso, J.A.; Naume, B. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. 2018, 20, 120. [Google Scholar] [CrossRef]
- Klein, C.A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 2020, 20, 681–694. [Google Scholar] [CrossRef]
- Bushnell, G.G.; Deshmukh, A.P.; den Hollander, P.; Luo, M.; Soundararajan, R.; Jia, D.; Levine, H.; Mani, S.A.; Wicha, M.S. Breast cancer dormancy: Need for clinically relevant models to address current gaps in knowledge. NPJ Breast Cancer. 2021, 7, 66. [Google Scholar] [CrossRef]
- Suva, L.J.; Washam, C.; Nicholas, R.W.; Griffin, R.J. Bone metastasis: Mechanisms and therapeutic opportunities. Nat. Rev. Endocrinol. 2011, 7, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.; Whiting-Theobald, N.; Kawai, T.; Linton, G.F.; Rudikoff, A.G.; Choi, U.; Ryser, M.F.; Murphy, P.M.; Sechler, J.M.; Malech, H.L. CXCR4-transgene expression significantly improves marrow engraftment of cultured hematopoietic stem cells. Stem Cells 2004, 22, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Sun, S.; Guo, Z.; Xiao, X.; Liu, B.; Liu, X.; Tang, P.H.; Mao, N. Isolation of mouse marrow mesenchymal progenitors by a novel and reliable method. Stem Cells. 2003, 21, 527–535. [Google Scholar] [CrossRef]
- Wang, J.; Loberg, R.; Taichman, R.S. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006, 25, 573–587. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem. 2008, 105, 370–380. [Google Scholar] [CrossRef]
- Jung, Y.; Wang, J.; Song, J.; Shiozawa, Y.; Wang, J.; Havens, A.; Wang, Z.; Sun, Y.-X.; Emerson, S.G.; Krebsbach, P.H.; et al. Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood 2007, 110, 82–90. [Google Scholar] [CrossRef][Green Version]
- Jung, Y.; Shiozawa, Y.; Wang, J.; Patel, L.R.; Havens, A.M.; Song, J.; Krebsbach, P.H.; Roodman, G.D.; Taichman, R.S. Annexin-2 is a regulator of stromal cell-derived factor-1/cxcl12 function in the hematopoietic stem cell endosteal niche. Exp. Hematol. 2011, 39, 151–166.e1. [Google Scholar] [CrossRef]
- Jung, Y.; Wang, J.; Lee, E.; McGee, S.; Berry, J.E.; Yumoto, K.; Dai, J.; Keller, E.T.; Shiozawa, Y.; Taichman, R.S. Annexin 2-cxcl12 interactions regulate metastatic cell targeting and growth in the bone marrow. Mol. Cancer Res. MCR 2015, 13, 197–207. [Google Scholar] [CrossRef]
- Chu, K.; Cheng, C.J.; Ye, X.; Lee, Y.C.; Zurita, A.J.; Chen, D.T.; Yu-Lee, L.Y.; Zhang, S.; Yeh, E.T.; Hu, M.C.; et al. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol. Cancer Res. 2008, 6, 1259–1267. [Google Scholar] [CrossRef]
- Tamura, D.; Hiraga, T.; Myoui, A.; Yoshikawa, H.; Yoneda, T. Cadherin-11-mediated Interactions with Bone Marrow Stromal/Osteoblastic Cells Support Selective Colonization of Breast Cancer Cells in Bone. Int. J. Oncol. 2008, 33, 17–24. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Lu, J.T.; Tan, C.C.; Wang, Q.S.; Feng, Y.M. RUNX2 promotes breast cancer bone metastasis by increasing integrin alpha5-mediated colonization. Cancer Lett. 2016, 380, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. Gas6/axl axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Wieder, R. Stromal Co-Cultivation for Modeling Breast Cancer Dormancy in the Bone Marrow. Cancers 2022, 14, 3344. [Google Scholar] [CrossRef]
- Beerman, I.; Luis, T.C.; Singbrant, S.; Lo Celso, C.; Mendez-Ferrer, S. The evolving view of the hematopoietic stem cell niche. Exp. Hematol. 2017, 50, 22–26. [Google Scholar] [CrossRef]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef]
- Lo Celso, C.; Fleming, H.E.; Wu, J.W.; Zhao, C.X.; Miake-Lye, S.; Fujisaki, J.; Cote, D.; Rowe, D.W.; Lin, C.P.; Scadden, D.T. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 2009, 457, 92–96. [Google Scholar] [CrossRef]
- Ho, Y.H.; Mendez-Ferrer, S. Microenvironmental contributions to hematopoietic stem cell aging. Haematologica 2020, 105, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Kent, D.G.; Copley, M.R.; Benz, C.; Wohrer, S.; Dykstra, B.J.; Ma, E.; Cheyne, J.; Zhao, Y.; Bowie, M.B.; Zhao, Y.; et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood 2009, 113, 6342–6350. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Sanjuan-Pla, A.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 11075. [Google Scholar] [CrossRef] [PubMed]
- Carrelha, J.; Meng, Y.; Kettyle, L.M.; Luis, T.C.; Norfo, R.; Alcolea, V.; Boukarabila, H.; Grasso, F.; Gambardella, A.; Grover, A.; et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 2018, 554, 106–111. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Li, Z.; Li, L. Understanding hematopoietic stem-cell microenvironments. Trends Biochem. Sci. 2006, 31, 589–595. [Google Scholar] [CrossRef]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef]
- Chitteti, B.R.; Cheng, Y.H.; Kacena, M.A.; Srour, E.F. Hierarchical organization of osteoblasts reveals the significant role of CD166 in hematopoietic stem cell maintenance and function. Bone 2013, 54, 58–67. [Google Scholar] [CrossRef]
- Hooker, R.A.; Chitteti, B.R.; Egan, P.H.; Cheng, Y.H.; Himes, E.R.; Meijome, T.; Srour, E.F.; Fuchs, R.K.; Kacena, M.A. Activated leukocyte cell adhesion molecule (ALCAM or CD166) modulates bone phenotype and hematopoiesis. J. Musculoskelet. Neuronal Interact. 2015, 15, 83–94. [Google Scholar]
- Panaroni, C.; Fulzele, K.; Saini, V.; Chubb, R.; Pajevic, P.D.; Wu, J.Y. PTH Signaling in Osteoprogenitors Is Essential for B-Lymphocyte Differentiation and Mobilization. J. Bone Miner. Res. 2015, 30, 2273–2286. [Google Scholar] [CrossRef]
- He, Q.; Scott Swindle, C.; Wan, C.; Flynn, R.J.; Oster, R.A.; Chen, D.; Zhang, F.; Shu, Y.; Klug, C.A. Enhanced Hematopoietic Stem Cell Self-Renewal-Promoting Ability of Clonal Primary Mesenchymal Stromal/Stem cells Versus Their Osteogenic Progeny. Stem Cells 2017, 35, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, L.; Liu, J.; Goldstein, A.; Bado, I.; Zhang, W.; Arenkiel, B.R.; Li, Z.; Yang, M.; Du, S.; et al. The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell 2018, 34, 823–839.e7. [Google Scholar] [CrossRef]
- Khurana, S.; Schouteden, S.; Manesia, J.K.; Santamaria-Martinez, A.; Huelsken, J.; Lacy-Hulbert, A.; Verfaillie, C.M. Outside-in integrin signalling regulates haematopoietic stem cell function via Periostin-Itgav axis. Nat. Commun. 2016, 7, 13500. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, Y.; Katayama, Y.; Asada, N.; Minagawa, K.; Sato, M.; Okamura, A.; Shimoyama, M.; Nakagawa, K.; Okano, T.; Tanimoto, M.; et al. Role for vitamin D receptor in the neuronal control of the hematopoietic stem cell niche. Blood 2010, 116, 5528–5535. [Google Scholar] [CrossRef]
- Yeh, S.A.; Hou, J.; Wu, J.W.; Yu, S.; Zhang, Y.; Belfield, K.D.; Camargo, F.D.; Lin, C.P. Quantification of bone marrow interstitial pH and calcium concentration by intravital ratiometric imaging. Nat. Commun. 2022, 13, 393. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Kim, M.; Kim, C.; Choi, Y.S.; Kim, M.; Park, C.; Suh, Y. Age-related alterations in mesenchymal stem cells related to shift in differentiation from osteogenic to adipogenic potential: Implication to age-associated bone diseases and defects. Mech. Ageing Dev. 2012, 133, 215–225. [Google Scholar] [CrossRef]
- Lai, P.; Song, Q.; Yang, C.; Li, Z.; Liu, S.; Liu, B.; Li, M.; Deng, H.; Cai, D.; Jin, D.; et al. Loss of Rictor with aging in osteoblasts promotes age-related bone loss. Cell Death Dis. 2016, 7, e2408. [Google Scholar] [CrossRef]
- Sui, B.; Hu, C.; Liao, L.; Chen, Y.; Zhang, X.; Fu, X.; Zheng, C.; Li, M.; Wu, L.; Zhao, X.; et al. Mesenchymal progenitors in osteopenias of diverse pathologies: Differential characteristics in the common shift from osteoblastogenesis to adipogenesis. Sci. Rep. 2016, 6, 30186. [Google Scholar] [CrossRef]
- Nehlin, J.O.; Jafari, A.; Tencerova, M.; Kassem, M. Aging and lineage allocation changes of bone marrow skeletal (stromal) stem cells. Bone 2019, 123, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Trotter, T.N.; Gibson, J.T.; Sherpa, T.L.; Gowda, P.S.; Peker, D.; Yang, Y. Adipocyte-Lineage Cells Support Growth and Dissemination of Multiple Myeloma in Bone. Am. J. Pathol. 2016, 186, 3054–3063. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef]
- Bowers, M.; Zhang, B.; Ho, Y.; Agarwal, P.; Chen, C.C.; Bhatia, R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 2015, 125, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Krevvata, M.; Silva, B.C.; Manavalan, J.S.; Galan-Diez, M.; Kode, A.; Matthews, B.G.; Park, D.; Zhang, C.A.; Galili, N.; Nickolas, T.L.; et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood 2014, 124, 2834–2846. [Google Scholar] [CrossRef]
- Gabbianelli, M.; Sargiacomo, M.; Pelosi, E.; Testa, U.; Isacchi, G.; Peschle, C. “Pure” human hematopoietic progenitors: Permissive action of basic fibroblast growth factor. Science 1990, 249, 1561–1564. [Google Scholar] [CrossRef]
- Yeoh, J.S.; van Os, R.; Weersing, E.; Ausema, A.; Dontje, B.; Vellenga, E.; de Haan, G. Fibroblast growth factor-1 and -2 preserve long-term repopulating ability of hematopoietic stem cells in serum-free cultures. Stem Cells 2006, 24, 1564–1572. [Google Scholar] [CrossRef]
- Itkin, T.; Kaufmann, K.B.; Gur-Cohen, S.; Ludin, A.; Lapidot, T. Fibroblast growth factor signaling promotes physiological bone remodeling and stem cell self-renewal. Curr. Opin. Hematol. 2013, 20, 237–244. [Google Scholar] [CrossRef]
