
Analytical Considerations of Large-Scale Aptamer-Based Datasets for Translational Applications


 ,
, 
Simple Summary
Abstract
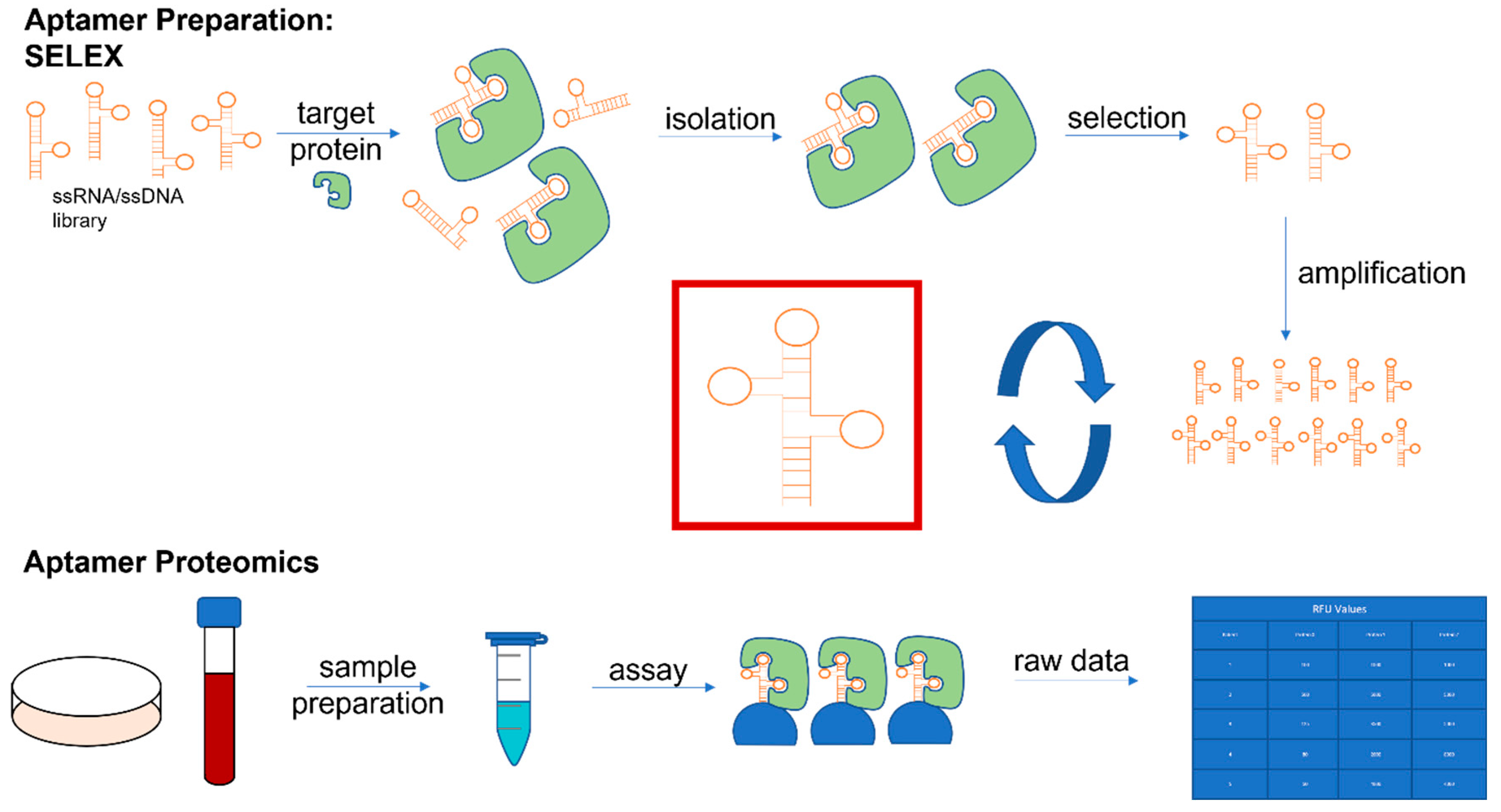
1. Introduction
2. Setting Up Aptamer Studies for Clinical Translation
2.1. Power, Power Analysis, and Protein Effect Size
2.2. Samples and Study Designs: Case–Control and Cohort Studies
3. Statistical Strategies for Analyzing Differential Expression
3.1. Quality Control and Basic Statistics
3.2. Machine Learning Approaches
3.3. Pathway Analysis
4. Multi-Omics Approaches and Verification
4.1. Proteomic Quantitative Trait Loci (pQTL)
4.2. GWAS and PWAS
5. Translational Challenges of Aptamer Proteomics
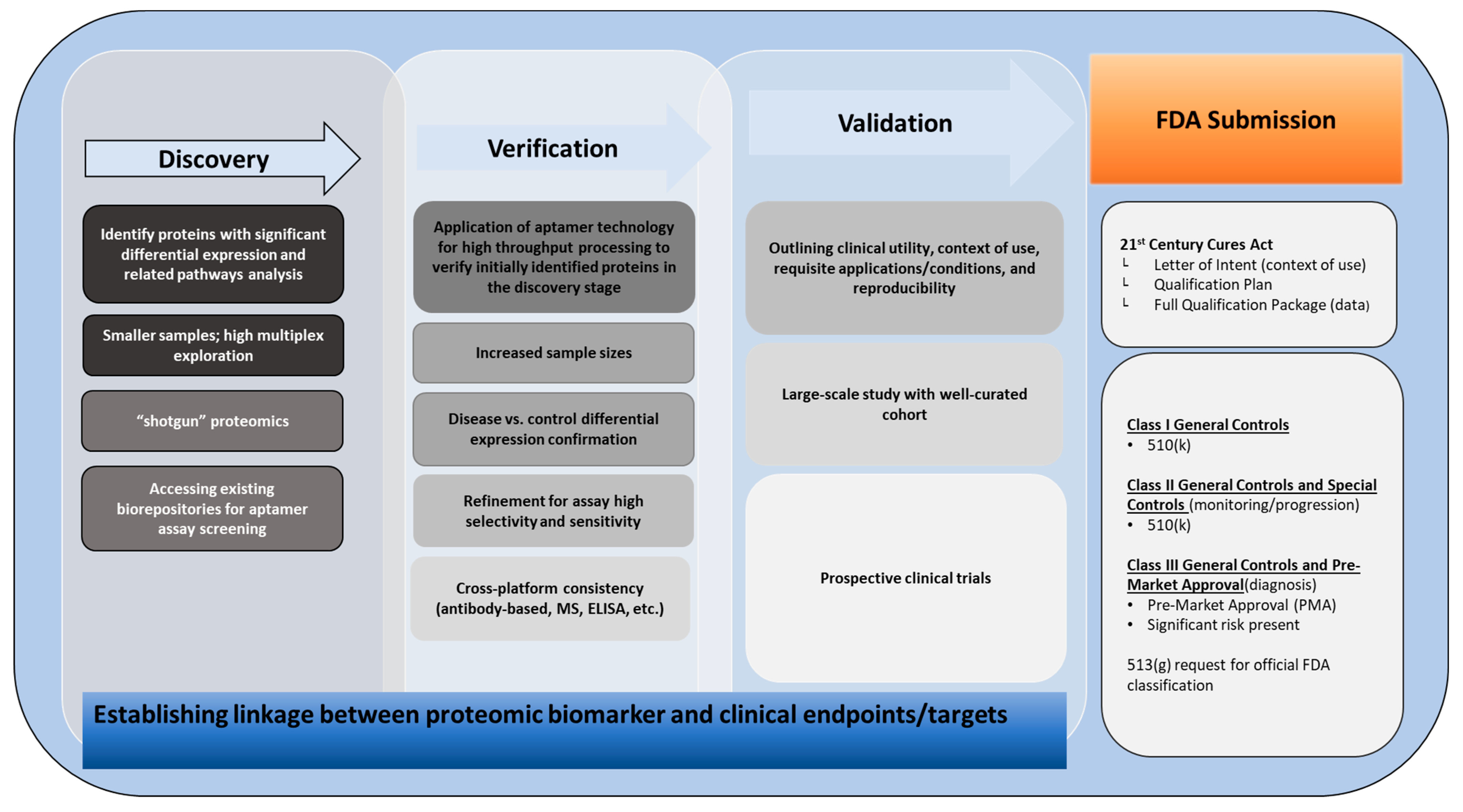
5.1. FDA Approval and Clinical Translation
5.2. Cross-Platform Consistency
5.3. Intra-Platform Consistency
6. Conclusions
- (1)
- Study designThe study design for aptamer-derived data is understandably contingent on the disease and clinical context. Maximal benefit is elicited if the study benefits from maximal data capture and annotation to ensure that potential confounders in the proteomic data signals can be addressed down the line. Controls are crucial for meaningful comparison. Controls may not necessarily represent a normal population but rather a population whose proteome in comparison to that of the intervention will allow the researcher to derive conclusions that will address the hypothesis being tested. Cohort studies may be employed with a “before” and “after” sample being obtained from the same patient, thus using the patient as their own control. Provided robust large-scale data are available and, following a thorough review, are sufficiently comparable to the study population, this is a reasonable option, with the study design in this context benefitting greatly from collaboration amongst researchers.
- (2)
- Statistical analysisStatistical analysis for aptamer-derived data is actively evolving. Traditional approaches described here may be employed. ML approaches may be used with semi-supervised approaches likely to be the most successful and contingent on annotation of the data.
- (3)
- An established team that combines researchers, statisticians, and clinicians will need to maintain a close relationship in the planned acquisition, curation, annotation and analysis of data to allow for meaningful translation into the clinic and advancement in relation to patient outcomes.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of Variance |
| AI | Artificial Intelligence |
| CV | Coefficient of Variations |
| CNN | Convolutional Neural Networks |
| DDA-MS | Data-Dependent Mass Spectrometry |
| DL | Deep Learning |
| DIA-MS | Data-Independent Acquisition Mass Spectrometry |
| ELISA | Enzyme-Linked Immunosorbent Assays |
| eQTL | Expression Quantitative Trait Loci |
| FDA | U.S. Food and Drug Administration |
| FDR | False Discovery Rate |
| FCS | Functional Class Scoring |
| GAGE | Generally Applicable Gene set Enrichment |
| GSEA | Gene Set Enrichment Analysis |
| GWAS | Genome-Wide Association Studies |
| HEIDI | Heterogeneity in Dependent Instruments |
