
Comparing the Secretomes of Chemorefractory and Chemoresistant Ovarian Cancer Cell Populations


Abstract
Simple Summary
Abstract
1. Introduction
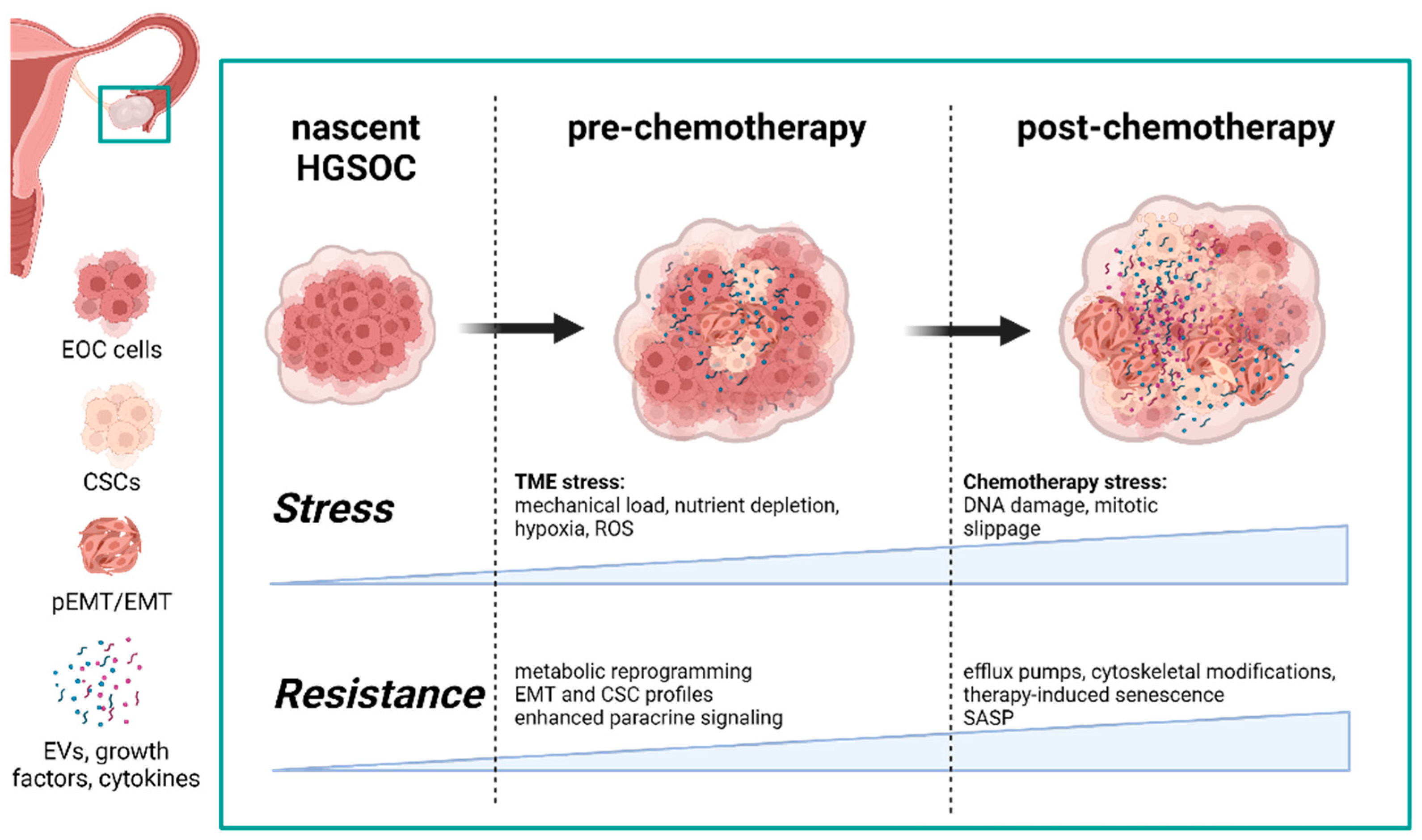
1.1. Extrinsic Tme Stressors Promote Intra- and Intercellular Adaptations
1.2. Metabolic Reprogramming as a Priming Mechanism in Response to TME-Associated Stress
1.3. Chemorefractory HGSOC Highlights the Priming Capabilities of the TME
2. Differences in the Development of Chemorefractory and Chemoresistant HGSOC Populations
2.1. Hypoxia Confers Resistance in Refractory HGSOC
2.2. Hypoxia Alters HGSOC Secretome Profile
2.2.1. Cytokines
2.2.2. Growth Factors
2.2.3. EVs
2.3. Hypoxia Alters HGSOC Exosomes
2.4. Acquired Chemoresistance Is Achieved through Drug-Specific Adaptations and Induction of Therapy-Induced Senescence
2.5. Therapy-Induced Senescence and Escape as a Mechanism for Recurrent Disease
2.6. Therapy-Induced Chemoresistance Alters the HGSOC Secretome
2.6.1. Cytokines
2.6.2. Growth Factors
2.6.3. EVs
2.7. Role of Exosomes in Developing Chemoresistance
2.8. Changes in miRNAs
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Christie, E.L.; Bowtell, D.D.L. Acquired Chemotherapy Resistance in Ovarian Cancer. Ann. Oncol. 2017, 28, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Robert, C.; Bast, J. Epithelial Ovarian Cancer. In Holland-Frei Cancer Medicine, 6th ed.; Oxford University Press: Melbourne, Australia, 2003. [Google Scholar]
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian Cancer; Lancet: London, UK, 2009; pp. 1371–1382. [Google Scholar]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S. Induction of Apoptosis as Well as Necrosis by Hypoxia and Predominant Prevention of Apoptosis by Bcl-2 and Bcl-X. Cancer Res. 1996, 56, 2161–2166. [Google Scholar] [PubMed]
- Le Maout, V.; Alessandri, K.; Gurchenkov, B.; Bertin, H.; Nassoy, P.; Sciumè, G. Role of Mechanical Cues and Hypoxia on the Growth of Tumor Cells in Strong and Weak Confinement: A Dual in Vitro–in Silico Approach. Sci. Adv. 2020, 6, eaaz7130. [Google Scholar] [CrossRef]
- McGrail, D.J.; Kieu, Q.M.N.; Dawson, M.R. The Malignancy of Metastatic Ovarian Cancer Cells Is Increased on Soft Matrices through a Mechanosensitive Rho-ROCK Pathway. J. Cell Sci. 2014, 127, 2621–2626. [Google Scholar] [CrossRef]
- Novak, C.; Horst, E.; Mehta, G. Review: Mechanotransduction in Ovarian Cancer: Shearing into the Unknown. APL Bioeng. 2018, 2, 031701. [Google Scholar] [CrossRef]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17. [Google Scholar] [CrossRef]
- Bregenzer, M.E.; Horst, E.N.; Mehta, P.; Novak, C.M.; Repetto, T.; Mehta, G. The Role of Cancer Stem Cells and Mechanical Forces in Ovarian Cancer Metastasis. Cancers 2019, 11, 1008. [Google Scholar] [CrossRef]
- Kipps, E.; Tan, D.S.P.; Kaye, S.B. Meeting the Challenge of Ascites in Ovarian Cancer: New Avenues for Therapy and Research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Sakai, R. Direct Interaction between Carcinoma Cells and Cancer Associated Fibroblasts for the Regulation of Cancer Invasion. Cancers 2015, 7, 2054–2062. [Google Scholar] [CrossRef]