- Abdallah, B.M.; Alzahrani, A.M.; Abdel-Moneim, A.M.; Ditzel, N.; Kassem, M. A simple and reliable protocol for long-term culture of murine bone marrow stromal (mesenchymal) stem cells that retained their in vitro and in vivo stemness in long-term culture. Biol. Proced. Online 2019, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Gabrilove, J.L.; Wong, G.; Bollenbacher, E.; White, K.; Kojima, S.; Wilson, E.L. Basic fibroblast growth factor counteracts the suppressive effect of transforming growth factor-beta 1 on human myeloid progenitor cells. Blood 1993, 81, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Gabrilove, J.L.; White, K.; Rahman, Z.; Wilson, E.L. Stem cell factor and basic fibroblast growth factor are synergistic in augmenting committed myeloid progenitor cell growth. Blood 1994, 83, 907–910. [Google Scholar] [CrossRef]
- Buono, M.; Visigalli, I.; Bergamasco, R.; Biffi, A.; Cosma, M.P. Sulfatase modifying factor 1-mediated fibroblast growth factor signaling primes hematopoietic multilineage development. J. Exp. Med. 2010, 207, 1647–1660. [Google Scholar] [CrossRef]
- Wilson, E.L.; Rifkin, D.B.; Kelly, F.; Hannocks, M.J.; Gabrilove, J.L. Basic fibroblast growth factor stimulates myelopoiesis in long-term human bone marrow cultures. Blood 1991, 77, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Weidenfeld, K.; Schif-Zuck, S.; Abu-Tayeh, H.; Kang, K.; Kessler, O.; Weissmann, M.; Neufeld, G.; Barkan, D. Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget 2016, 7, 71362–71377. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.P.; George, A.; Schewe, D.; Bragado, P.; Iglesias, B.V.; Ranganathan, A.C.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Computational identification of a p38sapk-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009, 69, 5664–5672. [Google Scholar] [CrossRef]
- Kim, R.S.; Avivar-Valderas, A.; Estrada, Y.; Bragado, P.; Sosa, M.S.; Aguirre-Ghiso, J.A.; Segall, J.E. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 2012, 7, e35569. [Google Scholar] [CrossRef]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nature Rev. Mo. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef]
- Wieder, R. Fibroblasts as Turned Agents in Cancer Progression. Cancers 2023, 15, 2014. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Liu, X.; Cheng, K.; Sheng, J.; Kong, J.; Liu, T. Pre-metastatic niche formation in different organs induced by tumor extracellular vesicles. Front. Cell Dev. Biol. 2021, 9, 733627. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wen, A.; Lin, J. Pro-inflammatory cytokines in the formation of the pre-metastatic niche. Cancers 2020, 12, 3752. [Google Scholar] [CrossRef] [PubMed]
- Sanmartin, M.C.; Borzone, F.R.; Giorello, M.B.; Pacienza, N.; Yannarelli, G.; Chasseing, N.A. Bone marrow/bone pre-metastatic niche for breast cancer cells colonization: The role of mesenchymal stromal cells. Crit. Rev. Oncol. Hematol. 2021, 164, 103416. [Google Scholar] [CrossRef]
- Bakhshandeh, S.; Werner, C.; Fratzl, P.; Cipitria, A. Microenvironment-mediated cancer dormancy: Insights from metastability theory. Proc. Natl. Acad. Sci. USA 2022, 119, 46118. [Google Scholar] [CrossRef]
- Johnson, R.W.; Sowder, M.E.; Giaccia, A.J. Hypoxia and Bone Metastatic Disease. Curr. Osteoporos. Rep. 2017, 15, 231–238. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Fang, C.; Kang, Y. Cellular Plasticity in Bone Metastasis. Bone 2020, 158, 115693. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef]
- White, A.C.; Khuu, J.K.; Dang, C.Y.; Hu, J.; Tran, K.V.; Liu, A.; Gomez, S.; Zhang, Z.; Yi, R.; Scumpia, P.; et al. Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat. Cell Biol. 2014, 16, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef]
- Risson, E.; Nobre, A.R.; Maguer-Satta, V.; Aguirre-Ghiso, J.A. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer 2020, 1, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Nobre, A.R.; Risson, E.; Singh, D.K.; di Martino, J.; Cheung, J.F.; Wang, J.; Johnson, J.; Russnes, H.G.; Bravo-Cordero, J.J.; Birbrair, A.; et al. Bone Marrow NG2+/Nestin+ mesenchymal stem cells drive DTC dormancy via TGFβ2. Nat. Cancer 2021, 2, 327–339. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, M.; Tang, Y.; Liang, X. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. Onco Targets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Veeriah, V.; Xi, P.; Labella, R.; Chen, J.; Romeo, S.G.; Ramasamy, S.K.; Kusumbe, A.P. Angiocrine signals regulate quiescence and therapy resistance in bone metastasis. JCI Insight 2019, 4, 125679. [Google Scholar] [CrossRef]
- Korah, R.; Boots, M.; Wieder, R. Integrin α5β1 promotes survival of growth-arrested breast cancer cells: An in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res. 2004, 64, 4514–4522. [Google Scholar] [CrossRef]
- Brunner, G.; Nguyen, H.; Gabrilove, J.; Rifkin, D.B.; Wilson, E.L. Basic fibroblast growth factor expression in human bone marrow and peripheral blood cells. Blood 1993, 81, 631–638. [Google Scholar] [CrossRef]
- Hassan, N.; Greve, B.; Espinoza-Sanchez, N.A.; Gotte, M. Cell-surface heparan sulfate proteoglycans as multifunctional integrators of signaling in cancer. Cell. Signal. 2021, 77, 109822. [Google Scholar] [CrossRef]
- Guereno, M.; Delgado Pastore, M.; Lugones, A.C.; Cercato, M.; Todaro, L.; Urtreger, A.; Peters, M.G. Glypican-3 (GPC3) inhibits metastasis development promoting dormancy in breast cancer cells by p38 MAPK pathway activation. Eur. J. Cell Biol. 2020, 99, 151096. [Google Scholar] [CrossRef]
- Nilsson, S.K.; Debatis, M.E.; Dooner, M.S.; Madri, J.A.; Quesenberry, P.J.; Becker, P.S. Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J. Histochem. Cytochem. 1998, 46, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Barney, L.E.; Hall, C.L.; Schwartz, A.D.; Parks, A.N.; Sparages, C.; Galarza, S.; Platt, M.O.; Mercurio, A.M.; Peyton, S.R. Tumor cell-organized fibronectin maintenance of a dormant breast cancer population. Sci. Adv. 2020, 6, eaaz4157. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Elevated levels of the alpha 5 beta 1 fibronectin receptor suppress the transformed phenotype of Chinese hamster ovary cells. Cell 1990, 60, 849–859. [Google Scholar] [CrossRef]
- Montagner, M.; Dupont, S. Mechanical Forces as Determinants of Disseminated Metastatic Cell Fate. Cells 2020, 9, 250. [Google Scholar] [CrossRef]
- Klein, C.A.; Blankenstein, T.J.; Schmidt-Kittler, O.; Petronio, M.; Polzer, B.; Stoecklein, N.H.; Riethmuller, G. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet 2002, 360, 683–689. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Rugo, H.S.; Hauranieh, L.; Roy, R.; Scott, J.H.; Lee, J.C.; Hsiao, F.; Sosa, E.V.; Van’t Veer, L.; Esserman, L.J.; et al. Genomic and expression profiling reveal molecular heterogeneity of disseminated tumor cells in bone marrow of early breast cancer. NPJ Breast Cancer 2018, 4, 31. [Google Scholar] [CrossRef]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most Early Disseminated Cancer Cells Detected in Bone Marrow of Breast Cancer Patients Have a Putative Breast Cancer Stem Cell Phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning Cancer Fate: Tumor Microenvironment’s Role in Cancer Stem Cell Quiescence and Reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef]
- Huang, Y.; Hamana, T.; Liu, J.; Wang, C.; An, L.; You, P.; Chang, J.Y.; Xu, J.; Jin, C.; Zhang, Z.; et al. Type 2 Fibroblast Growth Factor Receptor Signaling Preserves Stemness and Prevents Differentiation of Prostate Stem Cells from the Basal Compartment. J. Biol. Chem. 2015, 290, 17753–17761. [Google Scholar] [CrossRef]
- Quan, M.Y.; Guo, Q.; Liu, J.; Yang, R.; Bai, J.; Wang, W.; Cai, Y.; Han, R.; Lv, Y.Q.; Ding, L.; et al. An FGFR/AKT/SOX2 Signaling Axis Controls Pancreatic Cancer Stemness. Front. Cell Dev. Biol. 2020, 8, 287. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Watt, A.C.; Cejas, P.; DeCristo, M.J.; Metzger-Filho, O.; Lam, E.Y.N.; Qiu, X.; BrinJones, H.; Kesten, N.; Coulson, R.; Font-Tello, A.; et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nat. Cancer 2021, 2, 34–48. [Google Scholar] [CrossRef]
- Yoon, K.A.; Son, Y.; Choi, Y.J.; Kim, J.H.; Cho, J.Y. Fibroblast growth factor 2 supports osteoblastic niche cells during hematopoietic homeostasis recovery after bone marrow suppression. Cell Commun. Signal. 2017, 15, 25. [Google Scholar] [CrossRef]
- Bae, S.H.; Ryu, H.; Rhee, K.J.; Oh, J.E.; Baik, S.K.; Shim, K.Y.; Kong, J.H.; Hyun, S.Y.; Pack, H.S.; Im, C.; et al. L-ascorbic acid 2-phosphate and fibroblast growth factor-2 treatment maintains differentiation potential in bone marrow-derived mesenchymal stem cells through expression of hepatocyte growth factor. Growth Factors 2015, 33, 71–78. [Google Scholar] [CrossRef]
- Ito, T.; Sawada, R.; Fujiwara, Y.; Tsuchiya, T. FGF-2 increases osteogenic and chondrogenic differentiation potentials of human mesenchymal stem cells by inactivation of TGF-beta signaling. Cytotechnology 2008, 56, 1–7. [Google Scholar] [CrossRef]
- Najmi, S.; Korah, R.; Chandra, R.; Abdellatif, M.; Wieder, R. Flavopiridol blocks integrin-mediated survival in dormant breast cancer cells. Clin. Cancer Res. 2005, 11, 2038–2046. [Google Scholar] [CrossRef]
- Barrios, J.; Wieder, R. Dual FGF-2 and intergrin α5β1 signaling mediate GRAF-induced RhoA inactivation in a model of breast cancer dormancy. Cancer Microenviron. 2009, 2, 33–47. [Google Scholar] [CrossRef]
- Tivari, S.; Lu, H.; Dasgpta, T.; De Lorenzo, M.S.; Wieder, R. Reawakening of dormant estrogen-dependent human breast cancer cells by bone marrow stroma secretory senescence. Cell Commun. Signal. 2018, 16, 48. [Google Scholar] [CrossRef]
- Fenig, E.; Wieder, R.; Paglin, S.; Wang, H.; Persaud, R.; Haimovitz-Friedman, A.; Fuks, Z.; Yahalom, J. Basic fibroblast growth factor confers growth inhibition and Mitogen-activated Protein Kinase activation in human breast cancer cells. Clin. Cancer Res. 1997, 3, 135–142. [Google Scholar]
