
Neprilysin Inhibition in the Prevention of Anthracycline-Induced Cardiotoxicity

Abstract
Simple Summary
Abstract
1. Introduction
2. AIC Characteristics
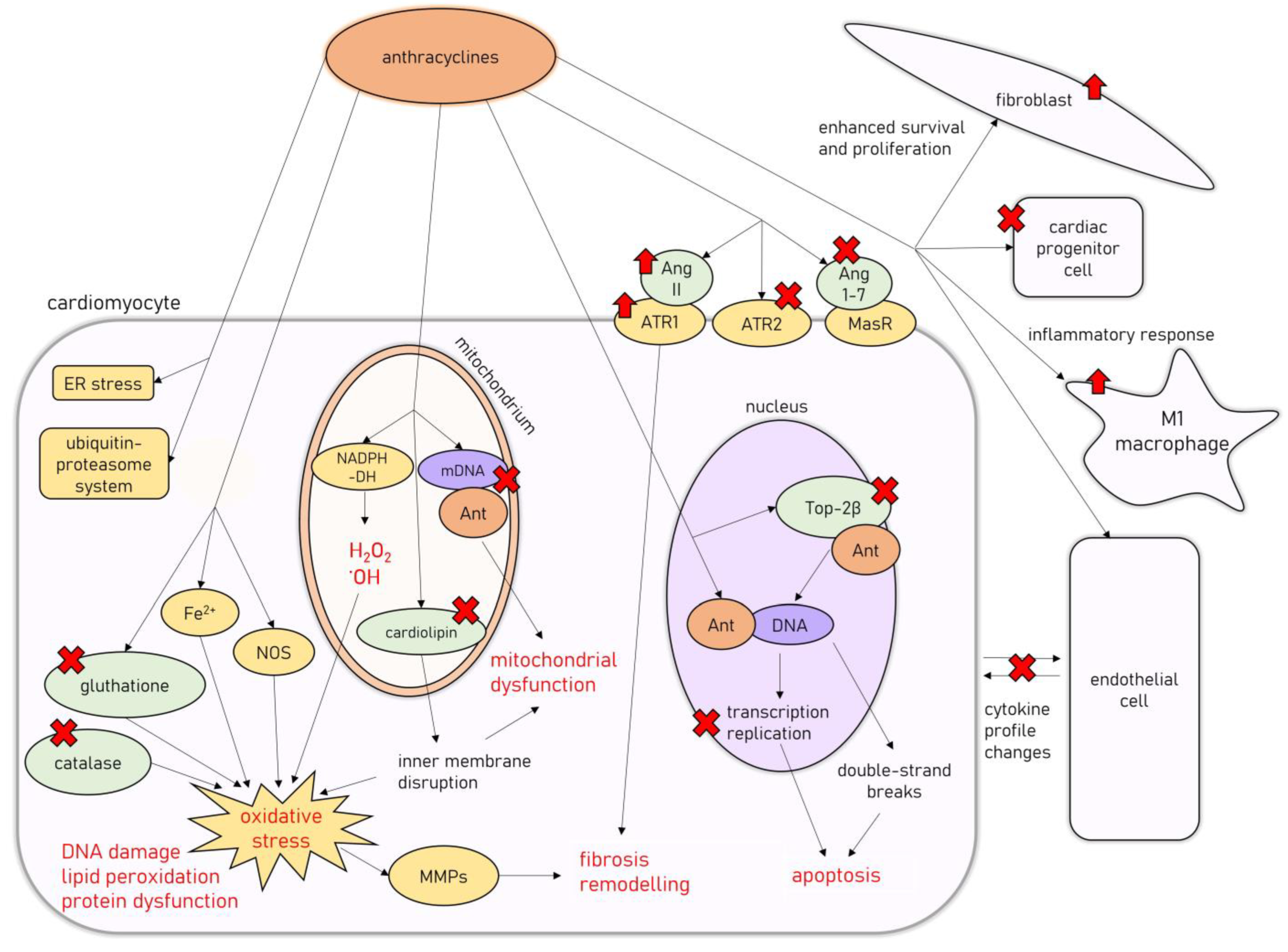
3. Molecular Mechanisms of AIC
4. Prevention of AIC
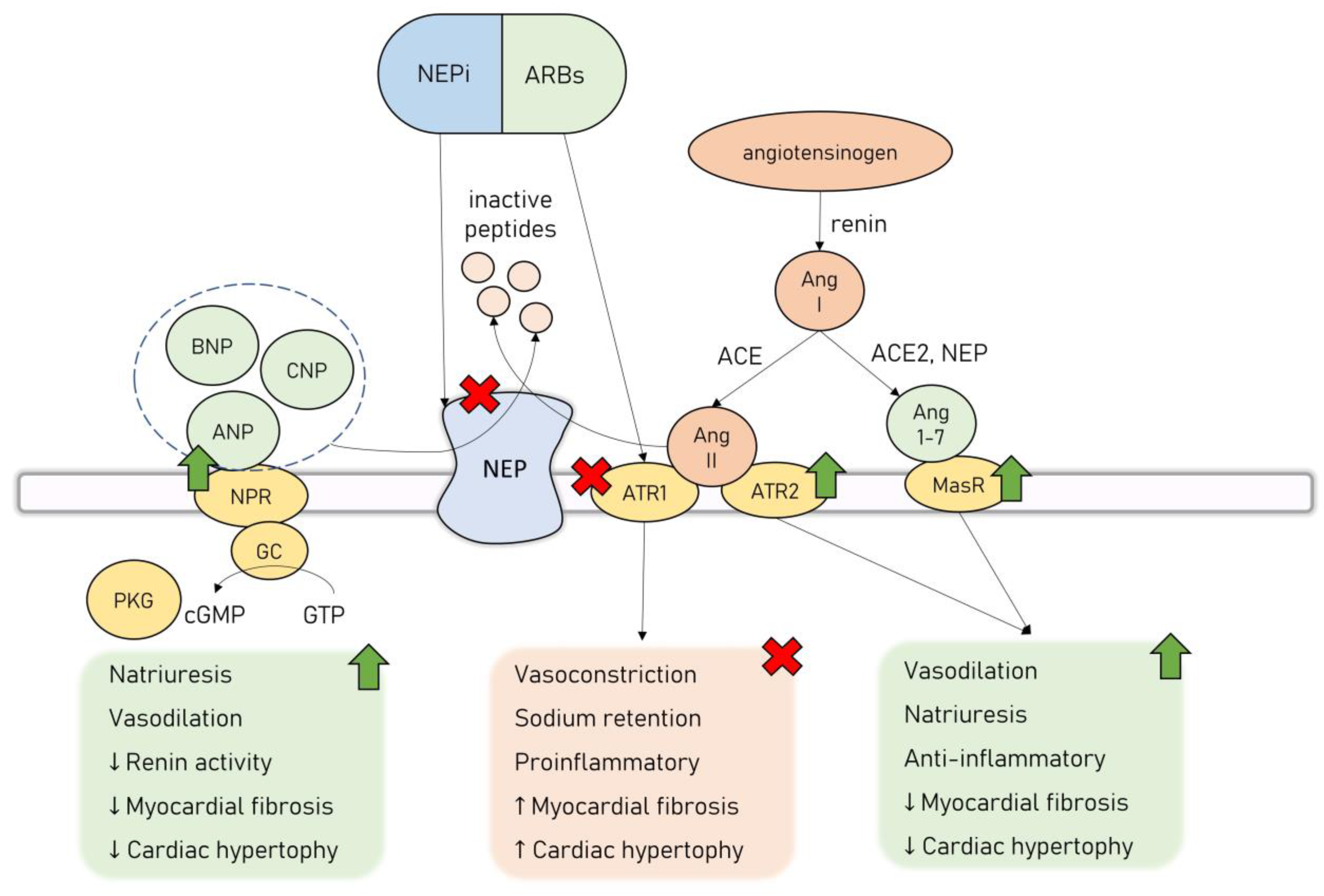
5. Neprilysin Inhibition in HF
6. ARNi in Preclinical Models of Myocardial Infarction
7. ARNi in Preclinical Models of Other CVDs
8. ARNi in Preclinical Models of AIC
9. ARNi in Human Studies on AIC
10. Summary and Future Perspectives
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tan, C.; Tasaka, H.; Yu, K.-P.; Murphy, M.L.; Karnofsky, D.A. Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease.Clinical evaluation with special reference to childhood leukemia. Cancer 1967, 20, 333–353. [Google Scholar] [CrossRef] [PubMed]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.-B.; Ewer, M.; et al. Prevention and Monitoring of Cardiac Dysfunction in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Lenihan, D.; Fradley, M.; Ganatra, S.; Barac, A.; Blaes, A.; Herrmann, J.; Porter, C.; Lyon, A.R.; Lancellotti, P.; et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann. Oncol. 2020, 31, 171–190. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Karnik, R.; Sambatakos, P.; Franco, V.I.; Ross, S.W.; Miller, T.L. Anthracycline-related cardiotoxicity in childhood cancer survivors. Curr. Opin. Cardiol. 2014, 29, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef]
- Sobczuk, P.; Czerwińska, M.; Kleibert, M.; Cudnoch-Jędrzejewska, A. Anthracycline-Induced Cardiotoxicity and Renin-Angiotensin-Aldosterone System-from Molecular Mechanisms to Therapeutic Applications. Heart Fail. Rev. 2022, 27, 295–319. [Google Scholar] [CrossRef]
- Ma, Y.; Bai, F.; Qin, F.; Li, J.; Liu, N.; Li, D.; Li, T.; Xie, H.; Liu, D.; Zhou, S.; et al. Beta-blockers for the primary prevention of anthracycline-induced cardiotoxicity: A meta-analysis of randomized controlled trials. BMC Pharmacol. Toxicol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Mcmurray, J.J.V.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. PARADIGM-HF Investigators and Committees. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Gregorietti, V.; Fernandez, T.L.; Costa, D.; Chahla, E.O.; Daniele, A.J. Use of Sacubitril/valsartan in patients with cardio toxicity and heart failure due to chemotherapy. Cardiooncology 2020, 6, 24. [Google Scholar] [CrossRef]
- Perez, I.E.; Alam, S.T.; Hernandez, G.A.; Sancassani, R. Cancer Therapy-Related Cardiac Dysfunction: An Overview for the Clinician. Clin. Med. Insights Cardiol. 2019, 13, 1179546819866445. [Google Scholar] [CrossRef]
- Herrmann, J.; Lenihan, D.; Armenian, S.; Barac, A.; Blaes, A.; Cardinale, D.; Carver, J.; Dent, S.; Ky, B.; Lyon, A.R.; et al. Defining cardiovascular toxicities of cancer therapies: An International Cardio-Oncology Society (IC-OS) consensus statement. Eur. Heart J. 2021, 43, 280–299. [Google Scholar] [CrossRef]
- Lyon, A.R.; López-Fernández, T.; Couch, L.; Asteggiano, R.; Aznar, M.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 Esc Guidelines on Cardio-Oncology Developed in Collaboration with the European Hematology Association (Eha), the European Society for Therapeutic Radiology and Oncology (Estro) and the International Cardio-Oncology Society (Ic-Os): Developed by the Task Force on Cardio-Oncology of the European Society of Cardiology (Esc). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [PubMed]
- Al-Malky, H.S.; Al Harthi, S.E.; Osman, A.M. Major Obstacles to Doxorubicin Therapy: Cardiotoxicity and Drug Resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.S.; Cohen, A.J.; Wasserman, A.G.; Cohen, P.; Ross, A.M. Acute Arrhythmogenicity of Doxorubicin Administration. Cancer 1987, 60, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Groarke, J.D.; Nohria, A. Anthracycline Cardiotoxicity. Circulation 2015, 131, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L., Jr.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk Factors for Doxorubicin-Induced Congestive Heart Failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive Heart Failure in Patients Treated with Doxorubicin: A Retrospective Analysis of Three Trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Armstrong, G.T.; Plana, J.C.; Zhang, N.; Srivastava, D.; Green, D.M.; Ness, K.K.; Donovan, F.D.; Metzger, M.L.; Arevalo, A.; Durand, J.-B.; et al. Screening Adult Survivors of Childhood Cancer for Cardiomyopathy: Comparison of Echocardiography and Cardiac Magnetic Resonance Imaging. J. Clin. Oncol. 2012, 30, 2876–2884. [Google Scholar] [CrossRef]
- Mancilla, T.R.; Iskra, B.; Aune, G.J. Doxorubicin-Induced Cardiomyopathy in Children. Compr. Physiol. 2019, 9, 905–931. [Google Scholar]
- Mulrooney, D.A.; Yeazel, M.W.; Leisenring, W.M.; Kawashima, T.; Mertens, A.C.; Mitby, P.; Stovall, M.; Donaldson, S.S.; Green, D.M.; Sklar, C.A.; et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: Retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ 2009, 339, b4606. [Google Scholar] [CrossRef]
