The Inhibitory Effects of Anti-ERC/Mesothelin Antibody 22A31 on Colorectal Adenocarcinoma Cells, within a Mouse Xenograft Model

 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cells and Antibodies
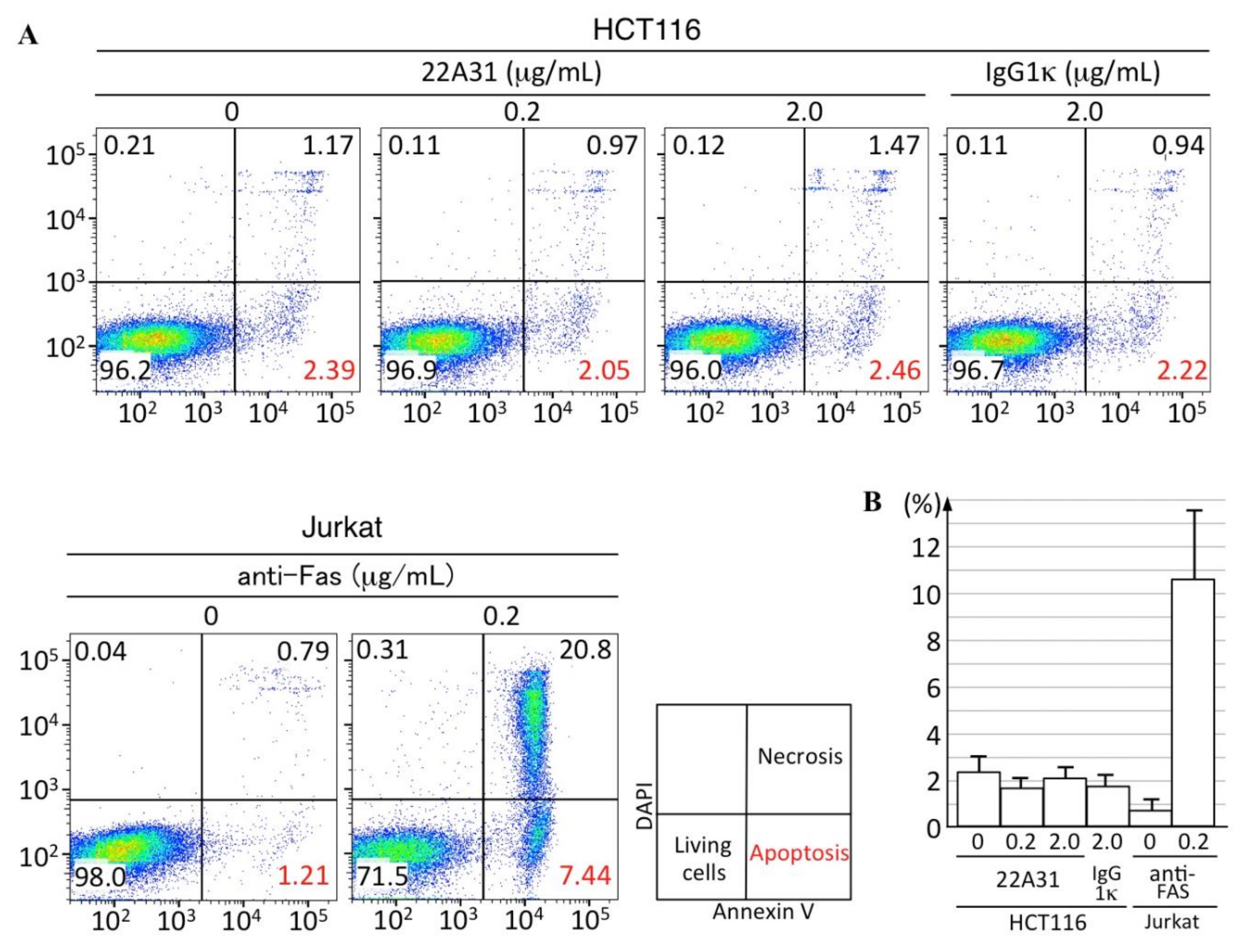
2.3. Flow Cytometric Analysis
2.4. Anti-Tumor Activity of 22A31 mAb in BALB⁄c nu⁄nu Mice
2.5. Immunohistochemistry
2.6. TdT-Mediated dUTP Nick End Labeling (TUNEL) Assay
2.7. Quantification
2.8. Statistical Analysis
3. Results
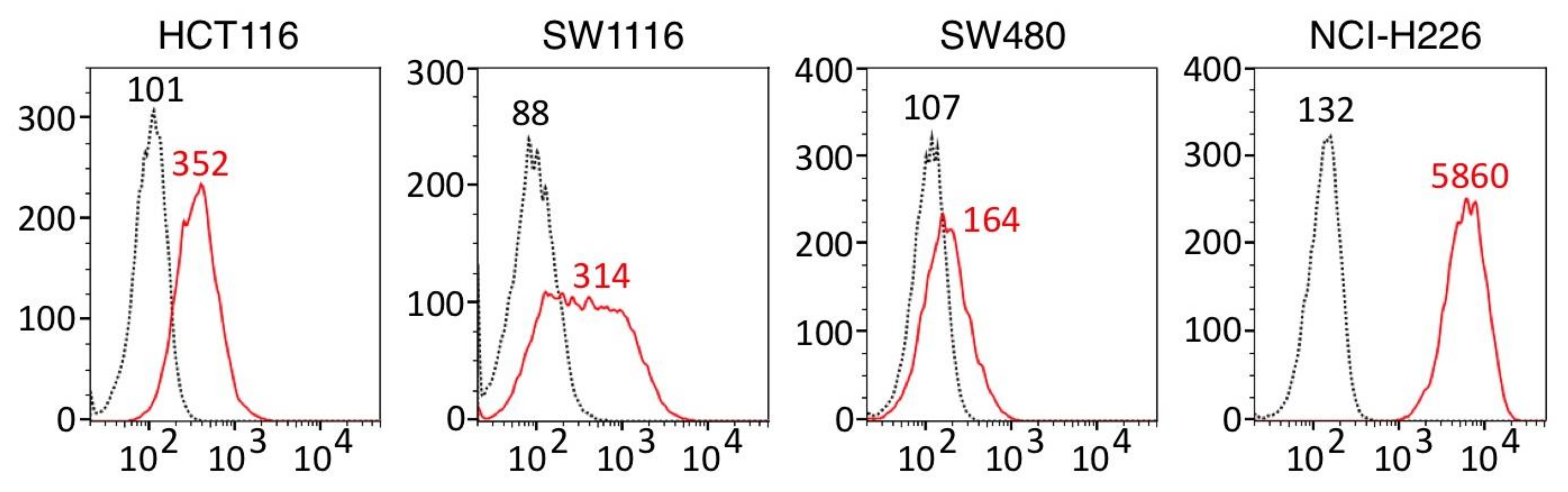
3.1. ERC⁄Mesothelin Expression in Colorectal Adenocarcinoma Cells
3.2. Anti-Tumor Activity of 22A31 in Mice Xenograft Model
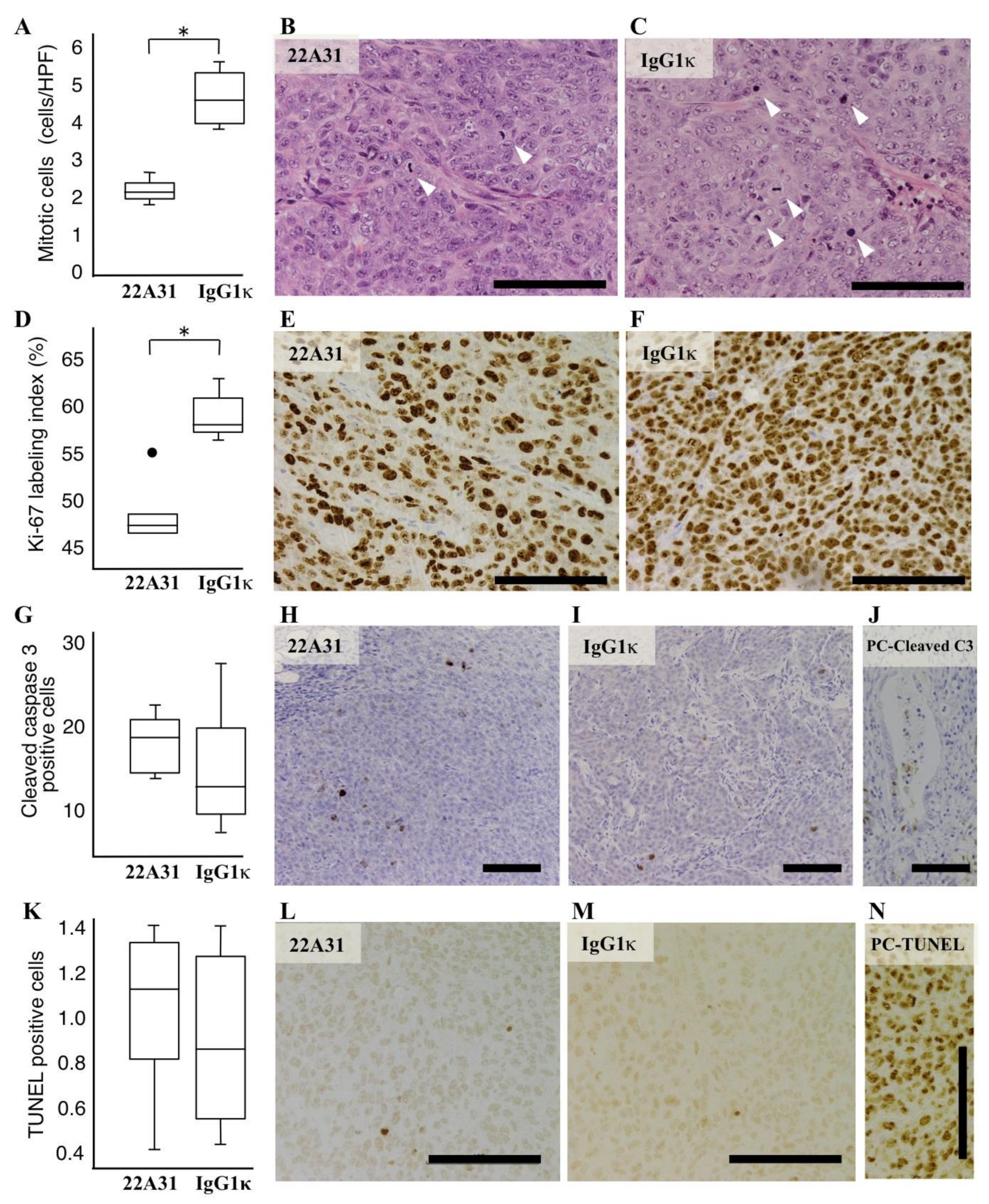
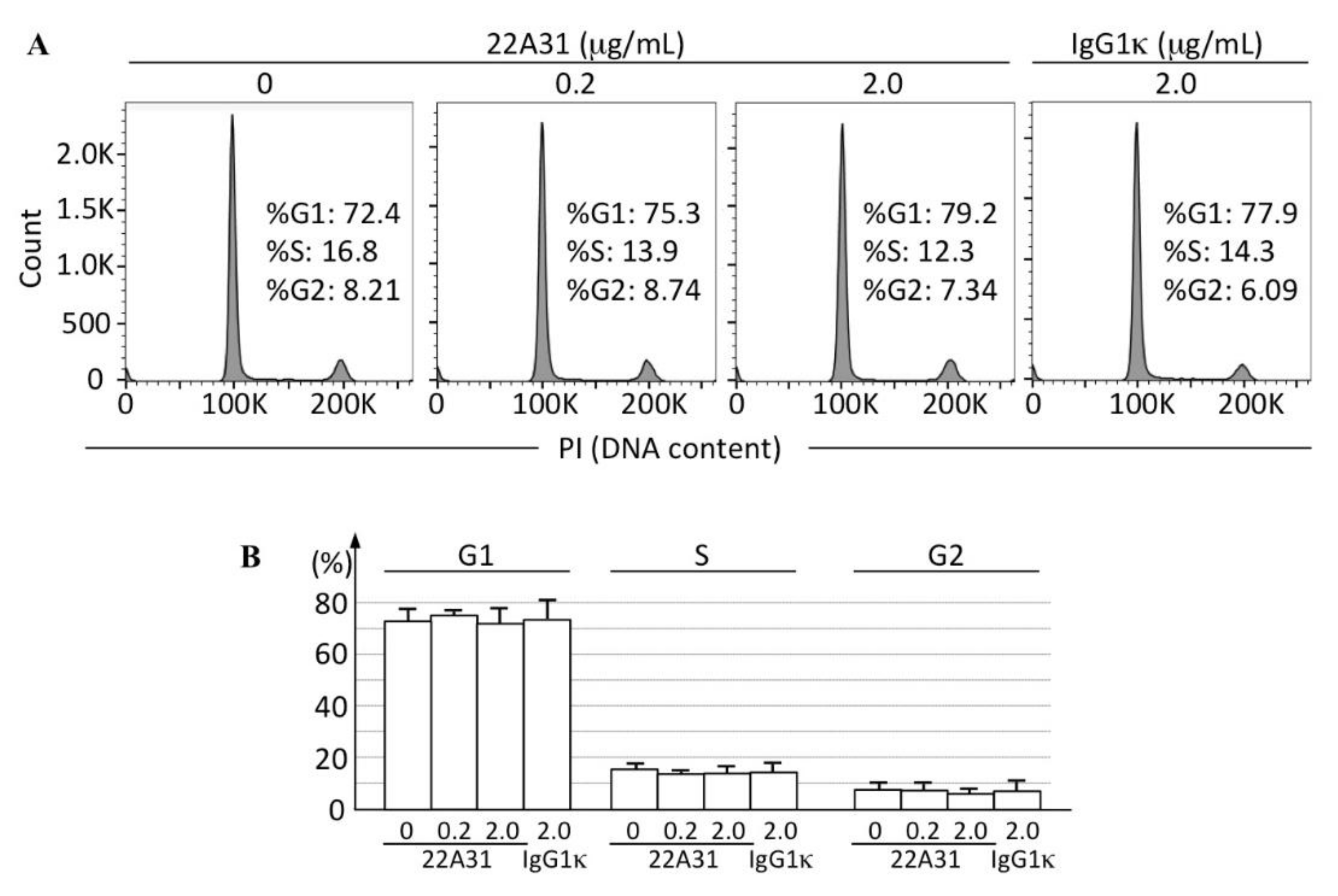
3.3. Characteristics of Xenograft Tumor Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Pastan, I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl. Acad. Sci. USA. 1996, 93, 136–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argani, P.; Iacobuzio-Donahue, C.; Ryu, B.; Rosty, C.; Goggins, M.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Jaffee, E.; Yeo, C.J.; et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer. Res. 2001, 7, 3862–3868. [Google Scholar] [PubMed]
- Hassan, R.; Kreitman, R.J.; Pastan, I.; Willingham, M.C. Localization of mesothelin in epithelial ovarian cancer. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 243–247. [Google Scholar] [CrossRef]
- Ho, M.; Bera, T.K.; Willingham, M.C.; Onda, M.; Hassan, R.; FitzGerald, D.; Pastan, I. Mesothelin expression in human lung cancer. Clin. Cancer. Res. 2007, 13, 1571–1575. [Google Scholar] [CrossRef] [Green Version]