- Wang, H.; Rubin, M.; Fenig, E.; DeBlasio, T.; Mendelsohn, J.; Yahalom, J.; Wieder, R. Basic FGF causes growth arrest in MCF-7 human breast cancer cells while inducing both mitogenic and inhibitory G1 events. Cancer Res. 1997, 57, 1750–1757. [Google Scholar] [PubMed]
- Fenig, E.; Kanfi, Y.; Wang, Q.; Beery, E.; Livnat, T.; Wasserman, L.; Lilling, G.; Yahalom, J.; Wieder, R.; Nordenberg, J. Role of transforming growth factor beta in the growth inhibition of human breast cancer cells by basic fibroblast growth factor. Breast Cancer Res. Treat. 2001, 70, 27–37. [Google Scholar] [CrossRef]
- Ono, M.; Kosala, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef]
- Greco, S.J.; Rameshwar, P. Analysis of the transfer of circulating microRNA between cells mediated by gap junction. Methods Mol. Biol. 2013, 1024, 87–96. [Google Scholar]
- Zhou, Y.; Zuo, D.; Wang, M.; Zhang, Y.; Yu, M.; Yang, J.; Yao, Z. Effect of truncated neurokinin-1 receptor expression changes on the interaction between human breast cancer and bone marrow-derived mesenchymal stem cells. Genes Cells 2014, 19, 676–691. [Google Scholar] [CrossRef]
- Di Martino, J.S.; Akhter, T.; Bravo-Cordero, J.J. Remodeling the ECM: Implications for metastasis and tumor dormancy. Cancers 2021, 13, 4916. [Google Scholar] [CrossRef]
- Kim, S.; Dubrovska, A.; Salamone, R.J.; Walker, J.R.; Grandinetti, K.B.; Bonamy, G.M.; Orth, A.P.; Elliott, J.; Porta, D.G.; Garcia-Echeverria, C.; et al. FGFR2 promotes breast tumorigenicity through maintenance of breast tumor-initiating cells. PLoS ONE 2013, 8, e51671. [Google Scholar] [CrossRef]
- Coller, H.A.; Sang, L.; Roberts, J.M. A new description of cellular quiescence. PLoS Biol. 2006, 4, e83. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Gawrzak, S.; Rinaldi, L.; Gregorio, S.; Arenas, E.J.; Salvador, F.; Urosevic, J.; Figueras-Puig, C.; Rojo, F.; Del Barco Barrantes, I.; Cejalvo, J.M.; et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nat. Cell Biol. 2018, 20, 211–221. [Google Scholar] [CrossRef]
- Marlow, R.; Honeth, G.; Lombardi, S.; Cariati, M.; Hessey, S.; Pipili, A.; Mariotti, V.; Buchupalli, B.; Foster, K.; Bonnet, D.; et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res. 2013, 73, 6886–6899. [Google Scholar] [CrossRef]
- Cackowski, F.C.; Eber, M.R.; Rhee, J.; Decker, A.M.; Yumoto, K.; Berry, J.E.; Lee, E.; Shiozawa, Y.; Jung, Y.; Aguirre-Ghiso, J.A.; et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J. Cell Biochem. 2017, 118, 891–902. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef]
- Qin, S.; Li, B.; Ming, H.; Nice, E.C.; Zou, B.; Huang, C. Harnessing redox signaling to overcome therapeutic-resistant cancer dormancy. Biochim. Et Biophys. Acta Rev. Cancer 2022, 1877, 188749. [Google Scholar] [CrossRef]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef]
- Ren, D.; Dai, Y.; Yang, Q.; Zhang, X.; Guo, W.; Ye, L.; Huang, S.; Chen, X.; Lai, Y.; Du, H.; et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J. Exp. Med. 2019, 216, 428–449. [Google Scholar] [CrossRef]
- Baksh, D.; Tuan, R.S. Canonical and non-canonical Wnts differentially affect the development potential of primary isolate of human bone marrow mesenchymal stem cells. J. Cell. Physiol. 2007, 212, 817–826. [Google Scholar] [CrossRef]
- Melzer, C.; Ohe, J.v.d.; Luo, T.; Hass, R. Spontaneous Fusion of MSC with Breast Cancer Cells Can Generate Tumor Dormancy. Int. J. Mol. Sci. 2021, 22, 5930. [Google Scholar] [CrossRef]
- Melzer, C.; Ohe, J.v.d.; Hass, R. Altered Tumor Plasticity after Different Cancer Cell Fusions with MSC. Int. J. Mol. Sci. 2020, 21, 8347. [Google Scholar] [CrossRef]
- Bartosh, T.J.; Ullah, M.; Zeitouni, S.; Beaver, J.; Prockop, D.J. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc. Natl. Acad. Sci. USA 2016, 113, E6447–E6456. [Google Scholar] [CrossRef]
- Bui, A.T.; Laurent, F.; Havard, M.; Dautry, F.; Tchénio, T. SMAD signaling and redox imbalance cooperate to induce prostate cancer cell dormancy. Cell Cycle 2015, 14, 1218–1231. [Google Scholar] [CrossRef][Green Version]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef]
- Capulli, M.; Hristova, D.; Valbret, Z.; Carys, K.; Arjan, R.; Maurizi, A.; Masedu, F.; Cappariello, A.; Rucci, N.; Teti, A. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br. J. Cancer 2019, 121, 157–171. [Google Scholar] [CrossRef]
- Zheng, H.; Bae, Y.; Kasimir-Bauer, S.; Tang, R.; Chen, J.; Ren, G.; Yuan, M.; Esposito, M.; Li, W.; Wei, Y.; et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell 2017, 32, 731–747. [Google Scholar] [CrossRef]
- Janghorban, M.; Yang, Y.; Zhao, N.; Hamor, C.; Nguyen, T.M.; Zhang, X.H.; Rosen, J.M. Single-cell analysis unveils the role of the tumor immune microenvironment and notch signaling in dormant minimal residual disease. Cancer Res. 2022, 82, 885–899. [Google Scholar] [CrossRef]
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Marini, F.C.; Bussard, K.M. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res. 2019, 21, 31. [Google Scholar] [CrossRef]
- Shupp, A.B.; Neupane, M.; Agostini, L.C.; Ning, G.; Brody, J.R.; Bussard, K.M. Stromal-derived extracellular vesicles suppress proliferation of bone metastatic cancer cells mediated by ERK2. Mol. Cancer Res. MCR 2021, 19, 1763–1777. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Zhan, R.; Chaudhuri, A.; Watabe, M.; Pai, S.K.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; Takano, Y.; et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat. Med. 2006, 12, 933–938. [Google Scholar] [CrossRef]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- El-Shennawy, L.; Dubrovskyi, O.; Kastrati, I.; Danes, J.M.; Zhang, Y.; Whiteley, H.E.; Creighton, C.J.; Frasor, J. Coactivation of estrogen receptor and iKKbeta induces a dormant metastatic phenotype in ER-positive breast cancer. Cancer Res. 2018, 78, 974–984. [Google Scholar] [CrossRef]
- Nobre, A.R.; Dalla, E.; Yang, J.; Huang, X.; Wullkopf, L.; Risson, E.; Razghandi, P.; Anton, M.L.; Zheng, W.; Seoane, J.A.; et al. ZFP281 drives a mesenchymal-like dormancy program in early disseminated breast cancer cells that prevents metastatic outgrowth in the lung. Nat. Cancer 2022, 3, 1165–1180. [Google Scholar] [CrossRef]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, A.; Nagasawa, T.; Nombela-Arrieta, C.; Bhatia, R. Mesenchymal niche-specific expression of Cxcl12 controls quiescence of treatment-resistant leukemia stem cells. Cell Stem Cell 2019, 24, 769–784. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Yan, G.N.; Yang, L.; Lv, Y.F.; Shi, Y.; Shen, L.L.; Yao, X.H.; Guo, Q.N.; Zhang, P.; Cui, Y.H.; Zhang, X.; et al. Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J. Pathol. 2014, 234, 11–22. [Google Scholar] [CrossRef]
- Kurebayashi, J.; Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T. Anti-cancer stem cell activity of a hedgehog inhibitor gant61 in estrogen receptor-positive breast cancer cells. Cancer Sci. 2017, 108, 918–930. [Google Scholar] [CrossRef]
- Arnold, K.M.; Pohlig, R.T.; Sims-Mourtada, J. Co-activation of hedgehog and wnt signaling pathways is associated with poor outcomes in triple negative breast cancer. Oncol. Lett. 2017, 14, 5285–5292. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Jeng, C.J.; Sheen, I.S.; Wu, S.H.; Lu, S.J.; Wang, C.H.; Chang, C.F. Glioma-associated oncogene homolog inhibitors have the potential of suppressing cancer stem cells of breast cancer. Int. J. Mol. Sci. 2018, 19, 1375. [Google Scholar] [CrossRef]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal–epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.; Anaparthy, N.; Memos, N.; Kelley, Z.L.; Gouronnec, A.; Yan, R.; Auffray, C.; Albrengues, J.; Egeblad, M.; Iacobuzio-Donahue, C.A.; et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018, 360, eaao4908. [Google Scholar] [CrossRef]
- Satcher, R.L.; Zhang, X.H. Evolving cancer-niche interactions and therapeutic targets during bone metastasis. Nat. Rev. Cancer 2022, 22, 85–101. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massague, J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef]
- Zhang, X.H.; Wang, Q.; Gerald, W.; Hudis, C.A.; Norton, L.; Smid, M.; Foekens, J.A.; Massague, J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009, 16, 67–78. [Google Scholar] [CrossRef]
- Horak, C.E.; Lee, J.H.; Marshall, J.C.; Shreeve, S.M.; Steeg, P.S. The role of metastasis suppressor genes in metastatic dormancy. APMIS 2008, 116, 586–601. [Google Scholar] [CrossRef]
- Drescher, F.; Juárez, P.; Arellano, D.L.; Serafín-Higuera, N.; Olvera-Rodriguez, F.; Jiménez, S.; Licea-Navarro, A.F.; Fournier, P.G. TIE2 Induces Breast Cancer Cell Dormancy and Inhibits the Development of Osteolytic Bone Metastases. Cancers 2020, 12, 868. [Google Scholar] [CrossRef]
- Yang, C.; Ng, C.T.; Li, D.; Zhang, L. Targeting indoleamine 2,3-dioxygenase 1: Fighting cancers via dormancy regulation. Front. Immunol. 2021, 12, 725204. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.M.; Mseeh, F.; McAfoos, T.J.; Leonard, P.G.; Reyna, N.J.; Harris, A.L.; Xu, A.; Han, M.; Soth, M.J.; Czako, B.; et al. Discovery of iacs-9779 and iacs-70465 as potent inhibitors targeting indoleamine 2,3-dioxygenase 1 (ido1) apoenzyme. J. Med. Chem. 2021, 64, 11302–11329. [Google Scholar] [CrossRef]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; Leroy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of cdk8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA methyltransferases in cancer: Biology, paradox, aberrations, and targeted therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bernstein, E.; Aguirre-Ghiso, J.A. Epigenetic Regulation of Cancer Dormancy as a Plasticity Mechanism for Metastasis Initiation. In Tumor Dormancy and Recurrence. Cancer Drug Discovery and Development; Wang, Y., Crea, F., Eds.; Humana Press: Totowa, NJ, USA, 2017. [Google Scholar]