- Buzdar, A.U.; Marcus, C.; Blumenschein, G.R.; Smith, T.L. Early and delayed clinical cardiotoxicity of doxorubicin. Cancer 1985, 55, 2761–2765. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Dainiak, N.; Berger, H.J.; Goldman, L.; Johnstone, D.; Reduto, L.; Duffy, T.; Schwartz, P.; Gottschalk, A.; Zaret, B.L.; et al. Serial Assessment of Doxorubicin Cardiotoxicity with Quantitative Radionuclide Angiocardiography. N. Engl. J. Med. 1979, 300, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin Cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Cove-Smith, L.; Woodhouse, N.; Hargreaves, A.; Kirk, J.; Smith, S.; Price, S.; Galvin, M.; Betts, C.J.; Brocklehurst, S.; Backen, A.; et al. An Integrated Characterization of Serological, Pathological, and Functional Events in Doxorubicin-Induced Cardiotoxicity. Toxicol. Sci. 2014, 140, 3–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pontes, J.C.D.V.; Júnior, J.F.G.; Da Silva, G.V.R.; Benfatti, R.A.; Dias, A.E.M.S.; Duarte, J.J.; Gardenal, N.; Odashiro, M.; Dos Santos, C.H.M. Anatomopathological study of cardiomyopathy induced by doxorubicin in rats. Acta Cir. Bras. 2010, 25, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Manish; Shandilya; Shrutika, S.; Prasad, D.P.; Sonika, C. Molecular-Level Understanding of the Anticancer Action Mechanism of Anthracyclines. In Advances in Precision Medicine Oncology; Hilal, A., Rasool, H.B.A., Eds.; IntechOpen: Rijeka, Yugoslavia, 2020. [Google Scholar]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Capranico, G.; Tinelli, S.; Austin, C.A.; Fisher, M.L.; Zunino, F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 1992, 1132, 43–48. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the Molecular Basis of Doxorubicin-Induced Cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Polegato, B.F.; Minicucci, M.F.; Azevedo, P.S.; Carvalho, R.F.; Chiuso-Minicucci, F.; Pereira, E.J.; Paiva, S.A.; Zornoff, L.A.; Okoshi, M.P.; Matsubara, B.B.; et al. Acute Doxorubicin-Induced Cardiotoxicity Is Associated with Matrix Metalloproteinase-2 Alterations in Rats. Cell Physiol. Biochem. 2015, 35, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Ivanová, M.; Dovinová, I.; Okruhlicová, L.; Tribulová, N.; Šimončíková, P.; Barte-ková, M.; Vlkovičová, J.; Barančík, M. Chronic Cardiotoxicity of Doxorubicin Involves Activation of Myocardial and Circulating Matrix Metalloproteinases in Rats. Acta Pharmacol. Sin. 2012, 33, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Osataphan, N.; Phrommintikul, A.; Chattipakorn, S.C.; Chattipakorn, N. Effects of doxorubicin-induced cardiotoxicity on cardiac mitochondrial dynamics and mitochondrial function: Insights for future interventions. J. Cell. Mol. Med. 2020, 24, 6534–6557. [Google Scholar] [CrossRef]
- Catanzaro, M.P.; Weiner, A.; Kaminaris, A.; Li, C.; Cai, F.; Zhao, F.; Kobayashi, S.; Kobayashi, T.; Huang, Y.; Sesaki, H.; et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. 2019, 33, 11096–11108. [Google Scholar] [CrossRef]
- Bagchi, A.K.; Malik, A.; Akolkar, G.; Zimmer, A.; Belló-Klein, A.; De Angelis, K.; Jassal, D.S.; Fini, M.A.; Stenmark, K.R.; Singal, P.K. Study of Er Stress and Apoptotic Proteins in the Heart and Tumor Exposed to Doxorubicin. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119039. [Google Scholar] [CrossRef]
- Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. The effects of doxorubicin on cardiac calcium homeostasis and contractile function. J. Cardiol. 2022, 80, 125–132. [Google Scholar] [CrossRef]
- Ranek, M.J.; Wang, X. Activation of the ubiquitin-proteasome system in doxorubicin cardiomyopathy. Curr. Hypertens. Rep. 2009, 11, 389–395. [Google Scholar] [CrossRef]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef]
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am. J. Physiol. Physiol. 2020, 318, C380–C391. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, A.; Piegari, E.; Cappetta, D.; Marino, L.; Filippelli, A.; Berrino, L.; Ferreira-Martins, J.; Zheng, H.; Hosoda, T.; Rota, M.; et al. Anthracycline Cardiomyopathy Is Mediated by Depletion of the Cardiac Stem Cell Pool and Is Rescued by Restoration of Progenitor Cell Function. Circulation 2010, 121, 276–292. [Google Scholar] [CrossRef]
- Luu, A.Z.; Chowdhury, B.; Al-Omran, M.; Teoh, H.; Hess, D.A.; Verma, S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl. Sci. 2018, 3, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; Soto-Pantoja, D.R.; Bi, J.; Hundley, W.G.; Widiapradja, A.; Manteufel, E.J.; Bradshaw, T.W.; Meléndez, G.C. Doxorubicin-Induced Myocardial Fibrosis Involves the Neurokinin-1 Receptor and Direct Effects on Cardiac Fibroblasts. Heart Lung Circ. 2019, 28, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Ma, W.; Li, X.; Jiang, C.; Sun, T.; Li, Y.; Zhang, B.; Li, W. Involvement of ROS/NLRP3 Inflammasome Signaling Pathway in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Toxicol. 2020, 20, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Reis-Mendes, A.; Padrão, A.I.; Duarte, J.A.; Gonçalves-Monteiro, S.; Duarte-Araújo, M.; Remião, F.; Carvalho, F.; Sousa, E.; Bastos, M.L.; Costa, V.M. Role of Inflammation and Redox Status on Doxorubicin-Induced Cardiotoxicity in Infant and Adult Cd-1 Male Mice. Biomolecules 2021, 11, 1725. [Google Scholar] [CrossRef]
- Bagchi, A.K.; Malik, A.; Akolkar, G.; Jassal, D.S.; Singal, P.K. Endoplasmic Reticulum Stress Promotes Inos/No and Influences Inflammation in the Development of Doxorubicin-Induced Cardiomyopathy. Antioxidants 2021, 10, 1897. [Google Scholar] [CrossRef]
- Nelu-Mihai, T.; Bordejevic, A.D.; Tomescu, M.C.; Petrescu, L.; Crisan, S.; Geavlete, O.; Mischie, A.; Onel, A.F.M.; Sasu, A.; Pop-Moldovan, A.L. Toll-Like Receptor 4 (Tlr4) Expression Is Correlated with T2* Iron Deposition in Response to Doxorubicin Treatment: Cardiotoxicity Risk Assessment. Sci. Rep. 2020, 10, 17013. [Google Scholar]
- Ye, S.; Su, L.; Shan, P.; Ye, B.; Wu, S.; Liang, G.; Huang, W. Lcz696 Attenuated Doxorubicin-Induced Chronic Cardiomyopathy through the Tlr2-Myd88 Complex Formation. Front. Cell Dev. Biol. 2021, 9, 654051. [Google Scholar] [CrossRef]
- Su, Q.; Li, L.; Sun, Y.; Yang, H.; Ye, Z.; Zhao, J. Effects of the Tlr4/Myd88/Nf-Κb Signaling Pathway on Nlrp3 Inflammasome in Coronary Microembolization-Induced Myocardial Injury. Cell Physiol. Biochem. 2018, 47, 1497–1508. [Google Scholar] [CrossRef]
- Chen, F.; Chen, Z.-Q.; Zhong, G.-L.; Zhu, J.-J. Nicorandil Inhibits Tlr4/Myd88/Nf-Κb/Nlrp3 Signaling Pathway to Reduce Pyroptosis in Rats with Myocardial Infarction. Exp. Biol. Med. 2021, 246, 1938–1947. [Google Scholar] [CrossRef]