- Dainty, L.A.; Risinger, J.I.; Morrison, C.; Chandramouli, G.V.R.; Bidus, M.A.; Zahn, C.; Rose, G.S.; Fowler, J.; Berchuck, A.; Maxwell, G.L. Overexpression of folate binding protein and mesothelin are associated with uterine serous carcinoma. Gynecol. Oncol. 2007, 105, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, D.; Onda, M.; Voigt, A.; Dawczynski, K.; Wittig, S.; Hassan, R.; Gruhn, B.; Pastan, I. Mesothelin, a possible target for immunotherapy, is expressed in primary AML cells. Eur. J. Haematol. 2007, 79, 281–286. [Google Scholar] [CrossRef]
- Yu, L.; Feng, M.; Kim, H.; Phung, Y.; Kleiner, D.E.; Gores, G.J.; Qian, M.; Wang, X.W.; Ho, M. Mesothelin as a potential therapeutic target in human cholangiocarcinoma. J. Cancer. 2010, 1, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Qian, M.; Ho, M. The role of mesothelin in tumor progression and targeted therapy. Anticancer. Agents. Med. Chem. 2013, 13, 276–280. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Li, M.; Chen, C.; Yal, Q. Mesothelin-induced pancreatic cancer cell proliferation involves alteration of cyclin E via activation of signal transducer and activator transcription protein 3. Mol. Cancer Res. 2008, 6, 1755–1765. [Google Scholar] [CrossRef] [Green Version]
- Faust, J.R.; Hamill, D.; Kolb, E.A.; Gopalakrishnapillai, A.; Barwe, S.P. Mesothelin: An immunotherapeutic target beyond solid tumors. Cancers 2022, 14, 1550. [Google Scholar] [CrossRef] [PubMed]