| ICA | Independent Component Analysis |
| IPA | QIAGEN’s Ingenuity Pathway Analysis |
| LC–MS | Liquid Chromatography–MS |
| ML | Machine Learning |
| MS | Mass Spectrometry |
| ORA | Over-Representation Analysis |
| PCA | Principal Component Analysis |
| PLS | Partial Least Squares |
| pQTL | Proteomic Quantitative Trait Loci |
| PT | Pathway Topology |
| PTM | Post-Translational Modification |
| PWAS | Protein-Wide Association Studies |
| RF | Random Forest |
| RFU | Relative Fluorescence Unit |
| SELEX | Systematic Evolution of Ligands by Exponential Enrichment |
| SVM | Support Vector Machine |
References
- Tighe, P.J.; Ryder, R.R.; Todd, I.; Fairclough, L.C. ELISA in the multiplex era: Potentials and pitfalls. Proteom. Clin. Appl. 2015, 9, 406–422. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Simon, G.M.; Yates, J.R., 3rd. The biological impact of mass-spectrometry-based proteomics. Nature 2007, 450, 991–1000. [Google Scholar] [CrossRef]
- Beer, I.; Barnea, E.; Ziv, T.; Admon, A. Improving large-scale proteomics by clustering of mass spectrometry data. Proteomics 2004, 4, 950–960. [Google Scholar] [CrossRef]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef]
- Krasny, L.; Huang, P.H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics 2021, 17, 29–42. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef]
- Courcelles, M.; Bridon, G.; Lemieux, S.; Thibault, P. Occurrence and detection of phosphopeptide isomers in large-scale phosphoproteomics experiments. J. Proteome Res. 2012, 11, 3753–3765. [Google Scholar] [CrossRef]
- Chick, J.M.; Kolippakkam, D.; Nusinow, D.P.; Zhai, B.; Rad, R.; Huttlin, E.L.; Gygi, S.P. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 2015, 33, 743–749. [Google Scholar] [CrossRef]
- SomaLogic. SOMAscan Proteomic Assay: Technical White Paper. Available online: https://www.somalogic.com/wp-content/uploads/2016/08/SSM-002-Rev-3-SOMAscan-Technical-White-Paper.pdf (accessed on 2 February 2022).
- Hong, J.M.; Gibbons, M.; Bashir, A.; Wu, D.; Shao, S.; Cutts, Z.; Chavarha, M.; Chen, Y.; Schiff, L.; Foster, M.; et al. ProtSeq: Toward high-throughput, single-molecule protein sequencing via amino acid conversion into DNA barcodes. iScience 2021, 25, 103586. [Google Scholar] [CrossRef]
- Wik, L.; Nordberg, N.; Broberg, J.; Björkesten, J.; Assarsson, E.; Henriksson, S.; Grundberg, I.; Pettersson, E.; Westerberg, C.; Liljeroth, E.; et al. Proximity Extension Assay in Combination with Next-Generation Sequencing for High-throughput Proteome-wide Analysis. Mol. Cell Proteom. 2021, 20, 100168. [Google Scholar] [CrossRef]
- Coarfa, C.; Grimm, S.L.; Rajapakshe, K.; Perera, D.; Lu, H.-Y.; Wang, X.; Christensen, K.R.; Mo, Q.; Edwards, D.P.; Huang, S. Reverse-Phase Protein Array: Technology, Application, Data Processing, and Integration. J. Biomol. Tech. 2021, 32, 15–29. [Google Scholar] [CrossRef]
- Gupta, V.; Zimmerman, R.; Zhan, T.; Hamilton, T.; Na, L.; Peng, J. Development and Validation of Bio-Plex Pro Human Chemokine Assays. 2014. Available online: https://www.bio-rad.com (accessed on 2 February 2022).