- Sauter, K.; Wood, L.; Wong, J.; Iordanov, M.; Magun, B.E. Doxorubicin and daunorubicin induce processing and release of interleukin-1β through activation of the NLRP3 inflammasome. Cancer Biol. Ther. 2011, 11, 1008–1016. [Google Scholar] [CrossRef]
- Arruda, F.D.S.; Tomé, F.D.; Miguel, M.P.; De Menezes, L.B.; Nagib, P.R.A.; Campos, E.C.; Soave, D.F.; Celes, M.R.N. Doxorubicin-induced Cardiotoxicity and Cardioprotective Agents: Classic and New Players in the Game. Curr. Pharm. Des. 2019, 25, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Visseren, F.L.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 Esc Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Adams, M.J.; Ganatra, S.; Colan, S.D.; Aggarwal, S.; Steiner, R.; Amdani, S.; Lipshultz, E.R.; Lipshultz, S.E. Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardiooncology 2019, 5, 18. [Google Scholar] [CrossRef]
- Lyu, Y.L.; Kerrigan, J.E.; Lin, C.-P.; Azarova, A.M.; Tsai, Y.-C.; Ban, Y.; Liu, L.F. Topoisomerase IIβ–Mediated DNA Double-Strand Breaks: Implications in Doxorubicin Cardiotoxicity and Prevention by Dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Herman, E.H. Dexrazoxane: How It Works in Cardiac and Tumor Cells. Is It a Prodrug or Is It a Drug? Cardiovasc. Toxicol. 2007, 7, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Kalam, K.; Marwick, T.H. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: A systematic review and meta-analysis. Eur. J. Cancer 2013, 49, 2900–2909. [Google Scholar] [CrossRef]
- van Dalen, E.C.; Caron, H.N.; Dickinson, H.O.; Kremer, L.C. Cardioprotective Interventions for Cancer Patients Receiving Anthracyclines. Cochrane Database Syst. Rev. 2008, 2, Cd003917. [Google Scholar]
- Smith, L.A.; Cornelius, V.R.; Plummer, C.J.; Levitt, G.; Verrill, M.; Canney, P.; Jones, A. Cardiotoxicity of anthracycline agents for the treatment of cancer: Systematic review and meta-analysis of randomised controlled trials. BMC Cancer 2010, 10, 337. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Gerber, M.C.; Weisberg, S.; York, M.; Spicer, D.; Jones, S.E.; Wadler, S.; Desai, A.; Vogel, C.; et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J. Clin. Oncol. 1997, 15, 1318–1332. [Google Scholar] [CrossRef]
- Macedo, A.V.; Hajjar, L.A.; Lyon, A.R.; Nascimento, B.R.; Putzu, A.; Rossi, L.; Costa, R.B.; Landoni, G.; Nogueira-Rodrigues, A.; Ribeiro, A.L. Efficacy of Dexrazoxane in Preventing Anthracycline Cardiotoxicity in Breast Cancer. JACC Cardiooncol. 2019, 1, 68–79. [Google Scholar] [CrossRef]
- Swain, S.M.; Vici, P. The current and future role of dexrazoxane as a cardioprotectant in anthracycline treatment: Expert panel review. J. Cancer Res. Clin. Oncol. 2004, 130, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van Tine, B.A.; Hirbe, A.C.; Oppelt, P.; Frith, A.E.; Rathore, R.; Mitchell, J.D.; Wan, F.; Berry, S.; Landeau, M.; Heberton, G.A.; et al. Interim Analysis of the Phase Ii Study: Noninferiority Study of Doxorubicin with Upfront Dexrazoxane Plus Olaratumab for Advanced or Metastatic Soft-Tissue Sarcoma. Clin. Cancer Res. 2021, 27, 3854–3860. [Google Scholar] [CrossRef] [PubMed]
- Gulati, G.; Heck, L.S.; Ree, A.H.; Hoffmann, P.; Schulz-Menger, J.; Fagerland, M.W.; Gravdehaug, B.; von Knobelsdorff-Brenkenhoff, F.; Bratland, Å.; Storås, T.H.; et al. Prevention of Cardiac Dysfunction During Adjuvant Breast Cancer Therapy (Prada): A 2 × 2 Factorial, Randomized, Placebo-Controlled, Double-Blind Clinical Trial of Candesartan and Metoprolol. Eur. Heart J. 2016, 37, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Bosch, X.; Rovira, M.; Sitges, M.; Domènech, A.; Ortiz-Pérez, J.T.; de Caralt, T.M.; Morales-Ruiz, M.; Perea, R.J.; Monzó, M.; Esteve, J. Enalapril and Carvedilol for Preventing Chemotherapy-Induced Left Ventricular Systolic Dysfunction in Patients with Malignant Hemopathies: The Overcome Trial (Prevention of Left Ventricular Dysfunction with Enalapril and Carvedilol in Patients Submitted to Intensive Chemotherapy for the Treatment of Malignant Hemopathies). J. Am. Coll. Cardiol. 2013, 61, 2355–2362. [Google Scholar] [PubMed]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-Induced Cardiotoxicity: An Update on the Molecular Mechanism and Novel Therapeutic Strategies for Effective Management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Quagliariello, V.; Vecchione, R.; Coppola, C.; Di Cicco, C.; De Capua, A.; Piscopo, G.; Paciello, R.; Narciso, V.; Formisano, C.; Taglialatela-Scafati, O.; et al. Cardioprotective Effects of Nanoemulsions Loaded with Anti-Inflammatory Nutraceuticals against Doxorubicin-Induced Cardiotoxicity. Nutrients 2018, 10, 1304. [Google Scholar] [CrossRef]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The Sglt-2 Inhibitor Empagliflozin Improves Myocardial Strain, Reduces Cardiac Fibrosis and Pro-Inflammatory Cytokines in Non-Diabetic Mice Treated with Doxorubicin. Cardiovasc. Diabetol. 2021, 20, 150. [Google Scholar] [CrossRef]
- Feygina, E.E.; Katrukha, A.G.; Semenov, A.G. Neutral Endopeptidase (Neprilysin) in Therapy and Diagnostics: Yin and Yang. Biochemistry 2019, 84, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, E.; Iacovoni, A.; Vaduganathan, M.; Lorini, F.L.; Perlini, S.; Senni, M. Neprilysin inhibition in heart failure: Mechanisms and substrates beyond modulating natriuretic peptides. Eur. J. Heart Fail. 2017, 19, 710–717. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Zhuravin, I.A.; Turner, A.J. Neprilysin Expression and Functions in Development, Ageing and Disease. Mech. Ageing Dev. 2020, 192, 111363. [Google Scholar] [CrossRef]