- Rump, A.; Morikawa, Y.; Tanaka, M.; Minami, S.; Umesaki, N.; Takeuchi, M.; Miyajima, A. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J. Biol. Chem. 2004, 279, 9190–9198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, A.; Hirono, S.; Tani, M.; Kawai, M.; Okada, I.; Miyazawa, M.; Kitahara, Y.; Nakamura, Y.; Noda, T.; Yokoyama, S.; et al. Coexpression of MUC16 and mesothelin is related to the invasion process in pancreatic ductal adenocarcinoma. Cancer. Sci. 2012, 103, 739–746. [Google Scholar] [CrossRef]
- Hassan, R.; Ebel, W.; Routhier, E.L.; Patel, R.; Kline, J.B.; Zhang, J.; Chao, Q.; Jacob, S.; Turchin, H.; Gibbs, L.; et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007, 7, 20. [Google Scholar]
- Hassan, R.; Cohen, S.J.; Phillips, M.; Pastan, I.; Sharon, E.; Kelly, R.J.; Schweizer, C.; Weil, S.; Laheru, D. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin. Cancer. Res. 2010, 16, 6132–6138. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, F.; Kamachi, H.; Mizukami, T.; Einama, T.; Kawamata, F.; Fujii, Y.; Fukai, M.; Kobayashi, N.; Hatanaka, Y.; Taketomi, A. Mesothelin blockage by Amatuximab suppresses cell invasiveness, enhances gemcitabine sensitivity and regulates cancer cell stemness in mesothelin-positive pancreatic cancer cells. BMC Cancer 2021, 21, 200. [Google Scholar] [CrossRef]