- Arandkar, S.; Furth, N.; Elisha, Y.; Nataraj, N.B.; van der Kuip, H.; Yarden, Y.; Aulitzky, W.; Ulitsky, I.; Geiger, B.; Oren, M. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc. Natl. Acad. Sci. USA 2018, 11, 6410–6415. [Google Scholar] [CrossRef]
- Suh, Y.A.; Lee, H.Y.; Virmani, A.; Wong, J.; Mann, K.K.; Miller, W.H.; Gazdar, A.; Kurie, J.M. Loss of retinoic acid receptor beta gene expression is linked to aberrant histone h3 acetylation in lung cancer cell lines. Cancer Res. 2002, 62, 3945–3949. [Google Scholar]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mavrakis, K.J.; Wendel, H.G.; Giancotti, F.G. Forward genetic screens in mice uncover mediators and suppressors of metastatic reactivation. Proc. Natl. Acad. Sci. USA 2014, 111, 16532–16537. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.J.; Parker, K.A.; Schiemann, W.P. Epigenetic plasticity in metastatic dormancy: Mechanisms and therapeutic implications. Ann. Transl. Med. 2020, 8, 903. [Google Scholar] [CrossRef]
- Parker, K.A.; Robinson, N.J.; Schiemann, W.P. The role of RNA processing and regulation in metastatic dormancy. Semin. Cancer Biol. 2022, 78, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.; Johansson, A.; Nordenskjöld, A.; Iftimi, A.; Yau, C.; Perez-Tenorio, G.; Benz, C.; Nordenskjöld, B.; Stål, O.; Esserman, L.J.; et al. Assessment of 25-Year Survival of Women with Estrogen Receptor-Positive/ERBB2-Negative Breast Cancer Treated With and Without Tamoxifen Therapy: A Secondary Analysis of Data From the Stockholm Tamoxifen Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e2114904. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Wieder, R.; Shafiq, B.; Adam, N. African American race is an independent risk factor in survival form initially diagnosed localized breast cancer. J. Cancer 2016, 7, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Wieder, R.; Shafiq, B.; Adam, N. Greater Survival Improvement in African American vs. Caucasian Women with Hormone Negative Breast Cancer. J. Cancer 2020, 11, 2808–2820. [Google Scholar] [CrossRef]
- Geurts, Y.M.; Witteveen, A.; Bretveld, R.; Poortmans, P.M.; Sonke, G.S.; Strobbe, L.J.A.; Siesling, S. Patterns and predictors of first and subsequent recurrence in women with early breast cancer. Breast Cancer Res. Treat. 2017, 165, 709–720. [Google Scholar] [CrossRef]
- Colleoni, M.; Sun, Z.; Price, K.N.; Karlsson, P.; Forbes, J.F.; Thürlimann, B.; Gianni, L.; Castiglione, M.; Gelber, R.D.; Coates, A.S.; et al. Annual Hazard Rates of Recurrence for Breast Cancer During 24 Years of Follow-Up: Results from the International Breast Cancer Study Group Trials I to V. J. Clin. Oncol. 2016, 34, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Massafra, R.; Latorre, A.; Fanizzi, A.; Bellotti, R.; Didonna, V.; Giotta, F.; La Forgia, D.; Nardone, A.; Pastena, M.; Ressa, C.M.; et al. A clinical decision support system for predicting invasive breast cancer recurrence: Preliminary results. Front. Oncol. 2021, 11, 576007. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, Y.S.; Yu, J.; Park, Y.; Lee, S.K.; Lee, M.; Lee, J.E.; Kim, S.W.; Nam, S.J.; Park, Y.H.; et al. Deep learning-based prediction model for breast cancer recurrence using adjuvant breast cancer cohort in tertiary cancer center registry. Front. Oncol. 2021, 11, 596364. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Garcia-Closas, M.; Olshan, A.F.; Perou, C.M.; Troester, M.A.; Love, M.I. A framework for transcriptome-wide association studies in breast cancer in diverse study populations. Genome Biol. 2020, 21, 42. [Google Scholar] [CrossRef]
- Stein, W.D.; Litman, T. Data on the recurrence of breast tumors fit a model in which dormant cells are subject to slow attrition but can randomly awaken to become malignant. Cell Cycle 2006, 5, 2348–2353. [Google Scholar] [CrossRef]
- Gelman, I.H. The genomic regulation of metastatic dormancy. Cancer Metastasis Rev. 2023, 42, 255–276. [Google Scholar] [CrossRef]
- Sanchez Calle, A.; Yamamoto, T.; Kawamura, Y.; Hironaka-Mitsuhashi, A.; Ono, M.; Tsuda, H.; Shimomura, A.; Tamura, K.; Takeshita, F.; Ochiya, T.; et al. Long non-coding nr2f1-as1 is associated with tumor recurrence in estrogen receptor-positive breast cancers. Mol. Oncol. 2020, 14, 2271–2287. [Google Scholar] [CrossRef] [PubMed]
- Aftimos, P.; Oliveira, M.; Irrthum, A.; Fumagalli, D.; Sotiriou, C.; Gal-Yam, E.N.; Robson, M.E.; Ndozeng, J.; Di Leo, A.; Ciruelos, E.M.; et al. Genomic and transcriptomic analyses of breast cancer primaries and matched metastases in aurora, the breast international group (big) molecular screening initiative. Cancer Discov. 2021, 11, 2796–2811. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Ichikawa, Y.; Osako, T.; Fujita, T.; Baba, S.; Takeuchi, K.; Tsunoda, N.; Ebata, T.; Ueno, T.; Ohno, S.; et al. The ELEANOR noncoding RNA expression contributes to cancer dormancy and predicts late recurrence of estrogen receptor-positive breast cancer. Cancer Sci. 2022, 113, 2336–2351. [Google Scholar] [CrossRef]
- De Cock, J.M.; Shibue, T.; Dongre, A.; Keckesova, Z.; Reinhardt, F.; Weinberg, R.A. Inflammation triggers zeb1-dependent escape from tumor latency. Cancer Res. 2016, 76, 6778–6784. [Google Scholar] [CrossRef]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.D.; Elias, M.; Guiro, K.; Bhatia, R.; Greco, S.J.; Bryan, M.; Gergues, M.; Sandiford, O.A.; Ponzio, N.M.; Leibovich, S.J.; et al. Exosomes from differentially activated macrophages influence dormancy or resurgence of breast cancer cells within bone marrow stroma. Cell Death Dis. 2019, 10, 59. [Google Scholar] [CrossRef]
- Zhang, M.; Di Martino, J.S.; Bowman, R.L.; Campbell, N.R.; Baksh, S.C.; Simon-Vermot, T.; Kim, I.S.; Haldeman, P.; Mondal, C.; Yong-Gonzales, V.; et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov. 2018, 8, 1006–1025. [Google Scholar] [CrossRef]
- Zhang, W.; Bado, I.L.; Hu, J.; Wan, Y.W.; Wu, L.; Wang, H.; Gao, Y.; Jeong, H.H.; Xu, Z.; Hao, X.; et al. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell 2021, 184, 2471–2486.e20. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef]
- Obradovic, M.M.S.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.M.; Munst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nature 2019, 567, 540–544. [Google Scholar] [CrossRef]
- Indraccolo, S.; Favaro, E.; Amadori, A. Dormant tumors awaken by a short-term angiogenic burst: The spike hypothesis. Cell Cycle 2006, 5, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Krall, J.A.; Reinhardt, F.; Mercury, O.A.; Pattabiraman, D.R.; Brooks, M.W.; Dougan, M.; Lambert, A.W.; Bierie, B.; Ploegh, H.L.; Dougan, S.K.; et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 2018, 10, 3464. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Guo, M.; Liu, Z.; Fu, Y.; Wu, H.; Wang, C.; Cao, M. Morphine promotes the angiogenesis of postoperative recurrent tumors and metastasis of dormant breast cancer cells. Pharmacology 2019, 104, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Decker, A.M.; Jung, Y.; Cackowski, F.C.; Yumoto, K.; Wang, J.; Taichman, R.S. Sympathetic Signaling Reactivates Quiescent Disseminated Prostate Cancer Cells in the Bone Marrow. Mol. Cancer Res. MCR 2017, 15, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.C.; Wang, X.; Zhou, X.; Wang, C.; Chen, L.; Yin, L.J.; He, B.C.; Deng, Z.L. Gut-derived serotonin induced by depression promotes breast cancer bone metastasis through the runx2/pthrp/rankl pathway in mice. Oncol. Rep. 2016, 35, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Radman, M. Cellular parabiosis and the latency of age-related diseases. Open Biol. 2019, 9, 180250. [Google Scholar] [CrossRef]
- Manjili, S.H.; Isbell, M.; Ghochaghi, N.; Perkinson, T.; Manjili, M.H. Multifaceted functions of chronic inflammation in regulating tumor dormancy and relapse. Semin. Cancer Biol. 2022, 78, 17–22. [Google Scholar] [CrossRef]
- Mukai, H.; Miki, N.; Yamada, H.; Goto, H.; Kawakami, T.; Suzuki, A.; Yamamoto, K.; Nakanishi, Y.; Takahashi, K. Pannexin1 channel-dependent secretome from apoptotic tumor cells shapes immune-escape microenvironment. Biochem. Biophys. Res. Commun. 2022, 628, 116–122. [Google Scholar] [CrossRef]
- Xu, Z.; Ni, B.; Cao, Z.; Zielonka, J.; Gao, J.; Chen, F.; Kalyanaraman, B.; White, G.C.; Ma, Y.Q. Kindlin-3 negatively regulates the release of neutrophil extracellular traps. J. Leukoc. Biol. 2018, 104, 597–602. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef]
- Munir, H.; Jones, J.O.; Janowitz, T.; Hoffmann, M.; Euler, M.; Martins, C.P.; Welsh, S.J.; Shields, J.D. Stromal-driven and amyloid beta-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat. Commun. 2021, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhan, J.; Luo, Q.; Hou, X.; Wang, S.; Xiao, D.; Xie, Z.; Liang, H.; Lin, S.; Zheng, M. Adrb3 induces mobilization and inhibits differentiation of both breast cancer cells and myeloid-derived suppressor cells. Cell Death Dis. 2022, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Lerman, I.; Hammes, S.R. Neutrophil elastase in the tumor microenvironment. Steroids 2018, 133, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Zhang, Y.; Huang, H.; Zhang, L.; Femel, J.; Dimberg, A.; Olsson, A.K. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res. 2015, 75, 2653–2662. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Bray, T.M.; Ho, E. Induction of proinflammatory response in prostate cancer epithelial cells by activated macrophages. Cancer Lett. 2009, 276, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, W.; Aldape, K.; He, J.; Lu, Z. Epidermal growth factor (EGF)-enhanced vascular cell adhesion molecule-1 (VCAM-1) expression promotes macrophage and glioblastoma cell interaction and tumor cell invasion. J. Biol. Chem. 2013, 288, 31488–31495. [Google Scholar] [CrossRef]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Francescangeli, F.; De Angelis, M.L.; Baiocchi, M.; Rossi, R.; Biffoni, M.; Zeuner, A. COVID-19-induced modifications in the tumor microenvironment: Do they affect cancer reawakening and metastatic relapse? Front. Oncol. 2020, 10, 592891. [Google Scholar] [CrossRef]