- Arrigo, M.; Vodovar, N.; Nougue, H.; Sadoune, M.; Pemberton, C.J.; Ballan, P.; Ludes, P.O.; Gendron, N.; Carpentier, A.; Cholley, B.; et al. The Heart Regulates the Endocrine Response to Heart Failure: Cardiac Contribution to Circulating Neprilysin. Eur. Heart J. 2018, 39, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Bayés-Genís, A.; Barallat, J.; Galán, A.; de Antonio, M.; Domingo, M.; Zamora, E.; Urrutia, A.; Lupón, J. Soluble Neprilysin Is Predictive of Cardiovascular Death and Heart Failure Hospitalization in Heart Failure Patients. J. Am. Coll. Cardiol. 2015, 65, 657–665. [Google Scholar] [CrossRef] [PubMed]
- McKinnie, S.M.K.; Fischer, C.; Tran, K.M.H.; Wang, W.; Mosquera, F.; Oudit, G.Y.; Vederas, J.C. The Metalloprotease Neprilysin Degrades and Inactivates Apelin Peptides. Chembiochem 2016, 17, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Meems, L.M.; Burnett, J.C. Innovative Therapeutics: Designer Natriuretic Peptides. JACC Basic Transl. Sci. 2016, 1, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.A. The natriuretic peptide system: Kidney and cardiovascular effects. Curr. Opin. Nephrol. Hypertens. 2006, 15, 14–21. [Google Scholar] [CrossRef]
- Volpe, M.; Carnovali, M.; Mastromarino, V. The natriuretic peptides system in the pathophysiology of heart failure: From molecular basis to treatment. Clin. Sci. 2016, 130, 57–77. [Google Scholar] [CrossRef]
- Curry, F.R. Atrial Natriuretic Peptide: An Essential Physiological Regulator of Transvascular Fluid, Protein Transport, and Plasma Volume. J. Clin. Investig. 2005, 115, 1458–1461. [Google Scholar] [CrossRef]
- Okamoto, R.; Ali, Y.; Hashizume, R.; Suzuki, N.; Ito, M. BNP as a Major Player in the Heart-Kidney Connection. Int. J. Mol. Sci. 2019, 20, 3581. [Google Scholar] [CrossRef]
- Kerkelä, R.; Ulvila, J.; Magga, J. Natriuretic Peptides in the Regulation of Cardiovascular Physiology and Metabolic Events. J. Am. Heart Assoc. 2015, 4, e002423. [Google Scholar] [CrossRef]
- Chappell, M.C. The Angiotensin-(1-7) Axis: Formation and Metabolism Pathways. In Angiotensin-(1-7); Springer: Cham, Switzerland, 2019; pp. 1–26. [Google Scholar] [CrossRef]
- Lang, C.C.; Motwani, J.; Coutie, W.J.; Struthers, A.D. Influence of candoxatril on plasma brain natriuretic peptide in heart failure. Lancet 1991, 338, 255. [Google Scholar] [CrossRef]
- Cleland, J.G.; Swedberg, K. Lack of Efficacy of Neutral Endopeptidase Inhibitor Ecadotril in Heart Failure. The International Ecadotril Multi-Centre Dose-Ranging Study Investigators. Lancet 1998, 351, 1657–1658. [Google Scholar] [CrossRef]
- Packer, M.; McMurray, J.J.V. Importance of endogenous compensatory vasoactive peptides in broadening the effects of inhibitors of the renin-angiotensin system for the treatment of heart failure. Lancet 2016, 389, 1831–1840. [Google Scholar] [CrossRef]
- Solomon Scott, D.; McMurray, J.J.; Anand, I.S.; Ge, J.; Lam, C.S.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- D’Adamio, L.; Shipp, M.A.; Masteller, E.L.; Reinherz, E.L. Organization of the Gene Encoding Common Acute Lymphoblastic Leukemia Antigen (Neutral Endopeptidase 24.11): Multiple Miniexons and Separate 5′ Untranslated Regions. Proc. Natl. Acad. Sci. USA 1989, 86, 7103–7107. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Myocardial ischaemia–reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Frantz, S.; Hundertmark, M.J.; Schulz-Menger, J.; Bengel, F.M.; Bauersachs, J. Left Ventricular Remodelling Post-Myocardial Infarction: Pathophysiology, Imaging, and Novel Therapies. Eur. Heart J. 2022, 43, 2549–2561. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-C.; Lin, S.-F.; Chu, Y.; Wo, H.-T.; Lee, H.-L.; Huang, Y.-C.; Wen, M.-S.; Chou, C.-C. LCZ696 Therapy Reduces Ventricular Tachyarrhythmia Inducibility in a Myocardial Infarction-Induced Heart Failure Rat Model. Cardiovasc. Ther. 2019, 2019, 6032631. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-C.; Wo, H.-T.; Lee, H.-L.; Lin, S.-F.; Chu, Y.; Wen, M.-S.; Chou, C.-C. Sacubitril/Valsartan Therapy Ameliorates Ventricular Tachyarrhythmia Inducibility in a Rabbit Myocardial Infarction Model. J. Card. Fail. 2020, 26, 527–537. [Google Scholar] [CrossRef]
- Ishii, M.; Kaikita, K.; Sato, K.; Sueta, D.; Fujisue, K.; Arima, Y.; Oimatsu, Y.; Mitsuse, T.; Onoue, Y.; Araki, S.; et al. Cardioprotective Effects of LCZ696 (Sacubitril/Valsartan) After Experimental Acute Myocardial Infarction. JACC Basic Transl. Sci. 2017, 2, 655–668. [Google Scholar] [CrossRef]
- Kompa, A.R.; Lu, J.; Weller, T.J.; Kelly, D.J.; Krum, H.; von Lueder, T.G.; Wang, B.H. Angiotensin receptor neprilysin inhibition provides superior cardioprotection compared to angiotensin converting enzyme inhibition after experimental myocardial infarction. Int. J. Cardiol. 2018, 258, 192–198. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, Y.; Li, J.; Chen, M.; Chen, A.; Yang, D.; Guan, X.; Cao, Y. Combination of LCZ696 and ACEI further improves heart failure and myocardial fibrosis after acute myocardial infarction in mice. Biomed. Pharmacother. 2021, 133, 110824. [Google Scholar] [CrossRef]
- Pfau, D.; Thorn, S.L.; Zhang, J.; Mikush, N.; Renaud, J.M.; Klein, R.; Dekemp, R.A.; Wu, X.; Hu, X.; Sinusas, A.J.; et al. Angiotensin Receptor Neprilysin Inhibitor Attenuates Myocardial Remodeling and Improves Infarct Perfusion in Experimental Heart Failure. Sci. Rep. 2019, 9, 5791. [Google Scholar] [CrossRef]
- Raj, P.; Sayfee, K.; Parikh, M.; Yu, L.; Wigle, J.; Netticadan, T.; Zieroth, S. Comparative and Combinatorial Effects of Resveratrol and Sacubitril/Valsartan alongside Valsartan on Cardiac Remodeling and Dysfunction in MI-Induced Rats. Molecules 2021, 26, 5006. [Google Scholar] [CrossRef]
- Shen, J.; Fan, Z.; Sun, G.; Qi, G. Sacubitril/Valsartan (Lcz696) Reduces Myocardial Injury Following Myocardial Infarction by Inhibiting Nlrp3-Induced Pyroptosis Via the Tak1/Jnk Signaling Pathway. Mol. Med. Rep. 2021, 24, 676. [Google Scholar] [CrossRef]