- Wang, X.; Walt, D.R. Simultaneous detection of small molecules, proteins and microRNAs using single molecule arrays. Chem. Sci. 2020, 11, 7896–7903. [Google Scholar] [CrossRef]
- Puscasu, A.; Zanchetta, M.; Posocco, B.; Bunka, D.; Tartaggia, S.; Toffoli, G. Development and validation of a selective SPR aptasensor for the detection of anticancer drug irinotecan in human plasma samples. Anal. Bioanal. Chem. 2021, 413, 1225–1236. [Google Scholar] [CrossRef]
- Adhikari, M.; Strych, U.; Kim, J.; Goux, H.; Dhamane, S.; Poongavanam, M.-V.; Hagström, A.E.V.; Kourentzi, K.; Conrad, J.C.; Willson, R.C. Aptamer-Phage Reporters for Ultrasensitive Lateral Flow Assays. Anal. Chem. 2015, 87, 11660–11665. [Google Scholar] [CrossRef]
- Hwang, B.B.; Engel, L.; Goueli, S.A.; Zegzouti, H. A homogeneous bioluminescent immunoassay to probe cellular signaling pathway regulation. Commun. Biol. 2020, 3, 8. [Google Scholar] [CrossRef]
- Bastarache, J.A.; Koyama, T.; Wickersham, N.E.; Ware, L.B. Validation of a multiplex electrochemiluminescent immunoassay platform in human and mouse samples. J. Immunol. Methods 2014, 408, 13–23. [Google Scholar] [CrossRef]
- Magdeldin, S.; Enany, S.; Yoshida, Y.; Xu, B.; Zhang, Y.; Zureena, Z.; Lokamani, I.; Yaoita, E.; Yamamoto, T. Basics and recent advances of two dimensional- polyacrylamide gel electrophoresis. Clin. Proteom. 2014, 11, 16. [Google Scholar] [CrossRef]
- Whiteaker, J.R.; Lin, C.; Kennedy, J.; Hou, L.; Trute, M.; Sokal, I.; Yan, P.; Schoenherr, R.M.; Zhao, L.; Voytovich, U.J.; et al. A targeted proteomics–based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 2011, 29, 625–634. [Google Scholar] [CrossRef]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.-C.; Held, J.M.; et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017, 8, 291. [Google Scholar] [CrossRef]
- Sandberg, A.; Branca, R.M.M.; Lehtiö, J.; Forshed, J. Quantitative accuracy in mass spectrometry based proteomics of complex samples: The impact of labeling and precursor interference. J. Proteom. 2014, 96, 133–144. [Google Scholar] [CrossRef]
- Chambers, A.G.; Percy, A.J.; Yang, J.; Borchers, C.H. Multiple Reaction Monitoring Enables Precise Quantification of 97 Proteins in Dried Blood Spots. Mol. Cell Proteom. 2015, 14, 3094–3104. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.S.C.; Welch, M.J. Development and Evaluation of a Candidate Reference Method for the Determination of Total Cortisol in Human Serum Using Isotope Dilution Liquid Chromatography/Mass Spectrometry and Liquid Chromatography/Tandem Mass Spectrometry. Anal. Chem. 2004, 76, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Ozyurt, C.; Canbay, Z.C.; Dinckaya, E.; Evran, S. A highly sensitive DNA aptamer-based fluorescence assay for sarcosine detection down to picomolar levels. Int. J. Biol. Macromol. 2019, 129, 91–97. [Google Scholar] [CrossRef] [PubMed]
- BasePair. Available online: https://www.basepairbio.com/ (accessed on 2 February 2022).
- Singh, K.; Cheung, B.M.; Xu, A. Ultrasensitive detection of blood biomarkers of Alzheimer’s and Parkinson’s diseases: A systematic review. Biomark. Med. 2021, 15, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, W.; Chen, S.; Zhuang, Z.; Zhang, Y.; Jiang, L.; Lin, J.S. SELEX tool: A novel and convenient gel-based diffusion method for monitoring of aptamer-target binding. J. Biol. Eng. 2020, 14, 1. [Google Scholar] [CrossRef]
- Brody, E.N.; Willis, M.C.; Smith, J.D.; Jayasena, S.; Zichi, D.; Gold, L. The use of aptamers in large arrays for molecular diagnostics. Mol. Diagn. 1999, 4, 381–388. [Google Scholar] [CrossRef]
- Dhiman, A.; Kalra, P.; Bansal, V.; Bruno, J.G.; Sharma, T.K. Aptamer-based point-of-care diagnostic platforms. Sens. Actuators B Chem. 2017, 246, 535–553. [Google Scholar] [CrossRef]
- Pietzner, M.; Wheeler, E.; Carrasco-Zanini, J.; Kerrison, N.D.; Oerton, E.; Koprulu, M.; Luan, J.; Hingorani, A.D.; Williams, S.A.; Wareham, N.J.; et al. Synergistic insights into human health from aptamer- and antibody-based proteomic profiling. Nat. Commun. 2021, 12, 6822. [Google Scholar] [CrossRef]
- Abatemarco, J.; Sarhan, M.F.; Wagner, J.M.; Lin, J.-L.; Liu, L.; Hassouneh, W.; Yuan, S.-F.; Alper, H.S.; Abate, A.R. RNA-aptamers-in-droplets (RAPID) high-throughput screening for secretory phenotypes. Nat. Commun. 2017, 8, 332. [Google Scholar] [CrossRef] [PubMed]
- Ambati, A.; Ju, Y.E.; Lin, L.; Olesen, A.N.; Koch, H.; Hedou, J.J.; Leary, E.B.; Sempere, V.P.; Mignot, E.; Taheri, S. Proteomic biomarkers of sleep apnea. Sleep 2020, 43, zsaa086. [Google Scholar] [CrossRef] [PubMed]
- Helms, L.; Marchiano, S.; Stanaway, I.B.; Hsiang, T.-Y.; Juliar, B.A.; Saini, S.; Zhao, Y.T.; Khanna, A.; Menon, R.; Alakwaa, F.; et al. Cross-validation of SARS-CoV-2 responses in kidney organoids and clinical populations. JCI Insight 2021, 6, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tully, B.; Balleine, R.L.; Hains, P.G.; Zhong, Q.; Reddel, R.R.; Robinson, P.J. Addressing the Challenges of High-Throughput Cancer Tissue Proteomics for Clinical Application: ProCan. Proteomics 2019, 19, e1900109. [Google Scholar] [CrossRef]
- Anderson, N.L. The clinical plasma proteome: A survey of clinical assays for proteins in plasma and serum. Clin. Chem. 2010, 56, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Boellner, S.; Becker, K.-F. Reverse Phase Protein Arrays-Quantitative Assessment of Multiple Biomarkers in Biopsies for Clinical Use. Microarrays 2015, 4, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Lu, Y.; Gonzalez-Angulo, A.M.; Carey, M.S.; Myhre, S.; Ju, Z.; Davies, M.A.; Liu, W.; Coombes, K.; Meric-Bernstam, F.; et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin. Proteom. 2010, 6, 129–151. [Google Scholar] [CrossRef]