- Correia, A.L.; Guimaraes, J.C.; Auf der Maur, P.; De Silva, D.; Trefny, M.P.; Okamoto, R.; Bruno, S.; Schmidt, A.; Mertz, K.; Volkmann, K.; et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 2021, 594, 566–571. [Google Scholar] [CrossRef]
- Malanchi, I.; Santamaria-Martinez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [PubMed]
- Cheleuitte, D.; Mizuno, S.; Glowacki, J. In vitro secretion of cytokines by human bone marrow: Effects of age and estrogen status. J. Clin. Endocrinol. Metab. 1998, 83, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Kaack, M.B.; Harrison, R.M.; Roberts, J.A. Effect of age and hormonal state on cytokine synthesis in the monkey. Cytokine 1998, 10, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Schellinger, D.; Lin, C.S.; Fertikh, D.; Lee, J.S.; Lauerman, W.C.; Henderson, F.; Davis, B. Normal lumbar vertebrae: Anatomic, age, and sex variance in subjects at proton MR spectroscopy--initial experience. Radiology 2000, 215, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, S.; Fumoto, T.; Naoe, Y.; Ikeda, K. Age-related marrow adipogenesis is linked to increased expression of RankL. J. Biol. Chem. 2014, 289, 16699–16710. [Google Scholar] [CrossRef]
- Moerman, E.J.; Teng, K.; Lipschitz, D.A.; Lecka-Czernik, B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: The role of PPAR-gamma2 transcription factor and TGF-beta/bmp signaling pathways. Aging Cell 2004, 3, 379–389. [Google Scholar] [CrossRef]
- Stenderup, K.; Justesen, J.; Clausen, C.; Kassem, M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 2003, 33, 919–926. [Google Scholar] [CrossRef]
- Li, H.; Liu, P.; Xu, S.; Li, Y.; Dekker, J.D.; Li, B.; Fan, Y.; Zhang, Z.; Hong, Y.; Yang, G.; et al. FoxP1 controls mesenchymal stem cell commitment and senescence during skeletal aging. J. Clin. Investig. 2017, 127, 1241–1253. [Google Scholar] [CrossRef]
- Candini, O.; Spano, C.; Murgia, A.; Grisendi, G.; Veronesi, E.; Piccinno, M.S.; Ferracin, M.; Negrini, M.; Giacobbi, F.; Bambi, F.; et al. Mesenchymal progenitors aging highlights a mir-196 switch targeting hoxb7 as master regulator of proliferation and osteogenesis. Stem Cells 2015, 33, 939–950. [Google Scholar] [CrossRef]
- Sarsour, E.H.; Son, J.M.; Kalen, A.L.; Xiao, W.; Du, J.; Alexander, M.S.; O’Leary, B.R.; Cullen, J.J.; Goswami, P.C. Arachidonate 12-lipoxygenase and 12-hydroxyeicosatetraenoic acid contribute to stromal aging-induced progression of pancreatic cancer. J. Biol. Chem. 2020, 295, 6946–6957. [Google Scholar] [CrossRef]
- Hay, E.; Dieudonne, F.X.; Saidak, Z.; Marty, C.; Brun, J.; Da Nascimento, S.; Sonnet, P.; Marie, P.J. N-cadherin/wnt interaction controls bone marrow mesenchymal cell fate and bone mass during aging. J. Cell. Physiol. 2014, 229, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.N.; Ouyang, H.; Ma, Y.L.; Zeng, Q.; Gerin, I.; Sousa, K.M.; Lane, T.F.; Krishnan, V.; Hankenson, K.D.; MacDougald, O.A. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J. Bone Miner. Res. 2007, 22, 1924–1932. [Google Scholar] [CrossRef]
- Bilkovski, R.; Schulte, D.M.; Oberhauser, F.; Gomolka, M.; Udelhoven, M.; Hettich, M.M.; Roth, B.; Heidenreich, A.; Gutschow, C.; Krone, W.; et al. Role of WNT-5a in the determination of human mesenchymal stem cells into preadipocytes. J. Biol. Chem. 2010, 285, 6170–6178. [Google Scholar] [CrossRef]
- Stevens, J.R.; Miranda-Carboni, G.A.; Singer, M.A.; Brugger, S.M.; Lyons, K.M.; Lane, T.F. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J. Bone Miner. Res. 2010, 25, 2138–2147. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [PubMed]
- Sriuranpong, V.; Park, J.I.; Amornphimoltham, P.; Patel, V.; Nelkin, B.D.; Gutkind, J.S. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003, 63, 2948–2956. [Google Scholar]
- Berishaj, M.; Gao, S.P.; Ahmed, S.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.L.; Bornmann, W.; Bromberg, J.F. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007, 9, R32. [Google Scholar] [CrossRef]
- Rega, G.; Kaun, C.; Demyanets, S.; Pfaffenberger, S.; Rychli, K.; Hohensinner, P.J.; Kastl, S.P.; Speidl, W.S.; Weiss, T.W.; Breuss, J.M.; et al. Vascular endothelial growth factor is induced by the inflammatory cytokines interleukin-6 and oncostatin M in human adipose tissue in vitro and in murine adipose tissue in vivo. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1587–1795. [Google Scholar] [CrossRef]
- Steiner, H.; Berger, A.P.; Godoy-Tundidor, S.; Bjartell, A.; Lilja, H.; Bartsch, G.; Hobisch, A.; Culig, Z. An autocrine loop for vascular endothelial growth factor is established in prostate cancer cells generated after prolonged treatment with interleukin 6. Eur. J. Cancer 2004, 40, 1066–1072. [Google Scholar] [CrossRef]
- Luppi, F.; Longo, A.M.; de Boer, W.I.; Rabe, K.F. Hiemstra PS. Interleukin-8 stimulates cell proliferation in non-small cell lung cancer through epidermal growth factor receptor transactivation. Lung Cancer 2007, 56, 25–33. [Google Scholar] [CrossRef]
- Fane, M.E.; Chhabra, Y.; Alicea, G.M.; Maranto, D.A.; Douglass, S.M.; Webster, M.R.; Rebecca, V.W.; Marino, G.E.; Almeida, F.; Ecker, B.L.; et al. Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature 2022, 606, 396–405. [Google Scholar] [CrossRef]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H.; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef]
- Xia, W.; Zhuang, L.; Deng, X.; Hou, M. Long noncoding RNA-p21 modulates cellular senescence via the wnt/beta-catenin signaling pathway in mesenchymal stem cells. Mol. Med. Rep. 2017, 16, 7039–7047. [Google Scholar] [CrossRef]
- Le Blanc, S.; Simann, M.; Jakob, F.; Schutze, N.; Schilling, T. Fibroblast growth factors 1 and 2 inhibit adipogenesis of human bone marrow stromal cells in 3d collagen gels. Exp. Cell Res. 2015, 338, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Asumda, F.Z.; Chase, P.B. Age-related changes in rat bone-marrow mesenchymal stem cell plasticity. BMC Cell Biol. 2011, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yi, X.C.; Guo, G.; Long, Q.F.; Wang, X.A.; Zhong, J.; Liu, W.P.; Fei, Z.; Wang, D.M.; Liu, J. Basic fibroblast growth factor increases the transplantation-mediated therapeutic effect of bone mesenchymal stem cells following traumatic brain injury. Mol. Med. Rep. 2014, 9, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Kamiya, H.; Himeno, T.; Naruse, K.; Nakashima, E.; Watarai, A.; Shibata, T.; Tosaki, T.; Kato, J.; Okawa, T.; et al. Therapeutic efficacy of bone marrow-derived mononuclear cells in diabetic polyneuropathy is impaired with aging or diabetes. J. Diabetes Investig. 2015, 6, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Sobue, T.; Esliger, A.; Kronenberg, M.S.; Coffin, J.D.; Doetschman, T.; Hurley, M.M. Disruption of the FGF2 gene activates the adipogenic and suppresses the osteogenic program in mesenchymal marrow stromal stem cells. Bone 2010, 47, 360–370. [Google Scholar] [CrossRef]
- Ou, G.; Charles, L.; Matton, S.; Rodner, C.; Hurley, M.; Kuhn, L.; Gronowicz, G. Fibroblast growth factor-2 stimulates the proliferation of mesenchyme-derived progenitor cells from aging mouse and human bone. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 1051–1059. [Google Scholar] [CrossRef]
- Kanawa, M.; Igarashi, A.; Ronald, V.S.; Higashi, Y.; Kurihara, H.; Sugiyama, M.; Saskianti, T.; Pan, H.; Kato, Y. Age-dependent decrease in the chondrogenic potential of human bone marrow mesenchymal stromal cells expanded with fibroblast growth factor-2. Cytotherapy 2013, 15, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Hu, K.; Ji, K.; Sun, Q.; Xiong, J.; Yang, L.; Liu, H. Directed differentiation of aged human bone marrow multipotent stem cells effectively generates dopamine neurons. Vitr. Cell. Dev. Biol. Anim. 2014, 50, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhao, Q.; Yu, X. Bone marrow adipocytes, adipocytokines, and breast cancer cells: Novel implications in bone metastasis of breast cancer. Front. Oncol. 2020, 10, 561595. [Google Scholar] [CrossRef] [PubMed]
- Scheller, E.L.; Doucette, C.R.; Learman, B.S.; Cawthorn, W.P.; Khandaker, S.; Schell, B. Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat. Commun. 2015, 6, 7808. [Google Scholar] [CrossRef]
- Scheller, E.L.; Cawthorn, W.P.; Burr, A.A.; Horowitz, M.C.; MacDougald, O.A. Marrow adipose tissue: Trimming the fat. Trends Endocrinol. Metab. 2016, 27, 392–403. [Google Scholar] [CrossRef]
- Fazeli, P.K.; Horowitz, M.C.; MacDougald, O.A.; Scheller, E.L.; Rodeheffer, M.S.; Rosen, C.J. Marrow fat and bone--new perspectives. J. Clin. Endocrinol. Metab. 2013, 98, 935–945. [Google Scholar] [CrossRef]
- Zhang, X.; Robles, H.; Magee, K.L.; Lorenz, M.R.; Wang, Z.; Harris, C.A.; Craft, C.S.; Scheller, E.L. A bone-specific adipogenesis pathway in fat-free mice defines key origins and adaptations of bone marrow adipocytes with age and disease. eLife 2021, 10, 66275. [Google Scholar] [CrossRef]
- Bensreti, H.; Alhamad, D.W.; Gonzalez, A.M.; Pizarro-Mondesir, M.; Bollag, W.B.; Isales, C.M.; McGee-Lawrence, M.E. Update on the role of glucocorticoid signaling in osteoblasts and bone marrow adipocytes during aging. Curr. Osteoporos. Rep. 2023, 21, 32–44. [Google Scholar] [CrossRef]