- Suematsu, Y.; Miura, S.-I.; Goto, M.; Matsuo, Y.; Arimura, T.; Kuwano, T.; Imaizumi, S.; Iwata, A.; Yahiro, E.; Saku, K. LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur. J. Heart Fail. 2016, 18, 386–393. [Google Scholar] [CrossRef]
- Torrado, J.; Cain, C.; Mauro, A.G.; Romeo, F.; Ockaili, R.; Chau, V.Q.; Nestler, J.A.; Devarakonda, T.; Ghosh, S.; Das, A.; et al. Sacubitril/Valsartan Averts Adverse Post-Infarction Ventricular Remodeling and Preserves Systolic Function in Rabbits. J. Am. Coll. Cardiol. 2018, 72, 2342–2356. [Google Scholar] [CrossRef]
- Trivedi, R.K.; Polhemus, D.J.; Li, Z.; Yoo, D.; Koiwaya, H.; Scarborough, A.; Goodchild, T.T.; Lefer, D.J. Combined Angiotensin Receptor-Neprilysin Inhibitors Improve Cardiac and Vascular Function Via Increased No Bioavailability in Heart Failure. J. Am. Heart Assoc. 2018, 7, e008268. [Google Scholar] [CrossRef]
- Vaskova, E.; Ikeda, G.; Tada, Y.; Wahlquist, C.; Mercola, M.; Yang, P.C. Sacubitril/Valsartan Improves Cardiac Function and Decreases Myocardial Fibrosis Via Downregulation of Exosomal miR-181a in a Rodent Chronic Myocardial Infarction Model. J. Am. Heart Assoc. 2020, 9, e015640. [Google Scholar] [CrossRef]
- von Lueder, T.G.; Wang, B.H.; Kompa, A.R.; Huang, L.; Webb, R.; Jordaan, P.; Atar, D.; Krum, H. Angiotensin Receptor Neprilysin Inhibitor LCZ696 Attenuates Cardiac Remodeling and Dysfunction After Myocardial Infarction by Reducing Cardiac Fibrosis and Hypertrophy. Circ. Heart Fail. 2015, 8, 71–78. [Google Scholar] [CrossRef]
- Aroor, A.R.; Mummidi, S.; Lopez-Alvarenga, J.C.; Das, N.; Habibi, J.; Jia, G.; Lastra, G.; Chandrasekar, B.; DeMarco, V.G. Sacubitril/Valsartan Inhibits Obesity-Associated Diastolic Dysfunction through Suppression of Ventricular-Vascular Stiffness. Cardiovasc. Diabetol. 2021, 20, 80. [Google Scholar] [CrossRef]
- Burke, R.M.; Lighthouse, J.K.; Mickelsen, D.M.; Small, E.M. Sacubitril/Valsartan Decreases Cardiac Fibrosis in Left Ventricle Pressure Overload by Restoring PKG Signaling in Cardiac Fibroblasts. Circ. Heart Fail. 2019, 12, e005565. [Google Scholar] [CrossRef]
- Hamano, G.; Yamamoto, K.; Takami, Y.; Takeshita, H.; Shimosato, T.; Moritani, T.; Rakugi, H. Effects of Low-Dose Sacubitril/Valsartan on Different Stages of Cardiac Hypertrophy in Salt-Loaded Hypertensive Rats. J. Cardiovasc. Pharmacol. 2019, 73, 282–289. [Google Scholar] [CrossRef]
- Kusaka, H.; Sueta, D.; Koibuchi, N.; Hasegawa, Y.; Nakagawa, T.; Lin, B.; Ogawa, H.; Kim-Mitsuyama, S. LCZ696, Angiotensin II Receptor-Neprilysin Inhibitor, Ameliorates High-Salt-Induced Hypertension and Cardiovascular Injury More Than Valsartan Alone. Am. J. Hypertens. 2015, 28, 1409–1417. [Google Scholar] [CrossRef]
- Li, S.N.; Zhang, J.R.; Zhou, L.; Xi, H.; Li, C.Y.; Zhao, L. Sacubitril/Valsartan Decreases Atrial Fibrillation Susceptibility by Inhibiting Angiotensin Ii-Induced Atrial Fibrosis through P-Smad2/3, P-Jnk, and P-P38 Signaling Pathways. J. Cardiovasc. Transl. Res. 2022, 15, 131–142. [Google Scholar] [CrossRef]
- Liang, W.; Xie, B.K.; Ding, P.W.; Wang, M.; Yuan, J.; Cheng, X.; Liao, Y.H.; Yu, M. Sacubitril/Valsartan Alleviates Experimental Autoimmune Myocarditis by Inhibiting Th17 Cell Differentiation Independently of the Nlrp3 Inflammasome Pathway. Front. Pharmacol. 2021, 12, 727838. [Google Scholar] [CrossRef]
- Lu, H.-I.; Tong, M.-S.; Chen, K.-H.; Lee, F.-Y.; Chiang, J.Y.; Chung, S.-Y.; Sung, P.-H.; Yip, H.-K. Entresto therapy effectively protects heart and lung against transverse aortic constriction induced cardiopulmonary syndrome injury in rat. Am. J. Transl. Res. 2018, 10, 2290–2305. [Google Scholar]
- Maslov, M.Y.; Foianini, S.; Mayer, D.; Orlov, M.V.; Lovich, M.A. Interaction Between Sacubitril and Valsartan in Preventing Heart Failure Induced by Aortic Valve Insufficiency in Rats. J. Card. Fail. 2019, 25, 921–931. [Google Scholar] [CrossRef]
- Maslov, M.Y.; Foianini, S.; Mayer, D.; Orlov, M.V.; Lovich, M.A. Synergy between sacubitril and valsartan leads to hemodynamic, antifibrotic, and exercise tolerance benefits in rats with preexisting heart failure. Am. J. Physiol. Circ. Physiol. 2019, 316, H289–H297. [Google Scholar] [CrossRef]
- Nordén, E.S.; Bendiksen, B.A.; Andresen, H.; Bergo, K.K.; Espe, E.K.; Hasic, A.; Hauge-Iversen, I.M.; Veras, I.; Hussain, R.I.; Sjaastad, I.; et al. Sacubitril/valsartan ameliorates cardiac hypertrophy and preserves diastolic function in cardiac pressure overload. ESC Heart Fail. 2021, 8, 918–927. [Google Scholar] [CrossRef]
- Seki, T.; Goto, K.; Kansui, Y.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Angiotensin II Receptor–Neprilysin Inhibitor Sacubitril/Valsartan Improves Endothelial Dysfunction in Spontaneously Hypertensive Rats. J. Am. Heart Assoc. 2017, 6, e006617. [Google Scholar] [CrossRef]
- Suematsu, Y.; Jing, W.; Nunes, A.; Kashyap, M.L.; Khazaeli, M.; Vaziri, N.D.; Moradi, H. LCZ696 (Sacubitril/Valsartan), an Angiotensin-Receptor Neprilysin Inhibitor, Attenuates Cardiac Hypertrophy, Fibrosis, and Vasculopathy in a Rat Model of Chronic Kidney Disease. J. Card. Fail. 2018, 24, 266–275. [Google Scholar] [CrossRef]
- Sung, Y.L.; Lin, T.T.; Syu, J.Y.; Hsu, H.J.; Lin, K.Y.; Liu, Y.B.; Lin, S.F. Reverse Electromechanical Modelling of Diastolic Dysfunction in Spontaneous Hypertensive Rat after Sacubitril/Valsartan Therapy. ESC Heart Fail. 2020, 7, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- Suo, Y.; Yuan, M.; Li, H.; Zhang, Y.; Li, Y.; Fu, H.; Han, F.; Ma, C.; Wang, Y.; Bao, Q.; et al. Sacubitril/Valsartan Improves Left Atrial and Left Atrial Appendage Function in Patients With Atrial Fibrillation and in Pressure Overload-Induced Mice. Front. Pharmacol. 2019, 10, 1285. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, K.; Kuwano, T.; Ideishi, A.; Morita, H.; Idemoto, Y.; Goto, M.; Suematsu, Y.; Miura, S.I. Sacubitril/Valsartan Inhibits Cardiomyocyte Hypertrophy in Angiotensin Ii-Induced Hypertensive Mice Independent of a Blood Pressure-Lowering Effect. Cardiol. Res. 2020, 11, 376–385. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, R.; Yu, X.; Li, N.; Zhao, X.; Yu, J. AHU377+Valsartan (LCZ696) Modulates Renin–Angiotensin System (RAS) in the Cardiac of Female Spontaneously Hypertensive Rats Compared With Valsartan. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 450–459. [Google Scholar] [CrossRef]