- Byron, A. Reproducibility and Crossplatform Validation of Reverse-Phase Protein Array Data. In Reverse Phase Protein Arrays: From Technical and Analytical Fundamentals to Applications; Yamada, T., Nishizuka, S.S., Mills, G.B., Liotta, L.A., Eds.; Springer: Singapore, 2019; pp. 181–201. [Google Scholar] [CrossRef]
- Brase, J.C.; Mannsperger, H.; Fröhlich, H.; Gade, S.; Schmidt, C.; Wiemann, S.; Beissbarth, T.; Schlomm, T.; Sültmann, H.; Korf, U. Increasing the sensitivity of reverse phase protein arrays by antibody-mediated signal amplification. Proteome Sci. 2010, 8, 36. [Google Scholar] [CrossRef]
- Houser, B. Bio-Rad’s Bio-Plex® suspension array system, xMAP technology overview. Arch. Physiol. Biochem. 2012, 118, 192–196. [Google Scholar] [CrossRef]
- Wilson, D.H.; Rissin, D.M.; Kan, C.W.; Fournier, D.R.; Piech, T.; Campbell, T.G.; Meyer, R.E.; Fishburn, M.W.; Cabrera, C.; Patel, P.P.; et al. The Simoa HD-1 Analyzer:A Novel Fully Automated Digital Immunoassay Analyzer with Single-Molecule Sensitivity and Multiplexing. J. Lab. Autom. 2016, 21, 533–547. [Google Scholar] [CrossRef]
- Schubert, S.M.; Arendt, L.M.; Zhou, W.; Baig, S.; Walter, S.R.; Buchsbaum, R.J.; Kuperwasser, C.; Walt, D.R. Ultra-sensitive protein detection via Single Molecule Arrays towards early stage cancer monitoring. Sci. Rep. 2015, 5, 11034. [Google Scholar] [CrossRef] [PubMed]
- Chirinos, J.A.; Cohen, J.B.; Zhao, L.; Hanff, T.; Sweitzer, N.; Fang, J.; Corrales-Medina, V.; Ammar, R.; Morley, M.; Zamani, P.; et al. Clinical and Proteomic Correlates of Plasma ACE2 (Angiotensin-Converting Enzyme 2) in Human Heart Failure. Hypertension 2020, 76, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- George, M.J.; Kleveland, O.; Garcia-Hernandez, J.; Palmen, J.; Lovering, R.; Wiseth, R.; Aukrust, P.; Engmann, J.; Damås, J.K.; Hingorani, A.D.; et al. Novel Insights Into the Effects of Interleukin 6 Antagonism in Non–ST-Segment&–Elevation Myocardial Infarction Employing the SOMAscan Proteomics Platform. J. Am. Heart Assoc. 2020, 9, e015628. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.G.; Chan, N.Y.; Dillon, S.T.; Zhou, W.; Tripp, B.; Ngo, L.H.; Otu, H.H.; Inouye, S.K.; Vasunilashorn, S.M.; Cooper, Z.; et al. Identification of Plasma Proteome Signatures Associated With Surgery Using SOMAscan. Ann. Surg. 2021, 273, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Xiao, Z.; Kalantar-Zadeh, K.; Moradi, H.; Shafi, T.; Waikar, S.S.; Quarles, L.D.; Yu, Z.; Tin, A.; Coresh, J.; et al. Validation of a Novel Modified Aptamer-Based Array Proteomic Platform in Patients with End-Stage Renal Disease. Diagnostics 2018, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- MescoScale. Available online: https://www.bioxpedia.com/meso-scale-discovery-immunoassays/ (accessed on 2 February 2022).
- Günther, A.; Becker, M.; Göpfert, J.; Joos, T.; Schneiderhan-Marra, N. Comparison of Bead-Based Fluorescence Versus Planar Electrochemiluminescence Multiplex Immunoassays for Measuring Cytokines in Human Plasma. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Overview of ELISA in Relation to Other Disciplines. In The ELISA Guidebook; Humana Press: Totowa, NJ, USA, 2009; pp. 1–8. [CrossRef]
- Bastarache, J.A.; Koyama, T.; Wickersham, N.E.; Mitchell, D.B.; Mernaugh, R.L.; Ware, L.B. Accuracy and reproducibility of a multiplex immunoassay platform: A validation study. J. Immunol. Methods 2011, 367, 33–39. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Wu, Y.; Williams, E.G.; Aebersold, R. Application of SWATH Proteomics to Mouse Biology. Curr. Protoc. Mouse Biol. 2017, 7, 130–143. [Google Scholar] [CrossRef]
- Aggarwal, K.; Choe, L.H.; Lee, K.H. Shotgun proteomics using the iTRAQ isobaric tags. Brief. Funct. Genom. 2006, 5, 112–120. [Google Scholar] [CrossRef]
- Picotti, P.; Rinner, O.; Stallmach, R.; Dautel, F.; Farrah, T.; Domon, B.; Wenschuh, H.; Aebersold, R. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 2010, 7, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Su, D.; Liu, T.; Tang, K.; Camp, D.G., 2nd; Qian, W.-J.; Smith, R.D. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 2012, 12, 1074–1092. [Google Scholar] [CrossRef] [PubMed]
- Collett, J.R.; Cho, E.J.; Ellington, A.D. Production and processing of aptamer microarrays. Methods 2005, 37, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Tworoger, S.S.; Stampfer, M.J.; Dillon, S.T.; Gu, X.; Sawyer, S.J.; Chan, A.T.; Libermann, T.A.; Eliassen, A.H. Stability and reproducibility of proteomic profiles measured with an aptamer-based platform. Sci. Rep. 2018, 8, 8382. [Google Scholar] [CrossRef] [PubMed]
- MacLean, E.; Broger, T.; Yerlikaya, S.; Fernandez-Carballo, B.L.; Pai, M.; Denkinger, C.M. A systematic review of biomarkers to detect active tuberculosis. Nat. Microbiol. 2019, 4, 748–758. [Google Scholar] [CrossRef]
- Vidal, M.; Chan, D.W.; Gerstein, M.; Mann, M.; Omenn, G.S.; Tagle, D.; Sechi, S.; Workshop, P. The human proteome-a scientific opportunity for transforming diagnostics, therapeutics, and healthcare. Clin. Proteom. 2012, 9, 6. [Google Scholar] [CrossRef]
- Skates, S.J.; Gillette, M.A.; LaBaer, J.; Carr, S.A.; Anderson, L.; Liebler, D.C.; Ransohoff, D.; Rifai, N.; Kondratovich, M.; Težak, Ž.; et al. Statistical design for biospecimen cohort size in proteomics-based biomarker discovery and verification studies. J. Proteome Res. 2013, 12, 5383–5394. [Google Scholar] [CrossRef]
- Hortin, G.L.; Sviridov, D.; Anderson, N.L. High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin. Chem. 2008, 54, 1608–1616. [Google Scholar] [CrossRef]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.J.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009, 27, 633–641. [Google Scholar] [CrossRef]