- Song, L.; Liu, M.; Ono, N.; Bringhurst, F.R.; Kronenberg, H.M.; Guo, J. Loss of wnt/beta-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J. Bone Miner. Res. 2012, 27, 2344–2358. [Google Scholar] [CrossRef]
- De Bisschop, E.; Luypaert, R.; Louis, O.; Osteaux, M. Fat fraction of lumbar bone marrow using in vivo proton nuclear magnetic resonance spectroscopy. Bone 1993, 14, 133–136. [Google Scholar] [CrossRef]
- Kugel, H.; Jung, C.; Schulte, O.; Heindel, W. Age- and sex-specific differences in the 1h-spectrum of vertebral bone marrow. J. Magn. Reson. Imaging 2001, 13, 263–268. [Google Scholar] [CrossRef]
- Griffith, J.F.; Yeung, D.K.; Ma, H.T.; Leung, J.C.; Kwok, T.C.; Leung, P.C. Bone marrow fat content in the elderly: A reversal of sex difference seen in younger subjects. J. Magn. Reson. Imaging 2012, 36, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor cd36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, 5758. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tao, B.; Fan, Q.; Wang, S.; Chen, L.; Zhang, J.; Hao, Y.; Dong, S.; Wang, Z.; Wang, W.; et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its cys333-cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMed. 2019, 45, 108–123. [Google Scholar] [CrossRef]
- Panigrahy, D.; Edin, M.L.; Lee, C.R.; Huang, S.; Bielenberg, D.R.; Butterfield, C.E.; Barnes, C.M.; Mammoto, A.; Mammoto, T.; Luria, A.; et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Investig. 2012, 122, 178–191. [Google Scholar] [CrossRef] [PubMed]
- La Camera, G.; Gelsomino, L.; Malivindi, R.; Barone, I.; Panza, S.; De Rose, D.; Giordano, F.; D’Esposito, V.; Formisano, P.; Bonofiglio, D.; et al. Adipocyte-derived extracellular vesicles promote breast cancer cell malignancy through hif-1alpha activity. Cancer Letters. 2021, 521, 155–168. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, J.; Tian, X.; Fang, Z.; Gao, Y.; Ping, Z.; Liu, L. Body mass index increases the recurrence risk of breast cancer: A dose-response meta-analysis from 21 prospective cohort studies. Public Health 2022, 210, 26–33. [Google Scholar] [CrossRef]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Ando, S.; Catalano, S. The weight of obesity in breast cancer progression and metastasis: Clinical and molecular perspectives. Semin. Cancer Biol. 2020, 60, 274–284. [Google Scholar] [CrossRef]
- Sheinboim, D.; Parikh, S.; Manich, P.; Markus, I.; Dahan, S.; Parikh, R.; Stubbs, E.; Cohen, G.; Zemser-Werner, V.; Bell, R.E.; et al. An exercise-induced metabolic shield in distant organs blocks cancer progression and metastatic dissemination. Cancer Res. 2022, 82, 4164–4178. [Google Scholar] [CrossRef]
- Han, H.H.; Kim, B.G.; Lee, J.H.; Kang, S.; Kim, J.E.; Cho, N.H. Angiopoietin-2 promotes ER+ breast cancer cell survival in bone marrow niche. Endocr. Relat. Cancer 2016, 23, 609–623. [Google Scholar] [CrossRef]
- Kampeerawipakorn, O.; Navasumrit, P.; Settachan, D.; Promvijit, J.; Hunsonti, P.; Parnlob, V.; Nakngam, N.; Choonvisase, S.; Chotikapukana, P.; Chanchaeamsai, S.; et al. Health risk evaluation in a population exposed to chemical releases from a petrochemical complex in Thailand. Environ. Res. 2017, 152, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Jaguin, M.; Fardel, O.; Lecureur, V. Exposure to diesel exhaust particle extracts (DEPe) impairs some polarization markers and functions of human macrophages through activation of AhR and Nrf2. PLoS ONE 2015, 10, e0116560. [Google Scholar] [CrossRef]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence by Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol. Cancer Res. 2017, 15, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Huang, D.; Yuan, F.; Xie, N.; Li, Q.; Sun, X.; Zhou, X.; Li, G.; Tong, T.; Zhang, Y. Traf-interacting protein with forkhead-associated domain (tifa) transduces dna damage-induced activation of nf-κb. J. Biol. Chem. 2018, 293, 7268–7280. [Google Scholar] [CrossRef]
- Krisnawan, V.E.; Stanley, J.A.; Schwarz, J.K.; DeNardo, D.G. Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance. Cancers 2020, 12, 2916. [Google Scholar] [CrossRef]
- Ragunathan, K.; Upfold, N.L.E.; Oksenych, V. Interaction between Fibroblasts and Immune Cells Following DNA Damage Induced by Ionizing Radiation. Int. J. Mol. Sci. 2020, 21, 8635. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Yoshida, Y.; Ohtani, N. Cellular senescence and the tumour microenvironment. Mol. Oncol. 2022, 16, 3333–3351. [Google Scholar] [CrossRef]
- Luo, X.; Fu, Y.; Loza, A.J.; Murali, B.; Leahy, K.M.; Ruhland, M.K.; Gang, M.; Su, X.; Zamani, A.; Shi, Y.; et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep. 2016, 14, 82–92. [Google Scholar] [CrossRef]
- Alexander, K.; Hinds, P.W. Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol. Cell Biol. 2001, 21, 3616–3631. [Google Scholar] [CrossRef]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Baumann, Z.; Auf der Maur, P.; Bentires-Alj, M. Feed-forward loops between metastatic cancer cells and their microenvironment-the stage of escalation. EMBO Mol. Med. 2022, 14, e14283. [Google Scholar] [CrossRef] [PubMed]
- Nierodzik, M.L.; Plotkin, A.; Kajumo, F.; Karpatkin, S. Thrombin stimulates tumor-platelet adhesion in vitro and metastasis in vivo. J. Clin. Investig. 1991, 87, 229–236. [Google Scholar] [CrossRef]
- Nierodzik, M.L.; Bain, R.M.; Liu, L.X.; Shivji, M.; Takeshita, K.; Karpatkin, S. Presence of the seven transmembrane thrombin receptor on human tumour cells: Effect of activation on tumour adhesion to platelets and tumor tyrosine phosphorylation. Br. J. Haematol. 1996, 92, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Nierodzik, M.L.; Kajumo, F.; Karpatkin, S. Effect of thrombin treatment of tumor cells on adhesion of tumor cells to platelets in vitro and tumor metastasis in vivo. Cancer Res. 1992, 52, 3267–3272. [Google Scholar]
- Klepfish, A.; Greco, M.A.; Karpatkin, S. Thrombin stimulates melanoma tumor-cell binding to endothelial cells and subendothelial matrix. Int. J. Cancer 1993, 53, 978–982. [Google Scholar] [CrossRef]
- Green, D.; Karpatkin, S. Role of thrombin as a tumor growth factor. Cell Cycle 2010, 9, 656–661. [Google Scholar] [CrossRef]
- Chen, L.B.; Buchanan, J.M. Mitogenic activity of blood components. I. thrombin and prothrombin. Proc. Natl. Acad. Sci. USA 1975, 72, 131–135. [Google Scholar] [CrossRef]
- Carney, D.H.; Stiernberg, J.; Fenton, J.W. Initiation of proliferative events by human alpha-thrombin requires both receptor binding and enzymic activity. J. Cell. Biochem. 1984, 26, 181–195. [Google Scholar] [CrossRef]
- Karpatkin, S. Does hypercoagulability awaken dormant tumor cells in the host? J. Thromb. Haemost. 2004, 2, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Giaccherini, C.; Masci, G.; Verzeroli, C.; Russo, L.; Celio, L.; Sarmiento, R.; Gamba, S.; Tartari, C.J.; Diani, E.; et al. Thrombin generation predicts early recurrence in breast cancer patients. J. Thromb. Haemost. 2020, 18, 2220–2231. [Google Scholar] [CrossRef]
- Gomez-Rosas, P.; Pesenti, M.; Verzeroli, C.; Giaccherini, C.; Russo, L.; Sarmiento, R.; Masci, G.; Celio, L.; Minelli, M.; Gamba, S.; et al. Validation of the role of thrombin generation potential by a fully automated system in the identification of breast cancer patients at high risk of disease recurrence. TH Open Companion J. Thromb. Haemost. 2021, 5, e56–e65. [Google Scholar]
- Mandoj, C.; Pizzuti, L.; Sergi, D.; Sperduti, I.; Mazzotta, M.; Di Lauro, L.; Amodio, A.; Carpano, S.; Di Benedetto, A.; Botti, C.; et al. Observational study of coagulation activation in early breast cancer: Development of a prognostic model based on data from the real world setting. J. Transl. Med. 2018, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J.M.; et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J. Thromb. Haemost. 2021, 19, 2546–2553. [Google Scholar] [CrossRef]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in long COVID/post-acute sequelae of COVID-19 (pasc) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef]
- Alvarado-Moreno, J.A.; Davila-Moreno, J.; Dominguez-Reyes, V.; Arreola-Diaz, R.; Isordia-Salas, I.; Chavez-Gonzalez, A.; Majluf-Cruz, A. Morphological and functional alterations in endothelial colony-forming cells from recovered COVID-19 patients. Thromb. Res. 2021, 206, 55–59. [Google Scholar] [CrossRef]
- Nicolai, L.; Kaiser, R.; Stark, K. Thrombo-inflammation in long COVID—The elusive key to post-infection sequelae? J. Thromb. Haemost. 2023; in press. [Google Scholar]
- Ahamed, J.; Laurence, J. Long COVID endotheliopathy: Hypothesized mechanisms and potential therapeutic approaches. J. Clin. Investig. 2022, 132, I161167. [Google Scholar] [CrossRef]
- Takatsuka, H.; Wakae, T.; Mori, A.; Okada, M.; Fujimori, Y.; Takemoto, Y.; Okamoto, T.; Kanamaru, A.; Kakishita, E. Endothelial damage caused by cytomegalovirus and human herpesvirus-6. Bone Marrow Transplant. 2003, 31, 475–479. [Google Scholar] [CrossRef]
- Hyakutake, M.T.; Steinberg, E.; Disla, E.; Heller, M. Concomitant infection with epstein-barr virus and cytomegalovirus infection leading to portal vein thrombosis. J. Emerg. Med. 2019, 57, e49–e51. [Google Scholar] [CrossRef] [PubMed]
- Goeijenbier, M.; van Wissen, M.; van de Weg, C.; Jong, E.; Gerdes, V.E.; Meijers, J.C.; Brandjes, D.P.; van Gorp, E.C. Review: Viral infections and mechanisms of thrombosis and bleeding. J. Med. Virol. 2012, 84, 1680–1696. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Murdoch, C. Macrophage responses to hypoxia: Implications for tumor progression and anti-cancer therapies. Am. J. Pathol. 2005, 167, 627–635. [Google Scholar] [CrossRef]