- Ge, Q.; Zhao, L.; Liu, C.; Ren, X.; Yu, Y.-H.; Pan, C.; Hu, Z. LCZ696, an Angiotensin Receptor-Neprilysin Inhibitor, Improves Cardiac Hypertrophy and Fibrosis and Cardiac Lymphatic Remodeling in Transverse Aortic Constriction Model Mice. BioMed Res. Int. 2020, 2020, 7256862. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Q.; Wang, Q.; Zhang, Q.; Zheng, Y.; Wang, L.; Jin, Q. Protection of Sacubitril/Valsartan against Pathological Cardiac Remodeling by Inhibiting the NLRP3 Inflammasome after Relief of Pressure Overload in Mice. Cardiovasc. Drugs Ther. 2020, 34, 629–640. [Google Scholar] [CrossRef]
- Peng, S.; Lu, X.F.; Qi, Y.D.; Li, J.; Xu, J.; Yuan, T.Y.; Wu, X.Y.; Ding, Y.; Li, W.H.; Zhou, G.Q.; et al. Lcz696 Ameliorates Oxidative Stress and Pressure Overload-Induced Pathological Cardiac Remodeling by Regulating the Sirt3/Mnsod Pathway. Oxid. Med. Cell Longev. 2020, 2020, 9815039. [Google Scholar] [CrossRef]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.-H.; Jiang, H.; Kim, H.-S.; Flynn, C.R.; Hill, S.; McDonald, W.H.; et al. Sirt3-Mediated Deacetylation of Evolutionarily Conserved Lysine 122 Regulates MnSOD Activity in Response to Stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef]
- Chen, W.J.; Cheng, Y.; Li, W.; Dong, X.K.; Wei, J.L.; Yang, C.H.; Jiang, Y.H. Quercetin Attenuates Cardiac Hypertrophy by Inhibiting Mitochondrial Dysfunction through Sirt3/Parp-1 Pathway. Front. Pharmacol. 2021, 12, 739615. [Google Scholar] [CrossRef]
- Schauer, A.; Adams, V.; Augstein, A.; Jannasch, A.; Draskowski, R.; Kirchhoff, V.; Goto, K.; Mittag, J.; Galli, R.; Männel, A.; et al. Sacubitril/Valsartan Improves Diastolic Function But Not Skeletal Muscle Function in a Rat Model of HFpEF. Int. J. Mol. Sci. 2021, 22, 3570. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Zhao, L.; Ren, X.-M.; Ye, P.; Hu, Z.-Y. LCZ696, an angiotensin receptor-neprilysin inhibitor, ameliorates diabetic cardiomyopathy by inhibiting inflammation, oxidative stress and apoptosis. Exp. Biol. Med. 2019, 244, 1028–1039. [Google Scholar] [CrossRef]
- Yang, C.-C.; Chen, Y.-T.; Chen, C.-H.; Li, Y.-C.; Shao, P.-L.; Huang, T.-H.; Chen, Y.-L.; Sun, C.-K.; Yip, H.-K. The therapeutic impact of entresto on protecting against cardiorenal syndrome-associated renal damage in rats on high protein diet. Biomed. Pharmacother. 2019, 116, 108954. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.-N.; Yue, Y.; Chu, Y.-C.; Huang, C.-R.; Yang, C.-C.; Chiang, J.Y.; Yip, H.-K.; Guo, J. Entresto protected the cardiomyocytes and preserved heart function in cardiorenal syndrome rat fed with high-protein diet through regulating the oxidative stress and Mfn2-mediated mitochondrial functional integrity. Biomed. Pharmacother. 2021, 144, 112244. [Google Scholar] [CrossRef]
- Boutagy, N.E.; Feher, A.; Pfau, D.; Liu, Z.; Guerrera, N.M.; Freeburg, L.A.; Womack, S.J.; Hoenes, A.C.; Zeiss, C.; Young, L.H.; et al. Dual Angiotensin Receptor-Neprilysin Inhibition With Sacubitril/Valsartan Attenuates Systolic Dysfunction in Experimental Doxorubicin-Induced Cardiotoxicity. JACC Cardiooncol. 2020, 2, 774–787. [Google Scholar] [CrossRef]
- Dindaş, F.; Güngör, H.; Ekici, M.; Akokay, P.; Erhan, F.; Doğduş, M.; Yılmaz, M.B. Angiotensin Receptor-Neprilysin Inhibition by Sacubitril/Valsartan Attenuates Doxorubicin-Induced Cardiotoxicity in a Pretreatment Mice Model by Interfering with Oxidative Stress, Inflammation, and Caspase 3 Apoptotic Pathway. Anatol. J. Cardiol. 2021, 25, 821–828. [Google Scholar] [CrossRef]
- Kim, B.S.; Park, I.-H.; Lee, A.-H.; Kim, H.-J.; Lim, Y.-H.; Shin, J.-H. Sacubitril/valsartan reduces endoplasmic reticulum stress in a rat model of doxorubicin-induced cardiotoxicity. Arch. Toxicol. 2022, 96, 1065–1074. [Google Scholar] [CrossRef]
- Miyoshi, T.; Nakamura, K.; Amioka, N.; Hatipoglu, O.F.; Yonezawa, T.; Saito, Y.; Yoshida, M.; Akagi, S.; Ito, H. LCZ696 ameliorates doxorubicin-induced cardiomyocyte toxicity in rats. Sci. Rep. 2022, 12, 4930. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, Z.; Chen, A.; Fu, M.; Dong, Z.; Hu, K.; Yang, X.; Zou, Y.; Sun, A.; Qian, J.; et al. LCZ696 improves cardiac function via alleviating Drp1-mediated mitochondrial dysfunction in mice with doxorubicin-induced dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 108, 138–148. [Google Scholar] [CrossRef]
- Yu, C.; Li, D.; Li, Z.; Yu, D.; Zhai, G. Effect of sacubitril/valsartan on inflammation and oxidative stress in doxorubicin-induced heart failure model in rabbits. Acta Pharm. 2021, 71, 473–484. [Google Scholar] [CrossRef]
- Maurea, N.; Canale, M.L.; Buccolo, S.; Bisceglia, I.; Paccone, A.; Maurea, F.; Scherillo, M.; Quagliariello, V. Abstract 12402: Sacubitril-Valsartan Increases Pampk and Reduces Inflammasome, Myddosome, Il-6 and Galectin-3 Levels in Short-Term Doxorubicin-Treated Mice Improving Longitudinal Strain and Ejection Fraction. Circulation 2022, 146, A12402. [Google Scholar]
- Maurea, N.; Quagliariello, V.; Iovine, M.; Buccolo, S.; Paccone, A.; Bisceglia, I.; De Laurentiis, M. Dapagliflozin associated to sacubitril/valsartan and relationship with cardioprotection in human cardiac cells exposed to doxorubicin and HER2-blocking agents through MyD88, NLRP3 mediated pathways. J. Clin. Oncol. 2022, 40, 587. [Google Scholar] [CrossRef]