- Jang, I.; Beningo, K.A. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers 2019, 11, 721. [Google Scholar] [CrossRef] [PubMed]
- Carey, S.P.; Martin, K.E.; Reinhart-King, C.A. Three-Dimensional Collagen Matrix Induces a Mechanosensitive Invasive Epithelial Phenotype. Sci. Rep. 2017, 7, 42088. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Bird, D.; Baker, A.-M.; Barker, H.E.; Ho, M.W.-Y.; Lang, G.; Erler, J.T. LOX-Mediated Collagen Crosslinking Is Responsible for Fibrosis-Enhanced Metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-Dependent Matrix Remodelling Is Required for the Generation and Maintenance of Cancer-Associated Fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, S.; Zhou, L.; Seyhan, A.A.; Hernandez Borrero, L.; Zhang, Y.; El-Deiry, W.S. Targeting the Integrated Stress Response in Cancer Therapy. Front. Pharmacol. 2021, 12, 747837. [Google Scholar] [CrossRef]
- Reich, S.; Nguyen, C.D.L.; Has, C.; Steltgens, S.; Soni, H.; Coman, C.; Freyberg, M.; Bichler, A.; Seifert, N.; Conrad, D.; et al. A Multi-Omics Analysis Reveals the Unfolded Protein Response Regulon and Stress-Induced Resistance to Folate-Based Antimetabolites. Nat. Commun. 2020, 11, 2936. [Google Scholar] [CrossRef]
- Heaster, T.M.; Landman, B.A.; Skala, M.C. Quantitative Spatial Analysis of Metabolic Heterogeneity Across in Vivo and in Vitro Tumor Models. Front. Oncol. 2019, 9, 1144. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of Glycolysis by the Hypoxia-Inducible Factor (HIF): Implications for Cellular Physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.-H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173.e10. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione Biosynthesis Is a Metabolic Vulnerability in PI(3)K/Akt-Driven Breast Cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic Shifts toward Glutamine Regulate Tumor Growth, Invasion and Bioenergetics in Ovarian Cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine Reliance in Cell Metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Ding, X.; Allmeroth, K.; Biggs, L.C.; Kolenc, O.I.; L’Hoest, N.; Chacón-Martínez, C.A.; Edlich-Muth, C.; Giavalisco, P.; Quinn, K.P.; et al. Glutamine Metabolism Controls Stem Cell Fate Reversibility and Long-Term Maintenance in the Hair Follicle. Cell Metab. 2020, 32, 629–642.e8. [Google Scholar] [CrossRef]
- Pacifico, F.; Badolati, N.; Mellone, S.; Stornaiuolo, M.; Leonardi, A.; Crescenzi, E. Glutamine Promotes Escape from Therapy-Induced Senescence in Tumor Cells. Aging (Albany NY) 2021, 13, 20962–20991. [Google Scholar] [CrossRef]
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Karaminejadranjbar, M.; Hu, Z.; et al. An Evolving Story of the Metastatic Voyage of Ovarian Cancer Cells: Cellular and Molecular Orchestration of the Adipose-Rich Metastatic Microenvironment. Oncogene 2019, 38, 2885–2898. [Google Scholar] [CrossRef]