- Retsky, M.; Demicheli, R.; Hrushesky, W.J. Does surgery induce angiogenesis in breast cancer? indirect evidence from relapse pattern and mammography paradox. Int. J. Surg. 2005, 3, 179–187. [Google Scholar] [CrossRef]
- O’Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y.; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar] [CrossRef]
- Georgiou, G.K.; Igglezou, M.; Sainis, I.; Vareli, K.; Batsis, H.; Briasoulis, E.; Fatouros, M. Impact of breast cancer surgery on angiogenesis circulating biomarkers: A prospective longitudinal study. World J. Surg. Oncol. 2013, 11, 213. [Google Scholar] [CrossRef]
- Retsky, M.W.; Demicheli, R.; Gukas, I.D.; Hrushesky, W.J. Enhanced surgery-induced angiogenesis among premenopausal women might partially explain excess breast cancer mortality of blacks compared to whites: An hypothesis. Int. J. Surg. 2007, 5, 300–304. [Google Scholar] [CrossRef]
- Retsky, M.; Demicheli, R.; Hrushesky, W.J. Authors respond to controversy surrounding breast cancer study. Int. J. Surg. 2005, 3, 235–239. [Google Scholar] [CrossRef][Green Version]
- Norton, L. Tumor dormancy: Separating observations from experimental science. Nat. Clin. Pract. Oncol. 2007, 4, 671. [Google Scholar] [CrossRef] [PubMed]
- Shaashua, L.; Eckerling, A.; Israeli, B.; Yanovich, G.; Rosenne, E.; Fichman-Horn, S.; Ben Zvi, I.; Sorski, L.; Haldar, R.; Satchi-Fainaro, R.; et al. Spontaneous regression of micro-metastases following primary tumor excision: A critical role for primary tumor secretome. BMC Biol. 2020, 18, 163. [Google Scholar]
- Hanin, L.; Rose, J. Suppression of metastasis by primary tumor and acceleration of metastasis following primary tumor resection: A natural law? Bull. Math. Biol. 2018, 80, 519–539. [Google Scholar] [CrossRef]
- Hanin, L.; Pavlova, L. A quantitative insight into metastatic relapse of breast cancer. J. Theor. Biol. 2016, 394, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Forget, P.; Vandenhende, J.; Berliere, M.; Machiels, J.P.; Nussbaum, B.; Legrand, C.; DeKock, M. Do intraoperative analgesics influence breast cancer recurrence after mastectomy? A retrospective analysis. Anesth. Analg. 2010, 110, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Vatner, R.E.; Cooper, B.T.; Vanpouille-Box, C.; Demaria, S.; Formenti, S.C. Combinations of immunotherapy and radiation in cancer therapy. Front. Oncol. 2014, 4, 325. [Google Scholar] [CrossRef]
- Hiller, J.; Schier, R.; Riedel, B. Perioperative Biologic Perturbation and Cancer Surgery: Targeting the Adrenergic-Inflammatory Response and Microcirculatory Dysregulation. In Perioperative Inflammation as a Triggering Origin of Metastasis Development; Retsky, M., Demicheli, R., Eds.; Springer: New York, NY, USA, 2017; pp. 83–107. [Google Scholar]
- Haldar, R.; Ben-Eliyahu, S. Reducing the risk of post-surgical cancer recurrence: A perioperative anti-inflammatory anti-stress approach. Future Oncol. 2018, 14, 1017–1021. [Google Scholar] [CrossRef]
- Haldar, R.; Berger, L.S.; Rossenne, E.; Radin, A.; Eckerling, A.; Sandbank, E.; Sloan, E.K.; Cole, S.W.; Ben-Eliyahu, S. Perioperative escape from dormancy of spontaneous micro-metastases: A role for malignant secretion of IL-6, IL-8, and vegf, through adrenergic and prostaglandin signaling. Brain Behav. Immun. 2023, 109, 175–187. [Google Scholar] [CrossRef]
- Huang, C.; Chen, Y.; Liu, H.; Yang, J.; Song, X.; Zhao, J.; He, N.; Zhou, C.J.; Wang, Y.; Huang, C.; et al. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin e2 and down-regulating the wnt pathway activity. Oncotarget 2017, 8, 115254–115269. [Google Scholar] [CrossRef]
- Kim, S.; Lee, E.S.; Lee, E.J.; Jung, J.Y.; Lee, S.B.; Lee, H.J.; Kim, J.; Kim, H.J.; Lee, J.W.; Son, B.H.; et al. Targeted eicosanoids profiling reveals a prostaglandin reprogramming in breast cancer by microrna-155. J. Exp. Clin. Cancer Res. 2021, 40, 43. [Google Scholar] [CrossRef]
- Shaashua, L.; Shabat-Simon, M.; Haldar, R.; Matzner, P.; Zmora, O.; Shabtai, M.; Sharon, E.; Allweis, T.; Barshack, I.; Hayman, L.; et al. Perioperative Cox-2 and beta-adrenergic blockade improves metastatic biomarkers in breast cancer patients in a phase-ii randomized trial. Clin. Cancer Res. 2017, 23, 4651–4661. [Google Scholar] [CrossRef]
- Haldar, R.; Ricon-Becker, I.; Radin, A.; Gutman, M.; Cole, S.W.; Zmora, O.; Ben-Eliyahu, S. Perioperative cox2 and beta-adrenergic blockade improves biomarkers of tumor metastasis, immunity, and inflammation in colorectal cancer: A randomized controlled trial. Cancer 2020, 126, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Dillekas, H.; Transeth, M.; Pilskog, M.; Assmus, J.; Straume, O. Differences in metastatic patterns in relation to time between primary surgery and first relapse from breast cancer suggest synchronized growth of dormant micrometastases. Breast Cancer Res. Treat. 2014, 146, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Miarka, L.; Hauser, C.; Helm, O.; Holdhof, D.; Beckinger, S.; Egberts, J.H.; Gundlach, J.P.; Lenk, L.; Rahn, S.; Mikulits, W.; et al. The hepatic microenvironment and TRAIL-R2 impact outgrowth of liver metastases in pancreatic cancer after surgical resection. Cancers 2019, 11, 745. [Google Scholar] [CrossRef] [PubMed]
- Retsky, M.; Demicheli, R.; Hrushesky, W.J.; Forget, P.; De Kock, M.; Gukas, I.; Rogers, R.A.; Baum, M.; Sukhatme, V.; Vaidya, J.S. Reduction of breast cancer relapses with perioperative non-steroidal anti-inflammatory drugs: New findings and a review. Curr. Med. Chem. 2013, 20, 4163–4176. [Google Scholar] [CrossRef] [PubMed]
- de Pedro, M.; Baeza, S.; Escudero, M.T.; Dierssen-Sotos, T.; Gomez-Acebo, I.; Pollan, M.; Llorca, J. Effect of COX-2 inhibitors and other non-steroidal inflammatory drugs on breast cancer risk: A meta-analysis. Breast Cancer Res. Treat. 2015, 149, 525–536. [Google Scholar] [CrossRef]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol reduces cancer risk: A population-based cohort study. Medicine 2015, 94, e1097. [Google Scholar] [CrossRef]
- Ben-Eliyahu, S. Tumor excision as a metastatic Russian roulette: Perioperative interventions to improve long-term survival of cancer patients. Trends Cancer 2020, 6, 951–959. [Google Scholar] [CrossRef]
- Johnson, J.D.; Barnard, D.F.; Kulp, A.C.; Mehta, D.M. Neuroendocrine regulation of brain cytokines after psychological stress. J. Endocr. Soc. 2019, 3, 1302–1320. [Google Scholar] [CrossRef]
- Mach, D.B.; Rogers, S.D.; Sabino, M.C.; Luger, N.M.; Schwei, M.J.; Pomonis, J.D.; Keyser, C.P.; Clohisy, D.R.; Adams, D.J.; O’Leary, P.; et al. Origins of skeletal pain: Sensory and sympathetic innervation of the mouse femur. Neuroscience 2002, 113, 155–166. [Google Scholar] [CrossRef]
- Dygai, A.M.; Skurikhin, E.G. Monoaminergic regulation of hemopoiesis under extreme conditions. Bull. Exp. Biol. Med. 2011, 151, 171–178. [Google Scholar] [CrossRef]
- Yanagawa, Y.; Matsumoto, M.; Togashi, H. Adrenoceptor-mediated enhancement of interleukin-33 production by dendritic cells. Brain Behav. Immun. 2011, 25, 1427–1433. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Hsieh, S.C.; Liu, C.W.; Lu, C.S.; Wu, C.H.; Liao, H.T.; Chen, M.H.; Li, K.J.; Shen, C.Y.; Kuo, Y.M.; et al. Cross-talk among polymorphonuclear neutrophils, immune, and non-immune cells via released cytokines, granule proteins, microvesicles, and neutrophil extracellular trap formation: A novel concept of biology and pathobiology for neutrophils. Int. J. Mol. Sci. 2021, 22, 3119. [Google Scholar] [CrossRef]
- Jin, J.; Wang, X.; Wang, Q.; Guo, X.; Cao, J.; Zhang, X.; Zhu, T.; Zhang, D.; Wang, W.; Wang, J.; et al. Chronic psychological stress induces the accumulation of myeloid-derived suppressor cells in mice. PLoS ONE 2013, 8, e74497. [Google Scholar] [CrossRef] [PubMed]
- Hanns, P.; Paczulla, A.M.; Medinger, M.; Konantz, M.; Lengerke, C. Stress and catecholamines modulate the bone marrow microenvironment to promote tumorigenesis. Cell Stress 2019, 3, 221–235. [Google Scholar] [CrossRef]
- Allawi, Z.; Cuzick, J.; Baum, M.; ATAC/LATTE Investigators. Does trauma or an intercurrent surgical intervention lead to a short-term increase in breast cancer recurrence rates? Ann. Oncol. 2012, 23, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Foertsch, S.; Haffner-Luntzer, M.; Kroner, J.; Gross, F.; Kaiser, K.; Erber, M.; Reber, S.O.; Ignatius, A. Chronic psychosocial stress disturbs long-bone growth in adolescent mice. Dis. Model. Mech. 2017, 10, 1399–1409. [Google Scholar] [CrossRef]
- Bible, L.E.; Pasupuleti, L.V.; Gore, A.V.; Sifri, Z.C.; Kannan, K.B.; Mohr, A.M. Chronic restraint stress after injury and shock is associated with persistent anemia despite prolonged elevation in erythropoietin levels. J. Trauma Acute Care Surg. 2015, 79, 91–96, discussion 96–97. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.; Schajnovitz, A.; Lapid, K.; Kalinkovich, A.; Itkin, T.; Ludin, A.; Kao, W.M.; Battista, M.; Tesio, M.; Kollet, O.; et al. Rapid mobilization of hematopoietic progenitors by amd3100 and catecholamines is mediated by cxcr4-dependent sdf-1 release from bone marrow stromal cells. Leukemia 2011, 25, 1286–1296. [Google Scholar] [CrossRef]