- Dankowski, R.; Sacharczuk, W.; Łojko-Dankowska, A.; Nowicka, A.; Szałek-Goralewska, A.; Szyszka, A. Sacubitril/Valsartan as First-Line Therapy in Anthracycline-Induced Cardiotoxicity. Kardiol. Pol. (Pol. Heart J.) 2021, 79, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Martín-García, A.; Díaz-Peláez, E.; Martín-García, A.C.; Sánchez-González, J.; Ibáñez, B.; Sánchez, P.L. Myocardial Function and Structure Improvement with Sacubitril/Valsartan in Cancer Therapy-Induced Cardiomyopathy. Rev. Española Cardiol. 2020, 73, 268–269. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Paper by | Animal Model | Gavage Initiation | Groups | LVEF (%) | Other Findings | |
|---|---|---|---|---|---|---|
| Chang [89] | SPRD rats | 1 wk after MI | Vehicle Enalapril ARNi | 38.5 ± 2.0 46.7 ± 9.1 ↑ 57.6 ± 5.5 ↑↑ | ↓↓ HW/BW ratio in ARNi ↓↓ Ventricular arrhythmias inducibility in ARNi ↑↑ Expression of K+ channel proteins in ARNi | |
| Chang [90] | New Zealand White rabbits | 1 wk after MI | Vehicle ARB ARNi | 37.1 ± 6.3 44.3 ± 6.3 ↑ 53.8 ± 10.0 ↑↑ | ↓↓ Ventricular arrhythmia inducibility in ARNi | |
| Ishii [91] | Mice | 1 day after MI | Vehicle Enalapril ARNi | NA | ↓↓ Post-MI mortality rate due to LV rupture in ARNi ↑↑ %FS 14 and 28 days post-MI in ARNi ↓↓ Myocardial expression of IL1β, IL6, and MMP-9 mRNA in ARNi No differences in myocardial fibrosis and inflammatory infiltration. | |
| Kompa [92] | SPRD rats | 1 wk after MI | Vehicle Perindopril ARNi | 40.46 ± 1.27 42.22 ± 1.16 46.65 ± 0.83 ↑↑ | ARNi improved end-systolic pressure-volume relationship compared with perindopril ↓ LV mass, cardiomyocyte CSA, and cardiac fibrosis in perindopril and ARNi ↓↓ ANP, MHC β, and TIMP2 expression in ARNi | |
| Liu [93] | C57BL/6J mice | Directly after MI | Vehicle Benazepril ARNi ARNi + Benazepril | = 58.7 ± 0.42 ↑ 62.35 ± 0.25 ↑↑ | HW/BW ratio ↓ in ARNi and benazepril and ↓↓ in ARNi + benazepril Myocardial fibrosis ↓ in ARNi and ↓↓ in ARNi + benazepril ↓↓ TGFβ1 expression in ARNi and ARNi + benazepril No differences in IL6 and TNFɑ expression. | |
| Pfau [94] | Lewis rats | 1 wk after MI | Vehicle ARB ARNi | 1 wk: 34 ± 2 38 ± 2 39 ± 2 ↑ | 5 wks: 35 ± 2 39 ± 2 42 ± 2 | ↓ HW/TL ratio and fibrosis in ARNi ↓ Myocyte CSA in ARNi and ARB ↓ Expression of CTGF, MHCβ, MHCβ/α, and ANP in ARNi and ARB |
| Raj [95] | SPRD rats | Directly after MI | Vehicle ARB ARNi | 56.60 ± 1.70 65.45 ± 2.70 ↑ 66.82 ± 1.43 ↑ | ↓ Oxidative stress in ARNi and ARB ↓ TNFα, collagen, and BNP in ARNi and ARB | |
| Shen [96] | SPRD rats | 1 wk after MI | Vehicle ARNi | 3 days: ↑ | 7 days: ↑ | ↓ Interstitial fibrosis in ARNi ↓ Serum IL1βa and IL18 levels in ARNi ↓ ROS and NLRP3 inflammasome activation in ARNi |
| Suematsu [97] | C57BL/6J diabetic mice | Day after MI | Vehicle ARB ARNi | 29 ± 3.2 = 43 ± 3.4 ↑ | ↓↓ LV fibrosis and expression of TGFβ mRNA in ARNi ↓ HW/BW ratio in ARB and ARNi ↓ ANP mRNA in ARNi | |
| Torrado [98] | New Zealand White rabbits | At reperfusion only | Vehicle ARB ARNi | ARB = ARNi ↑ | ↓ Infarct size in ARB and ARNi and ↓ cardiac troponin I serum concentration in ARNi | |
| At LVEF ≤ 40% | ARB = ARNi ↑↑ | |||||
| At reperfusion | 4 wks: ARB = ARNi ↑↑ | 10 wks: = ↑ | ↓ Infarct size in ARNi | |||
| Trivedi [99] | SHRs | 4 wks after reperfusion | Vehicle ARB ARNi | = = ↑↑ | ↓ Infarct border zone expansion in ARB and ARNi Aortic vasorelaxation responses to Ach and SNP ↑ in ARB and ↑↑ in ARNi ↑↑ Myocardial NO bioavailability in ARNi No differences in fibrosis between groups | |
| Vaskova [100] | SPRD rats | 1 wk after MI | Vehicle ARB ARNi | 36.79 ± 2.1 40.68 ± 4.8 ↑ 41.42 ± 3.4 ↑ | ↑ Production of plasma exosomes in ARB and ARNi ↓↓ Expression of rno-miR-181a in ARNi ↓ Fibrosis in ARB and ARNi | |
| Von Lueder [101] | Lewis rats | 1 wk after MI | Vehicle ARNi | 47 ± 5 60 ± 2 ↑ | Improved LV function in pressure-volume loops in ARNi ↓ LV mass and fibrosis in peri-infarct and remote myocardium in ARNi No differences in infarct size and perivascular fibrosis. | |
| Model of | Paper by | Species | Gavage Initiation | Groups | Findings * |
|---|---|---|---|---|---|
| HFpEF due to pressure overload | Burke [103] | C57Bl6/J mice | A day before TAC, cont. for 4 wks | Vehicle ARB ARNi | ↑↑ LVEF in ARNi ↓↓ Interstitial fibrosis and fibroblast population in ARNi ↓↓ Cardiomyocyte CSA and HW/TL in ARNi |
| Lu [108] | SPRD rats | 4 wks after TAC, cont. for 32 days | Vehicle Enalapril ARNi | ↓↓ Sarcomere-length, left ventricle fibrotic area, cardiomyocyte size and lung injury in ARNi ↓↓ Expressions of fibrotic, oxidative, apoptotic, DNA damage, mitochondrial damage, volume overload markers in LV in ARNi | |
| Norden [111] | SPRD rats | Cont. for 8 wks | Vehicle ARB ARNi | ↓↓ LV weight in ARNi ↓↓ Diastolic dysfunction in ARNi No differences in LVEF and myocardial fibrosis | |
| Suo [115] | C57BL/6J mice | 8 wks after TAC, cont. for 4 wks | Vehicle ARB ARNi | ↑↑ LVEF in ARNi ↓↓ Fibrosis in ARNi | |
| HT | Hamano [104] | SHRcp fed high-salt diet | I: 6 mos, with high-salt diet | Vehicle ARB ARNi | ↓ LV/BW ratio in ARNi in Plan I ↓↓ LV/BW and pulmonary edema in ARB in Plan II No differences in cardiomyocyte CSA and fibrosis |
| II: After 6 mos of high-salt diet, cont. for 6 mos | |||||
| Kusaka [105] | SHRcp fed high-salt diet | Cont. for 4 wks | Vehicle ARB ARNi | ↓ LV in ARNi ↓↓ Myocardial fibrosis in ARNi ↓↓ Impairment of acetylcholine-induced vascular relaxation in ARNi | |
| Seki [112] | SHRs | Cont. for 12 wks | Vehicle ARB ARNi | Endothelium-dependent hyperpolarization-mediated responses improved similarly in ARB and ARNi ↓ LV in ARNi | |
| Sung [114] | SHRs | Cont. for 2 wks | Vehicle ARB ARNi | ↓↓ Diastolic dysfunction and ↓↓ ventricular hypertrophy in ARNi ↓↓ Incidence of ventricular arrhythmias in ARNi No differences in LVEF | |