- Levin, Y. The role of statistical power analysis in quantitative proteomics. Proteomics 2011, 11, 2565–2567. [Google Scholar] [CrossRef] [PubMed]
- Tsim, S.; Kelly, C.; Alexander, L.; McCormick, C.; Thomson, F.; Woodward, R.; Foster, J.E.; Stobo, D.B.; Paul, J.; Maskell, N.A.; et al. Diagnostic and Prognostic Biomarkers in the Rational Assessment of Mesothelioma (DIAPHRAGM) study: Protocol of a prospective, multicentre, observational study. BMJ Open 2016, 6, e013324. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Dwivedi, A.K.; Deodhar, S.; Mishra, I.; Cistola, D.P. Aptamer-based search for correlates of plasma and serum water T2: Implications for early metabolic dysregulation and metabolic syndrome. Biomark. Res. 2018, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Obuchowski, N.A.; McClish, D.K. Sample size determination for diagnostic accuracy studies involving binormal ROC curve indices. Stat. Med. 1997, 16, 1529–1542. [Google Scholar] [CrossRef]
- Ngo, L.H.; Austin Argentieri, M.; Dillon, S.T.; Kent, B.V.; Kanaya, A.M.; Shields, A.E.; Libermann, T.A. Plasma protein expression profiles, cardiovascular disease, and religious struggles among South Asians in the MASALA study. Sci. Rep. 2021, 11, 961. [Google Scholar] [CrossRef] [PubMed]
- Diz, A.P.; Carvajal-Rodríguez, A.; Skibinski, D.O.F. Multiple hypothesis testing in proteomics: A strategy for experimental work. Mol. Cell Proteom. 2011, 10, M110.004374. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Ilkov, M.; Lamb, J.R.; Finkel, N.; Gudmundsson, E.F.; Pitts, R.; Hoover, H.; Gudmundsdottir, V.; Horman, S.R.; Aspelund, T.; et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 2018, 361, 769–773. [Google Scholar] [CrossRef]
- Kiddle, S.J.; Sattlecker, M.; Proitsi, P.; Simmons, A.; Westman, E.; Bazenet, C.; Nelson, S.K.; Williams, S.; Hodges, A.; Johnston, C.; et al. Candidate blood proteome markers of Alzheimer’s disease onset and progression: A systematic review and replication study. J. Alzheimers Dis. 2014, 38, 515–531. [Google Scholar] [CrossRef]
- Mischak, H.; Critselis, E.; Hanash, S.; Gallagher, W.M.; Vlahou, A.; Ioannidis, J.P.A. Epidemiologic Design and Analysis for Proteomic Studies: A Primer on -Omic Technologies. Am. J. Epidemiol. 2015, 181, 635–647. [Google Scholar] [CrossRef]
- Faresjö, T.; Faresjö, A. To match or not to match in epidemiological studies--same outcome but less power. Int. J. Environ. Res. Public Health 2010, 7, 325–332. [Google Scholar] [CrossRef]
- Kuo, C.-L.; Duan, Y.; Grady, J. Unconditional or Conditional Logistic Regression Model for Age-Matched Case–Control Data? Front. Public Health 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Ngo, D.; Sinha, S.; Shen, D.; Kuhn, E.W.; Keyes, M.J.; Shi, X.; Benson, M.D.; O’Sullivan, J.F.; Keshishian, H.; Farrell, L.A.; et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation 2016, 134, 270–285. [Google Scholar] [CrossRef] [PubMed]
- Karimpour-Fard, A.; Epperson, L.E.; Hunter, L.E. A survey of computational tools for downstream analysis of proteomic and other omic datasets. Hum. Genom. 2015, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.H.; Myers, S.L.; Ye, K.; Walpole, R.E. Probability and Statistics for Engineers and Scientists, 7th ed.; Prentice Hall: London, UK, 2002. [Google Scholar]
- Armstrong, R.A.; Slade, S.V.; Eperjesi, F. An introduction to analysis of variance (ANOVA) with special reference to data from clinical experiments in optometry. Ophthalmic Physiol. Opt. 2000, 20, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Hotelling, H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 417–441. [Google Scholar] [CrossRef]
- Hyvärinen, A.; Oja, E. Independent component analysis: Algorithms and applications. Neural. Netw. 2000, 13, 411–430. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Lloyd, S. Least squares quantization in PCM. IEEE Trans. Inf. Theory 1982, 28, 129–137. [Google Scholar] [CrossRef]
- Rokach, L.; Maimon, O. Clustering Methods; Springer: New York, NY, USA, 2005; pp. 321–352. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Mamidipalli, S.; Huan, T. HAPPI: An online database of comprehensive human annotated and predicted protein interactions. BMC Genom. 2009, 10, S16. [Google Scholar] [CrossRef] [PubMed]
- Swan, A.L.; Mobasheri, A.; Allaway, D.; Liddell, S.; Bacardit, J. Application of machine learning to proteomics data: Classification and biomarker identification in postgenomics biology. Omics 2013, 17, 595–610. [Google Scholar] [CrossRef]
- Bashir, A.; Yang, Q.; Wang, J.; Hoyer, S.; Chou, W.; McLean, C.; Davis, G.; Gong, Q.; Armstrong, Z.; Jang, J.; et al. Machine learning guided aptamer refinement and discovery. Nat. Commun. 2021, 12, 2366. [Google Scholar] [CrossRef]
- Krauze, A.V.; Zhuge, Y.; Zhao, R.; Tasci, E.; Camphausen, K. AI-Driven Image Analysis in Central Nervous System Tumors-Traditional Machine Learning, Deep Learning and Hybrid Models. J. Biotechnol. Biomed. 2022, 5, 1–19. [Google Scholar] [CrossRef]
- Odenkirk, M.T.; Reif, D.M.; Baker, E.S. Multiomic Big Data Analysis Challenges: Increasing Confidence in the Interpretation of Artificial Intelligence Assessments. Anal. Chem. 2021, 93, 7763–7773. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Dong, L.; Watson, J.; Cao, S.; Arregui, S.; Saxena, V.; Ketz, J.; Awol, A.K.; Cohen, D.M.; Caterino, J.M.; Hains, D.S.; et al. Aptamer based proteomic pilot study reveals a urine signature indicative of pediatric urinary tract infections. PLoS ONE 2020, 15, e0235328. [Google Scholar] [CrossRef] [PubMed]