- Saba, F.; Soleimani, M.; Atashi, A.; Mortaz, E.; Shahjahani, M.; Roshandel, E.; Jaseb, K.; Saki, N. The role of the nervous system in hematopoietic stem cell mobilization. Lab. Hematol. 2013, 19, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Decker, A.M.; Decker, J.T.; Jung, Y.; Cackowski, F.C.; Daignault-Newton, S.; Morgan, T.M.; Shea, L.D.; Taichman, R.S. Adrenergic blockade promotes maintenance of dormancy in prostate cancer through upregulation of gas6. Transl. Oncol. 2020, 13, 100781. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, N.; Zhong, L.; Wang, S.; Zheng, Y.; Yang, B.; Zhang, J.; Lin, Y.; Wang, Z. Prognostic value of depression and anxiety on breast cancer recurrence and mortality: A systematic review and meta-analysis of 282,203 patients. Mol. Psychiatry 2020, 25, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Krzeszinski, J.Y.; Wan, Y. New therapeutic targets for cancer bone metastasis. Trends Pharmacol. Sci. 2015, 36, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Damen, M.P.F.; van Rheenen, J.; Scheele, C.L.G.J. Targeting dormant tumor cells to prevent cancer recurrence. FEBS J. 2021, 288, 6286–6303. [Google Scholar] [CrossRef]
- Sauer, S.; Reed, D.R.; Ihnat, M.; Hurst, R.E.; Warshawsky, D.; Barkan, D. Innovative approaches in the battle against cancer recurrence: Novel strategies to combat dormant disseminated tumor cells. Front. Oncol. 2021, 11, 659963. [Google Scholar] [CrossRef]
- Dias, M.H.; Bernards, R. Playing cancer at its own game: Activating mitogenic signaling as a paradoxical intervention. Mol. Oncol. 2021, 15, 1975–1985. [Google Scholar] [CrossRef]
- Pajic, M.; Blatter, S.; Guyader, C.; Gonggrijp, M.; Kersbergen, A.; Kucukosmanoglu, A.; Sol, W.; Drost, R.; Jonkers, J.; Borst, P.; et al. Selected alkylating agents can overcome drug tolerance of g0-like tumor cells and eradicate brca1-deficient mammary tumors in mice. Clin. Cancer Res. 2017, 23, 7020–7033. [Google Scholar] [CrossRef]
- El Touny, L.H.; Vieira, A.; Mendoza, A.; Khanna, C.; Hoenerhoff, M.J.; Green, J.E. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Investig. 2014, 124, 156–168. [Google Scholar] [CrossRef]
- Cook, K.L.; Warri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in er+ breast cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef]
- Yang, H.; Yu, Z.; Ji, S.; Huo, Q.; Yan, J.; Gao, Y.; Niu, Y.; Xu, M.; Liu, Y. Targeting bone microenvironments for treatment and early detection of cancer bone metastatic niches. J. Control. Release 2022, 341, 443–456. [Google Scholar] [CrossRef]
- Sloan, E.K.; Pouliot, N.; Stanley, K.L.; Chia, J.; Moseley, J.M.; Hards, D.K.; Anderson, R.L. Tumor-specific expression of alphavbeta3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006, 8, R20. [Google Scholar] [CrossRef]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clement-Lacroix, P.; Clezardin, P. Tumor alphavbeta3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef] [PubMed]
- Pantano, F.; Croset, M.; Driouch, K.; Bednarz-Knoll, N.; Iuliani, M.; Ribelli, G.; Bonnelye, E.; Wikman, H.; Geraci, S.; Bonin, F.; et al. Integrin alpha5 in human breast cancer is a mediator of bone metastasis and a therapeutic target for the treatment of osteolytic lesions. Oncogene 2021, 40, 1284–1299. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Chambers, A.F. β1-integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. 2011, 17, 7219–7223. [Google Scholar] [CrossRef] [PubMed]
- Wieder, R. Insurgent micrometastases: Sleeper cells and harboring the enemy. J. Surg. Oncol. 2005, 89, 207–210. [Google Scholar] [CrossRef]
- Khalili, P.; Arakelian, A.; Chen, G.; Plunkett, M.L.; Beck, I.; Parry, G.C.; Donate, F.; Shaw, D.E.; Mazar, A.P.; Rabbani, S.A. A non-RGD-based integrin binding peptide (ATN-161) blocks breast cancer growth and metastasis in vivo. Mol. Cancer Ther. 2006, 5, 2271–2280. [Google Scholar] [CrossRef]
- Cianfrocca, M.E.; Kimmel, K.A.; Gallo, J.; Cardoso, T.; Brown, M.M.; Hudes, G.; Lewis, N.; Weiner, L.; Lam, G.N.; Brown, S.C.; et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSN-NH(2)), a beta integrin antagonist, in patients with solid tumours. Br. J. Cancer 2006, 94, 1621–1626. [Google Scholar] [CrossRef]
- Rack, B.; Juckstock, J.; Genss, E.M.; Schoberth, A.; Schindlbeck, C.; Strobl, B.; Heinrigs, M.; Rammel, G.; Zwingers, T.; Sommer, H.; et al. Effect of zoledronate on persisting isolated tumour cells in patients with early breast cancer. Anticancer. Res. 2010, 30, 1807–1813. [Google Scholar]
- ClinicalTrials.gov Identifier: NCT00127205. S0307 Phase III Trial of Bisphosphonates as Adjuvant Therapy for Primary Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00127205 (accessed on 2 July 2021).
- Zhang, H.; Xu, X.; Xu, R.; Ye, T. Drug repurposing of ivermectin abrogates neutrophil extracellular traps and prevents melanoma metastasis. Front. Oncol. 2022, 12, 989167. [Google Scholar] [CrossRef]
- Miller-Ocuin, J.L.; Liang, X.; Boone, B.A.; Doerfler, W.R.; Singhi, A.D.; Tang, D.; Kang, R.; Lotze, M.T.; Zeh, H.J. DNA released from neutrophil extracellular traps (nets) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology 2019, 8, e1605822. [Google Scholar] [CrossRef]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Bertrand, P. Inside HDACs with more selective hdac inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, T.; Wu, H.; Yang, Y.; Liu, N.; Chen, A.; Li, Q.; Li, J.; Qin, L.; Jiang, B.; et al. Design and optimization of novel hydroxamate-based histone deacetylase inhibitors of bis-substituted aromatic amides bearing potent activities against tumor growth and metastasis. J. Med. Chem. 2014, 57, 9357–9369. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, J.; Hu, Y.; Wang, J.; Yuan, C. Curcumin inhibits growth of human breast cancer cells through demethylation of DLC1 promoter. Mol. Cell. Biochem. 2017, 425, 47–58. [Google Scholar] [CrossRef]
- Khalil, B.D.; Sanchez, R.; Rahman, T.; Rodriguez-Tirado, C.; Moritsch, S.; Martinez, A.R.; Miles, B.; Farias, E.; Mezei, M.; Nobre, A.R.; et al. An NR2F1-specific agonist suppresses metastasis by inducing cancer cell dormancy. J. Exp. Med. 2022, 219, e20210836. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Zarei, M.; Manjili, S.H.; Guruli, G.; Wang, X.Y.; Manjili, M.H. Immunotherapy of cancer: Targeting cancer during active disease or during dormancy? Immunotherapy 2017, 9, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Ardavanis, A.; Litton, J.K.; Shumway, N.M.; Hale, D.F.; Murray, J.L.; Perez, S.A.; Ponniah, S.; Baxevanis, C.N.; Papamichail, M.; et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the Her2 peptide GP2 vaccine in breast cancer patients to prevent recurrence. Oncotarget 2016, 7, 66192–66201. [Google Scholar] [CrossRef]
- Brown, T.A.; Mittendorf, E.A.; Hale, D.F.; Myers, J.W.; Peace, K.M.; Jackson, D.O.; Greene, J.M.; Vreeland, T.J.; Clifton, G.T.; Ardavanis, A.; et al. Prospective, randomized, single-blinded, multi-center phase ii trial of two her2 peptide vaccines, GP2 and AE37, in breast cancer patients to prevent recurrence. Breast Cancer Res. Treat. 2020, 181, 391–401. [Google Scholar] [CrossRef]
- Payne, K.K.; Keim, R.C.; Graham, L.; Idowu, M.O.; Wan, W.; Wang, X.Y.; Toor, A.A.; Bear, H.D.; Manjili, M.H. Tumor-reactive immune cells protect against metastatic tumor and induce immunoediting of indolent but not quiescent tumor cells. J. Leukoc. Biol. 2016, 100, 625–635. [Google Scholar] [CrossRef]
- Zeng, J.; Xu, H.; Fan, P.Z.; Xie, J.; He, J.; Yu, J.; Gu, X.; Zhang, C.J. Kaempferol blocks neutrophil extracellular traps formation and reduces tumour metastasis by inhibiting ros-pad4 pathway. J. Cell. Mol. Med. 2020, 24, 7590–7599. [Google Scholar] [CrossRef]
- Matzner, P.; Sandbank, E.; Neeman, E.; Zmora, O.; Gottumukkala, V.; Ben-Eliyahu, S. Harnessing cancer immunotherapy during the unexploited immediate perioperative period. Nat. Rev. Clin. Oncol. 2020, 17, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Ricon, I.; Hanalis-Miller, T.; Haldar, R.; Jacoby, R.; Ben-Eliyahu, S. Perioperative biobehavioral interventions to prevent cancer recurrence through combined inhibition of beta-adrenergic and cyclooxygenase 2 signaling. Cancer 2019, 125, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Matzner, P.; Sorski, L.; Haldar, R.; Shaashua, L.; Benbenishty, A.; Lavon, H.; Azan, Y.; Sandbank, E.; Melamed, R.; Rosenne, E.; et al. Deleterious synergistic effects of distress and surgery on cancer metastasis: Abolishment through an integrated perioperative immune-stimulating stress-inflammatory-reducing intervention. Brain Behav. Immun. 2019, 80, 170–178. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov Identifier: NCT03496805. Effects of Muscadine Grape Extract in Men with Prostate Cancer on Androgen Deprivation Therapy. Last Update Posted: 3 November 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03496805 (accessed on 9 February 2023).
- ClinicalTrials.gov Identifier: NCT00516243. Defined Green Tea Catechin Extract in Treating Women with Hormone Receptor Negative Stage I–III Breast Cancer. Last Update Posted: 12 September 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT00516243 (accessed on 9 February 2023).
- ClinicalTrials.gov Identifier: NCT03572387. A Pilot Study of 5-AZA and ATRA for Prostate Cancer with PSA-only Recurrence After Local Treatment. Last Update Posted 3 August 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03572387 (accessed on 8 February 2023).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieder, R. Awakening of Dormant Breast Cancer Cells in the Bone Marrow. Cancers 2023, 15, 3021. https://doi.org/10.3390/cancers15113021
Wieder R. Awakening of Dormant Breast Cancer Cells in the Bone Marrow. Cancers. 2023; 15(11):3021. https://doi.org/10.3390/cancers15113021
Chicago/Turabian StyleWieder, Robert. 2023. "Awakening of Dormant Breast Cancer Cells in the Bone Marrow" Cancers 15, no. 11: 3021. https://doi.org/10.3390/cancers15113021
APA StyleWieder, R. (2023). Awakening of Dormant Breast Cancer Cells in the Bone Marrow. Cancers, 15(11), 3021. https://doi.org/10.3390/cancers15113021