| Tashiro [116] | C57BL/6J mice | Started on the 7th day of Ang II infusion, cont. for 2 wks | Vehicle Enalapril ARB ARNi | ↓↓ LV concentric hypertrophy in ARNi Myocyte CSA ↓ in ARB, ↓ in enalapril, and ↓↓ in ARNi No differences in fibrosis and TGFβ expression | |
| Zhao [117] | SHRs | Cont. for 12 wks | Vehicle ARB ARNi | LVEF ↑↑ in ARNi and ↑ in ARB ↓↓ LV mass in ARNi ↓ Fibrosis, TGFβ expression and nNOS, eNOS protein expression in ARB and ARNi ↓↓ ACE, ATR1, and ↑↑ ACE2, MasR, ATR2 cardiac protein expression in ARNi | |
| HF due to volume overload (by AVI) | Maslow [109] | SPRD rats | 4 wks after AVI, cont. for 4 wks | Vehicle ARB NEPi ARNi | Improved load-dependent indexes of left ventricle contractility and relaxation only in ARNi Improved load-independent index of contractility in ARB and ARNi ↑↑ Exercise tolerance in ARNi ↓↓ Myocardial fibrosis in ARNi |
| Maslow [110] | SPRD rats | On the day of AVI, cont. for 8 wks | Vehicle ARB NEPi ARNi | ↑ LVEF in ARNi ↓ Myocardial fibrosis in ARB, NEPi, and ARNi ↑ Exercise tolerance in ARB and ARNi | |
| HFpEF due to obesity | Aroor [102] | Zucker Obese rats | At 16 wks of age, cont. for 10 wks | Vehicle ARB ARNi | ↑ LVEF, ↓ fibrosis, and ↓ oxidative stress in ARB and ARNi ↑ Endothelial-dependent aortic relaxation in ARB and ARNi ↑↑ E’/a’ ratio in ARNi |
| AF | Li [106] | SPRD rats | After AF induction, cont. for 4 wks | Vehicle ARB ARNi | ↑ LVEF in ARB and ARNi ↓↓ Atrial fibrosis and susceptibility to AF in ARNi |
| Myocarditis | Liang [107] | BALB/c mice | On the day of myocarditis, cont. for 3 wks | Vehicle ARB ARNi | ↓↓ HW/BW ratio, ↓↓ myocardial histopathologic scores, and ↓↓ cTnT levels in ARNi ↓↓ Serum hsCRP, IL6, and serum/myocardial IL17 levels in ARNi ↓↓ Th17 cells and their transcription factors in myocardial tissue in ARNi |
| CKD | Suematsu [113] | SPRD rats | 2 wks after nephrectomy, cont. for 8 wks | Vehicle ARB ARNi | HW/BW ratio, myocyte CSA, markers of oxidative stress, myocardial and aortic fibrosis ↓↓ in ARNi and ↓ in ARB ↓↓ expression of NF-κB, COX-2 in ARNi |
| Paper by | Animal Model | Groups + Dosage (mg/kg/d) | Other Findings (Presented in Comparison to DOX + Vehicle Groups) |
|---|---|---|---|
| Boutagy [127] | Wistar rats | DOX + Vehicle DOX + ARB 31 DOX + ARNi 68 | ↑ LVEF in ARNi ↓ Myocyte vacuolation in ARNi and ARB ↓ Myocardial fibrosis in ARNi and ARB at 4 wks (no longer seen at 6 wks) ↓ Capillary density in ARNi at 6 wks ↓ Matrix metalloproteinases activity in ARNi at 4 wks ↓ Myocyte CSA and heart weight in all DOX-receiving groups No differences in cellular apoptosis between groups |
| Dindas [128] | Balb-c mice | Vehicle ARNi 80 DOX + Vehicle DOX + ARNi 80 | ↓ Degenerative changes and streaking in cardiomyocytes in DOX + ARNi ↓ QRS duration, ST interval and QT/PQ index in DOX + ARNi ↓ NT-proBNP, TNFα, IL1β, IL6, and caspase 3 in DOX + ARNi ↓ Total oxidant status and ↑ total antioxidant status in DOX + ARNi |
| Kim [129] | Sprague Dawley rats | Vehicle DOX + Vehicle DOX + ARNi 60 | ↓ Cardiomyocyte apoptosis in ARNi ↓ Endoplasmic reticulum stress in ARNi ↓ Serum cardiac troponin I and NT-proBNP levels in ARNi group |
| Maurea [133] | C57Bl/6 mice | Sham Sac/Val 60 DOX DOX + Sac/Val 60 | ARNi improved EF and prevented the reduction of radial and longitudinal strain ↓ Cardiac expression of NLRP3, MyD88, DAMPs, and NF-kB in ARNi ↑ Expression of phosphorylated AMPK in ARNi ↓ Levels of Calgranulin S100 and galectine-3 in ARNi |
| Miyoshi [130] | Sprague Dawley rats | DOX + Vehicle DOX + Val 31 DOX + ARNi 68 | No differences in LVEF and FS between groups. ↑ Cardiomyocyte CSA and ↑ cardiac fibrosis in ARNi ↑ Cardiac TNFα and ANP mRNA expression in ARB and ARNi ↓ Myocardial collagen I mRNA expression in ARNi ↓ Cardiac troponin T and NT-proBNP levels in ARNi ↓ Cardiac reactive oxygen species levels in ARNi ↑ Phosphorylation of AMPK and ↑ Bax/Bcl-2 ratio in ARNi |
| Xia [131] | Balb-c mice | Vehicle DOX + Vehicle DOX + ARNi 80 | ↓ Cardiac hypertrophy, myocardial fibrosis and cellular apoptosis in ARNi ↓ Heart weight/body weight and ↑ heart weight/tibial length in ARNi ↑ Single cardiomyocyte contractile function in ARNi ↓ Pathologic changes to mitochondria and ↓ Drp1 expression in ARNi ↓ Cleaved caspase 3 in ARNi |
| Ye [48] | C57BL/6 mice | Vehicle DOX + Vehicle DOX + ARNi 60 TLR2 KO + Vehicle TLR2 KO + DOX | ↑ LVEF in ARNi, TLR2 KO, and TLR2 KO + DOX ↓ Ventricular wall thinning and ↓ heart cavity enlargement in ARNi and TLR2 KO ↓ Myocardial fibrosis in ARNi and TLR2 KO ↓ Myocardial collagen I and TGFβ protein levels in ARNi and TLR2 KO ↓ Myocardial TNFα and NF-κB levels in ARNi and TLR2 KO |
| Yu [132] | New Zealand white rabbits | Vehicle DOX + Vehicle DOX + ARNi 5 DOX + ARNi 10 | ↓ PR segment, QRC segment prolongation, QT interval and QT/PQ index in both ARNi groups ↓ Serum BNP level in ARNi groups ↑ Activity of superoxide dismutase and catalase and ↓ lipid peroxidation in both ARNi groups |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobiborowicz-Sadowska, A.M.; Kamińska, K.; Cudnoch-Jędrzejewska, A. Neprilysin Inhibition in the Prevention of Anthracycline-Induced Cardiotoxicity. Cancers 2023, 15, 312. https://doi.org/10.3390/cancers15010312
Sobiborowicz-Sadowska AM, Kamińska K, Cudnoch-Jędrzejewska A. Neprilysin Inhibition in the Prevention of Anthracycline-Induced Cardiotoxicity. Cancers. 2023; 15(1):312. https://doi.org/10.3390/cancers15010312
Chicago/Turabian StyleSobiborowicz-Sadowska, Aleksandra M., Katarzyna Kamińska, and Agnieszka Cudnoch-Jędrzejewska. 2023. "Neprilysin Inhibition in the Prevention of Anthracycline-Induced Cardiotoxicity" Cancers 15, no. 1: 312. https://doi.org/10.3390/cancers15010312
APA StyleSobiborowicz-Sadowska, A. M., Kamińska, K., & Cudnoch-Jędrzejewska, A. (2023). Neprilysin Inhibition in the Prevention of Anthracycline-Induced Cardiotoxicity. Cancers, 15(1), 312. https://doi.org/10.3390/cancers15010312