- Mc Ardle, A.; Kwasnik, A.; Szentpetery, A.; Hernandez, B.; Parnell, A.; de Jager, W.; de Roock, S.; FitzGerald, O.; Pennington, S.R. Identification and Evaluation of Serum Protein Biomarkers That Differentiate Psoriatic Arthritis From Rheumatoid Arthritis. Arthritis Rheumatol. 2022, 74, 81–91. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, L.J.; Hu, P.; Liu, Q.; Islam, M.M.; Spicer, V.; Rech, J.; Hueber, A.; Anaparti, V.; Smolik, I.; El-Gabalawy, H.S.; et al. Proteomic Approaches to Defining Remission and the Risk of Relapse in Rheumatoid Arthritis. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, L.; Mathews, J.A.; Devlin, M.; Schutte, C.; Lee, J.; German, D.C. Blood biomarker discovery for autism spectrum disorder: A proteomic analysis. PLoS ONE 2021, 16, e0246581. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Norman, K.C.; Xia, M.; Huang, Y.; Gurczynski, S.J.; Ashley, S.L.; White, E.S.; Flaherty, K.R.; Martinez, F.J.; Murray, S.; et al. The peripheral blood proteome signature of idiopathic pulmonary fibrosis is distinct from normal and is associated with novel immunological processes. Sci. Rep. 2017, 7, 46560. [Google Scholar] [CrossRef]
- Hung, J.-H.; Yang, T.-H.; Hu, Z.; Weng, Z.; DeLisi, C. Gene set enrichment analysis: Performance evaluation and usage guidelines. Brief. Bioinform. 2011, 13, 281–291. [Google Scholar] [CrossRef]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLOS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Gatti, D.M.; Barry, W.T.; Nobel, A.B.; Rusyn, I.; Wright, F.A. Heading down the wrong pathway: On the influence of correlation within gene sets. BMC Genom. 2010, 11, 574. [Google Scholar] [CrossRef]
- Pan, K.-H.; Lih, C.-J.; Cohen, S.N. Effects of threshold choice on biological conclusions reached during analysis of gene expression by DNA microarrays. Proc. Natl. Acad. Sci. USA 2005, 102, 8961–8965. [Google Scholar] [CrossRef]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef]
- Parolo, S.; Marchetti, L.; Lauria, M.; Misselbeck, K.; Scott-Boyer, M.-P.; Caberlotto, L.; Priami, C. Combined use of protein biomarkers and network analysis unveils deregulated regulatory circuits in Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0194225. [Google Scholar] [CrossRef] [PubMed]
- Çelik, H.; Lindblad, K.E.; Popescu, B.; Gui, G.; Goswami, M.; Valdez, J.; DeStefano, C.; Lai, C.; Thompson, J.; Ghannam, J.Y.; et al. Highly multiplexed proteomic assessment of human bone marrow in acute myeloid leukemia. Blood Adv. 2020, 4, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Volsky, D.J. PAGE: Parametric analysis of gene set enrichment. BMC Bioinform. 2005, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Maleki, F.; Ovens, K.L.; Hogan, D.J.; Rezaei, E.; Rosenberg, A.M.; Kusalik, A.J. Measuring consistency among gene set analysis methods: A systematic study. J. Bioinform. Comput. Biol. 2019, 17, 1940010. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-M.; Shafi, A.; Nguyen, T.; Draghici, S. Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biol. 2019, 20, 203. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Sarkar, R.R. Comparison of human cell signaling pathway databases—Evolution, drawbacks and challenges. Database 2015, 2015, 1–25. [Google Scholar] [CrossRef]
- García-Campos, M.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway Analysis: State of the Art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef]
- Norby, F.L.; Tang, W.; Pankow, J.S.; Lutsey, P.L.; Alonso, A.; Steffan, B.; Chen, L.Y.; Zhang, M.; Shippee, N.D.; Ballantyne, C.M.; et al. Proteomics and Risk of Atrial Fibrillation in Older Adults (From the Atherosclerosis Risk in Communities [ARIC] Study). Am. J. Cardiol. 2021, 161, 42–50. [Google Scholar] [CrossRef]
- Sathyan, S.; Ayers, E.; Gao, T.; Weiss, E.F.; Milman, S.; Verghese, J.; Barzilai, N. Plasma proteomic profile of age, health span, and all-cause mortality in older adults. Aging Cell 2020, 19, e13250. [Google Scholar] [CrossRef]
- Green, M.L.; Karp, P.D. The outcomes of pathway database computations depend on pathway ontology. Nucleic Acids Res. 2006, 34, 3687–3697. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Midford, P.E.; Caspi, R.; Khodursky, A. Pathway size matters: The influence of pathway granularity on over-representation (enrichment analysis) statistics. BMC Genom. 2021, 22, 191. [Google Scholar] [CrossRef] [PubMed]
- Battle, A.; Khan, Z.; Wang, S.H.; Mitrano, A.; Ford, M.J.; Pritchard, J.K.; Gilad, Y. Genomic variation. Impact of regulatory variation from RNA to protein. Science 2015, 347, 664–667. [Google Scholar] [CrossRef] [PubMed]
- Chick, J.M.; Munger, S.C.; Simecek, P.; Huttlin, E.L.; Choi, K.; Gatti, D.M.; Raghupathy, N.; Svenson, K.L.; Churchill, G.A.; Gygi, S.P. Defining the consequences of genetic variation on a proteome-wide scale. Nature 2016, 534, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Pietzner, M.; Wheeler, E.; Carrasco-Zanini, J.; Raffler, J.; Kerrison, N.D.; Oerton, E.; Auyeung, V.P.W.; Luan, J.a.; Finan, C.; Casas, J.P.; et al. Genetic architecture of host proteins involved in SARS-CoV-2 infection. Nat. Commun. 2020, 11, 6397. [Google Scholar] [CrossRef]
- Yao, C.; Chen, G.; Song, C.; Keefe, J.; Mendelson, M.; Huan, T.; Sun, B.B.; Laser, A.; Maranville, J.C.; Wu, H.; et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun. 2018, 9, 3268. [Google Scholar] [CrossRef]
- Ferkingstad, E.; Sulem, P.; Atlason, B.A.; Sveinbjornsson, G.; Magnusson, M.I.; Styrmisdottir, E.L.; Gunnarsdottir, K.; Helgason, A.; Oddsson, A.; Halldorsson, B.V.; et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 2021, 53, 1712–1721. [Google Scholar] [CrossRef]
- Sun, B.B.; Maranville, J.C.; Peters, J.E.; Stacey, D.; Staley, J.R.; Blackshaw, J.; Burgess, S.; Jiang, T.; Paige, E.; Surendran, P.; et al. Genomic atlas of the human plasma proteome. Nature 2018, 558, 73–79. [Google Scholar] [CrossRef]