- Lv, J.; Li, P. Mesothelin as a biomarker for targteted therapy. Biomark Res. 2019, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreitman, R.J.; Hassan, R.; Fitzgerald, D.J.; Pastan, I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin. Cancer. Res. 2009, 15, 5274–5279. [Google Scholar] [CrossRef] [Green Version]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab ravtansine: A novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Le, Q.; Castro, S.; Tang, T.; Loeb, A.M.; Hylkema, T.; McKay, C.N.; Perkins, L.; Srivastava, S.; Call, L.; Smith, J.; et al. Therapeutic Targeting of Mesothelin with Chimeric Antigen Receptor T Cells in Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 5718–5730. [Google Scholar] [CrossRef]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, F.; Homma, S.; Kamachi, H.; Einama, T.; Kato, Y.; Tsuda, M.; Tanaka, S.; Maeda, M.; Kajino, K.; Hino, O.; et al. C-ERC/mesothelin provokes lymphatic invasion of colorectal adenocarcinoma. J. Gastroenterol. 2014, 49, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Wang, Z.; Lasota, J.; Onda, M.; Czapiwski, P.; Langfort, R.; Rys, J.; Szpor, J.; Waloszczyk, P.; Okon, K.; et al. Comprehensive immunohistochemical study of mesothelin (MSLN) using different monoclonal antibodies 5B2 and MN-1 in 1562 tumors with evaluation of its prognostic value in malignant pleural mesothelioma. Oncotarget 2017, 8, 26744–26754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Tsunoda, T.; Riku, M.; Ito, H.; Inoko, A.; Murakami, H.; Ebi, M.; Ogasawara, N.; Pastan, I.; Kasugai, K.; et al. Diffuse mesothelin expression leads to worse prognosis through enhanced cellular proliferation in colorectal cancer. Oncol. Lett. 2020, 19, 1741–1750. [Google Scholar] [CrossRef]