- Di Narzo, A.F.; Telesco, S.E.; Brodmerkel, C.; Argmann, C.; Peters, L.A.; Li, K.; Kidd, B.; Dudley, J.; Cho, J.; Schadt, E.E.; et al. High-Throughput Characterization of Blood Serum Proteomics of IBD Patients with Respect to Aging and Genetic Factors. PLoS Genet. 2017, 13, e1006565. [Google Scholar] [CrossRef]
- Suhre, K.; Arnold, M.; Bhagwat, A.M.; Cotton, R.J.; Engelke, R.; Raffler, J.; Sarwath, H.; Thareja, G.; Wahl, A.; DeLisle, R.K.; et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 2017, 8, 14357. [Google Scholar] [CrossRef]
- Mosley, J.D.; Benson, M.D.; Smith, J.G.; Melander, O.; Ngo, D.; Shaffer, C.M.; Ferguson, J.F.; Herzig, M.S.; McCarty, C.A.; Chute, C.G.; et al. Probing the Virtual Proteome to Identify Novel Disease Biomarkers. Circulation 2018, 138, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Linial, N.; Linial, M. PWAS: Proteome-wide association study—linking genes and phenotypes by functional variation in proteins. Genome Biol. 2020, 21, 173. [Google Scholar] [CrossRef] [PubMed]
- Wingo, A.P.; Liu, Y.; Gerasimov, E.S.; Gockley, J.; Logsdon, B.A.; Duong, D.M.; Dammer, E.B.; Robins, C.; Beach, T.G.; Reiman, E.M.; et al. Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat. Genet. 2021, 53, 143–146. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- King, E.A.; Davis, J.W.; Degner, J.F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019, 15, e1008489. [Google Scholar] [CrossRef]
- Nguyen, V.-T.; Seo, H.B.; Kim, B.C.; Kim, S.K.; Song, C.-S.; Gu, M.B. Highly sensitive sandwich-type SPR based detection of whole H5Nx viruses using a pair of aptamers. Biosens. Bioelectron. 2016, 86, 293–300. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.; Ryu, S.; Han, D. Efficient isolation and elution of cellular proteins using aptamer-mediated protein precipitation assay. Biochem. Biophys. Res. Commun. 2014, 448, 114–119. [Google Scholar] [CrossRef][Green Version]
- Jung, Y.J.; Katilius, E.; Ostroff, R.M.; Kim, Y.; Seok, M.; Lee, S.; Jang, S.; Kim, W.S.; Choi, C.M. Development of a Protein Biomarker Panel to Detect Non-Small-Cell Lung Cancer in Korea. Clin. Lung. Cancer 2017, 18, e99–e107. [Google Scholar] [CrossRef]
- Müller, J.; Friedrich, M.; Becher, T.; Braunstein, J.; Kupper, T.; Berdel, P.; Gravius, S.; Rohrbach, F.; Oldenburg, J.; Mayer, G.; et al. Monitoring of plasma levels of activated protein C using a clinically applicable oligonucleotide-based enzyme capture assay. J. Thromb. Haemost. 2012, 10, 390–398. [Google Scholar] [CrossRef]
- Nakayasu, E.S.; Gritsenko, M.; Piehowski, P.D.; Gao, Y.; Orton, D.J.; Schepmoes, A.A.; Fillmore, T.L.; Frohnert, B.I.; Rewers, M.; Krischer, J.P.; et al. Tutorial: Best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 2021, 16, 3737–3760. [Google Scholar] [CrossRef]
- Füzéry, A.K.; Levin, J.; Chan, M.M.; Chan, D.W. Translation of proteomic biomarkers into FDA approved cancer diagnostics: Issues and challenges. Clin Proteom. 2013, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chan, D.W. The road from discovery to clinical diagnostics: Lessons learned from the first FDA-cleared in vitro diagnostic multivariate index assay of proteomic biomarkers. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2995–2999. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Morrow, D.A.; de Lemos, J.A.; Gibson, C.M.; Murphy, S.A.; Rifai, N.; McCabe, C.; Antman, E.M.; Cannon, C.P.; Braunwald, E. Multimarker approach to risk stratification in non-ST elevation acute coronary syndromes: Simultaneous assessment of troponin I, C-reactive protein, and B-type natriuretic peptide. Circulation 2002, 105, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Harlid, S.; Harbs, J.; Myte, R.; Brunius, C.; Gunter, M.J.; Palmqvist, R.; Liu, X.; Van Guelpen, B. A two-tiered targeted proteomics approach to identify pre-diagnostic biomarkers of colorectal cancer risk. Sci. Rep. 2021, 11, 5151. [Google Scholar] [CrossRef] [PubMed]
- Ueland, F.R.; Desimone, C.P.; Seamon, L.G.; Miller, R.A.; Goodrich, S.; Podzielinski, I.; Sokoll, L.; Smith, A.; van Nagell, J.R., Jr.; Zhang, Z. Effectiveness of a multivariate index assay in the preoperative assessment of ovarian tumors. Obs. Gynecol. 2011, 117, 1289–1297. [Google Scholar] [CrossRef]
- Argilés, Á.; Siwy, J.; Duranton, F.; Gayrard, N.; Dakna, M.; Lundin, U.; Osaba, L.; Delles, C.; Mourad, G.; Weinberger, K.M.; et al. CKD273, a new proteomics classifier assessing CKD and its prognosis. PLoS ONE 2013, 8, e62837. [Google Scholar] [CrossRef]
- Nkuipou-Kenfack, E.; Zürbig, P.; Mischak, H. The long path towards implementation of clinical proteomics: Exemplified based on CKD273. Proteomics–Clin. Appl. 2017, 11, 1600104. [Google Scholar] [CrossRef]
- Bratulic, S.; Gatto, F.; Nielsen, J. The Translational Status of Cancer Liquid Biopsies. Regen. Eng. Transl. Med. 2021, 7, 312–352. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, J.H.; Welsh, S.J.; Ahn, S.B.; Breen, E.; Khan, A.; Carlino, M.S.; Menzies, A.M.; Kefford, R.F.; Scolyer, R.A.; et al. Evaluation of two high-throughput proteomic technologies for plasma biomarker discovery in immunotherapy-treated melanoma patients. Biomark. Res. 2017, 5, 32. [Google Scholar] [CrossRef]
- Raffield, L.M.; Dang, H.; Pratte, K.A.; Jacobson, S.; Gillenwater, L.A.; Ampleford, E.; Barjaktarevic, I.; Basta, P.; Clish, C.B.; Comellas, A.P.; et al. Comparison of Proteomic Assessment Methods in Multiple Cohort Studies. Proteomics 2020, 20, e1900278. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.X.; Thiessen-Philbrook, H.R.; Vasan, R.S.; Coresh, J.; Ganz, P.; Bonventre, J.V.; Kimmel, P.L.; Parikh, C.R. Comparison of proteomic methods in evaluating biomarker-AKI associations in cardiac surgery patients. Transl. Res. 2021, 238, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Billing, A.M.; Ben Hamidane, H.; Bhagwat, A.M.; Cotton, R.J.; Dib, S.S.; Kumar, P.; Hayat, S.; Goswami, N.; Suhre, K.; Rafii, A.; et al. Complementarity of SOMAscan to LC-MS/MS and RNA-seq for quantitative profiling of human embryonic and mesenchymal stem cells. J. Proteom. 2017, 150, 86–97. [Google Scholar] [CrossRef]