- Inami, K.; Abe, M.; Takeda, K.; Hagiwara, Y.; Maeda, M.; Segawa, T.; Suyama, M.; Watanabe, S.; Hino, O. Antitumor activity of anti-C-ERC/mesothelin monoclonal antibody in vivo. Cancer. Sci. 2010, 101, 969–974. [Google Scholar] [CrossRef]
- Long, B.H.; Willson, J.K.V.; Brattain, D.E.; Musial, S.; Brattain, M.G. Effects of mitomycin on human colon carcinoma cells. J. Natl. Cancer. Inst. 1984, 73, 787–792. [Google Scholar]
- Phelps, R.M.; Johnson, B.E.; Ihde, D.C.; Gazdar, A.F.; Carbone, D.P.; McClintock, P.R.; Linnoila, R.I.; Matthews, M.J.; Bunn, P.A., Jr.; Carney, D.; et al. NCI-navy medical oncology branch cell line data base. J. Cell. Biochem. 1996, 63, 32–91. [Google Scholar] [CrossRef]
- Ishikawa, K.; Segawa, T.; Hagiwara, Y.; Maeda, M.; Abe, M.; Hino, O. Establishment of novel monoclonal antibody to human ERC/mesothelin useful for study and diagnosis of ERC/mesothelin-expressing cancers. Pathol. Int. 2009, 59, 161–166. [Google Scholar] [CrossRef]
- Chen, Q.; Zeng, Y.-N.; Zhang, K.; Zhao, Y.; Wu, Y.-Y.; Li, G.; Cheng, H.-Y.; Zhang, M.; Lai, F.; Wang, J.-B.; et al. Polydatin increases radiosensitivity by inducing apoptosis of stem cells in colorectal cancer. Int. J. Biol. Sci. 2019, 15, 430–440. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Bodempudi, V.; Liu, Z.; Borrego-Diaz, E.; Yamoutpoor, F.; Meyer, A.; Woo, R.A.; Pan, W.; Dudek, A.Z.; Olyaee, M.S.; et al. Inhibition of mesothelin as a novel strategy for targeting cancer cells. PLoS ONE 2012, 7, e33214. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, I.; Horejsi, V.; Ansotegui, I.J.; Knapp, W.; Stockinger, H. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science 1991, 254, 1016–1019. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taniguchi, G.; Kajino, K.; Momose, S.; Saeki, H.; Yue, L.; Ohtsuji, N.; Abe, M.; Shibuya, T.; Orimo, A.; Nagahara, A.; et al. The Inhibitory Effects of Anti-ERC/Mesothelin Antibody 22A31 on Colorectal Adenocarcinoma Cells, within a Mouse Xenograft Model. Cancers 2022, 14, 2198. https://doi.org/10.3390/cancers14092198
Taniguchi G, Kajino K, Momose S, Saeki H, Yue L, Ohtsuji N, Abe M, Shibuya T, Orimo A, Nagahara A, et al. The Inhibitory Effects of Anti-ERC/Mesothelin Antibody 22A31 on Colorectal Adenocarcinoma Cells, within a Mouse Xenograft Model. Cancers. 2022; 14(9):2198. https://doi.org/10.3390/cancers14092198
Chicago/Turabian StyleTaniguchi, Gentaro, Kazunori Kajino, Shuji Momose, Harumi Saeki, Liang Yue, Naomi Ohtsuji, Masaaki Abe, Tomoyoshi Shibuya, Akira Orimo, Akihito Nagahara, and et al. 2022. "The Inhibitory Effects of Anti-ERC/Mesothelin Antibody 22A31 on Colorectal Adenocarcinoma Cells, within a Mouse Xenograft Model" Cancers 14, no. 9: 2198. https://doi.org/10.3390/cancers14092198
APA StyleTaniguchi, G., Kajino, K., Momose, S., Saeki, H., Yue, L., Ohtsuji, N., Abe, M., Shibuya, T., Orimo, A., Nagahara, A., Watanabe, S., & Hino, O. (2022). The Inhibitory Effects of Anti-ERC/Mesothelin Antibody 22A31 on Colorectal Adenocarcinoma Cells, within a Mouse Xenograft Model. Cancers, 14(9), 2198. https://doi.org/10.3390/cancers14092198