- Berglund, L.; Bjorling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Szigyarto, C.A.; Persson, A.; Ottosson, J.; Wernerus, H.; Nilsson, P.; et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell Proteom. 2008, 7, 2019–2027. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Analytical Technique | Category | Protein Sample Literature Values └ | Accepted Biospecimen Types | Reported Dynamic Range ‡ | CV └└ | Protein Capacity Multiplex) |
|---|---|---|---|---|---|---|
| Proximity Extension Assay (Olink) [11] | Antibody | 1 µL | Plasma, tissue/cell, synovial fluid, CSF, plaque extract, and saliva | LLOQ = 0.25 pg/mL | 7.8% (intra) and 10.6% (inter) | *** |
| Reverse Phase Protein Arrays [12,39,40,41,42] | Antibody | 5 µg (1.0 to 1.5 mg/mL protein) | Tissue/cell, plasma, serum, biopsies, body fluids | LOD = 0.55 fg/mL | <15% | *** |
| Bio-Plex [13,43] | Antibody (bead) | 12.5 µL (serum/plasma) 50 µL (cell culture) | Plasma, serum, tissue/cell | LOD = 0.6–6.4 pg/mL | 2–15% | **** |
| Simoa [14,44,45] | Antibody (bead) | 25 µL | Plasma, serum, urine, tissue/cell, CSF, saliva | LOD = 0.005 pg/mL | <10% | *** |
| Aptamer Group (Optmer) [15] | Aptamer | 38 µL | Plasma (diagnostics and therapeutics), urine, tissue/cell, liquid matrices | LOD = 55 ng/mL | <5% | * |
| Base Pair Technologies [16,26] | Aptamer | 5–100 µL | Plasma, serum, tissue/cell | LOD = 1 pg/mL | ** | |
| SOMAscan [9,27,46,47,48,49] | Aptamer | 55–100 µL | Plasma, serum, CSF, urine, cell/tissue, synovial fluid, exosomes | LOD = 1.6 pg/mL | 4.6% | ****** |
| Electrochemiluminescence Immunoassay (Meso Scale and Lumit) [17,50] | ECLIA | 50 µL | Plasma, serum, tissue/cell, CSF, urine, blood spots, tears, synovial fluid, tissue extracts | LOD = fg/mL | 5–10% | ** |
| Multiplex ELISA [1,18,43,51,52] | ELISA | 25–50 µL | Plasma, serum, tissue/cell, urine, saliva, CSF | LOD = 0.61 to 18.90 pg/mL | 9.5–28.5% (inter/intra) | ** |
| Singleplex ELISA [53] | ELISA | 100 µL | Plasma, serum, tissue/cell, urine, saliva, CSF | LOD = pg/mL | 1.6–6.4% (intra) and 3.8–7.1% (inter) | * |
| 2D-PAGE [19] | Gel electrophoresis | ~100 µg (15–50 µL) | Plasma, serum, tissue/cell, urine | LOD = 10 ng to 100 ng | <20% | ****** |
| DDA-MS [20] | MS | 10 µL | Plasma, serum, tissue/cell | LOD = 157 ng/mL | 5.7% | ***** |
| SWATH-MS [21,54,55] | MS (DIA) | 5–10 µg | Plasma, serum tissue/cell, platelets, monocytes/neutrophils | LOD = 1 fmol | 13.7% | ***** |
| iTRAQ [22,56] | MS (labeling in LC–MS–MS) | 12 µg | Plasma, serum, tissue/cells, saliva | LOD = 1 fmol (50 µg/mL) | <0.53% | ***** |
| SRM/MRM [23,57,58] | MS (LC–MS–MS) | 15 µL | Plasma, tissue/cell, dried blood spots | LOD = µg/mL (no enrichment) | 6.1% (intra) and 11% (inter) | ** |
| Quality Control | ||
|---|---|---|
| Student’s t-test or nonparametric Wilcoxon | Mean difference between two groups | [80] |
| ANOVA or nonparametric Kruskal–Wallis | Variation between two or more groups | [81] |
| Visualization methods | Histogram, density plots, box and bar graphs | |
| Classification | ||
| Principle Component Analysis (PCA) | Dimension reduction, separates groups based upon commonality | [82] |
| Independent Component Analysis (ICA) | Dimension reduction, separates groups based upon correlation | [83] |
| Partial Least Squares (PLS) | Discriminant analysis that separates groups by maximum covariation ranks the important features | [84] |
| Random Forest (RF) | Separates groups by similarity, ranks important features | [85] |
| Support-Vector Machine (SVM) | Classifies the sample by kernel function | [86] |
| Clustering | ||
| K-means | Clustering of features or samples into user-specified numbers of clusters | [87] |
| Hierarchical | Unsupervised classification of features, samples, or any endpoint by dendrogram | [88] |
| Pathways | ||
| Gene Set Enrichment Analysis (GSEA) | Pathway analysis and functional annotation | [89] |
| Ingenuity Pathway Analysis (IPA) | Pathway and functional annotation from curated databases | |
| Database for Annotation, Visualization, and Integrated Discovery (DAVID) | Pathway and functional annotation using Gene Ontology (GO) | [90] |
| Cytoscape | Network analysis visualization | [91] |
| Kyoto Encyclopedia of Genes and Genomes (KEGG) | Pathway analysis | [92] |
| Human Annotated and Predicted Protein Interactions (HAPPI) | Protein interactions | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Jones, J.C.; Shankavaram, U.; Sproull, M.; Camphausen, K.; Krauze, A.V. Analytical Considerations of Large-Scale Aptamer-Based Datasets for Translational Applications. Cancers 2022, 14, 2227. https://doi.org/10.3390/cancers14092227
Jiang W, Jones JC, Shankavaram U, Sproull M, Camphausen K, Krauze AV. Analytical Considerations of Large-Scale Aptamer-Based Datasets for Translational Applications. Cancers. 2022; 14(9):2227. https://doi.org/10.3390/cancers14092227
Chicago/Turabian StyleJiang, Will, Jennifer C. Jones, Uma Shankavaram, Mary Sproull, Kevin Camphausen, and Andra V. Krauze. 2022. "Analytical Considerations of Large-Scale Aptamer-Based Datasets for Translational Applications" Cancers 14, no. 9: 2227. https://doi.org/10.3390/cancers14092227
APA StyleJiang, W., Jones, J. C., Shankavaram, U., Sproull, M., Camphausen, K., & Krauze, A. V. (2022). Analytical Considerations of Large-Scale Aptamer-Based Datasets for Translational Applications. Cancers, 14(9), 2227. https://doi.org/10.3390/cancers14092227