- Xu, Y. Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment. Cancers 2018, 10, 227. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.-C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling Cancer Persister Cells Arise from Lineages with Distinct Programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A Signature of Enhanced Lipid Metabolism, Lipid Peroxidation and Aldehyde Stress in Therapy-Induced Senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef]
- Zhao, G.; Cardenas, H.; Matei, D. Ovarian Cancer-Why Lipids Matter. Cancers 2019, 11, 1870. [Google Scholar] [CrossRef] [PubMed]
- Badolia, R.; Manne, B.K.; Dangelmaier, C.; Chernoff, J.; Kunapuli, S.P. Gq-Mediated Akt Translocation to the Membrane: A Novel PIP3-Independent Mechanism in Platelets. Blood 2015, 125, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, B.T.; Qamar, L.; Yamamoto, T.M.; McMellen, A.; Watson, Z.L.; Richer, J.K.; Behbakht, K.; Schlaepfer, I.R.; Bitler, B.G. Targeting Fatty Acid Oxidation to Promote Anoikis and Inhibit Ovarian Cancer Progression. Mol. Cancer Res. 2020, 18, 1088–1098. [Google Scholar] [CrossRef]
- Chen, R.R.; Yung, M.M.H.; Xuan, Y.; Zhan, S.; Leung, L.L.; Liang, R.R.; Leung, T.H.Y.; Yang, H.; Xu, D.; Sharma, R.; et al. Targeting of Lipid Metabolism with a Metabolic Inhibitor Cocktail Eradicates Peritoneal Metastases in Ovarian Cancer Cells. Commun. Biol. 2019, 2, 1–15. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and Cancer: Modulation of Cell Death Pathways and Cancer Cell Adaptations. J. Cell Biol. 2019, 219, e201909033. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and Oxidative Stress: The Role of Aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef]
- Grieco, J.P.; Allen, M.E.; Perry, J.B.; Wang, Y.; Song, Y.; Rohani, A.; Compton, S.L.E.; Smyth, J.W.; Swami, N.S.; Brown, D.A.; et al. Progression-Mediated Changes in Mitochondrial Morphology Promotes Adaptation to Hypoxic Peritoneal Conditions in Serous Ovarian Cancer. Front. Oncol. 2021, 10, 600113. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Sánchez-Rivera, F.J.; Ryan, J.; Soto-Feliciano, Y.M.; Clare Beytagh, M.; Xuan, L.; Feldser, D.M.; Hemann, M.T.; Zamudio, J.; Dimitrova, N.; Letai, A.; et al. Mitochondrial Apoptotic Priming Is a Key Determinant of Cell Fate upon P53 Restoration. Proc. Natl. Acad. Sci. USA 2021, 118, e2019740118. [Google Scholar] [CrossRef] [PubMed]
- Ni Chonghaile, T.; Sarosiek, K.A.; Vo, T.-T.; Ryan, J.A.; Tammareddi, A.; Moore, V.D.G.; Deng, J.; Anderson, K.C.; Richardson, P.; Tai, Y.-T.; et al. Pretreatment Mitochondrial Priming Correlates with Clinical Response to Cytotoxic Chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.; Macario, E.C. Chaperonopathies by Defect, Excess, or Mistake. Ann. N. Y. Acad. Sci. 2007, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Stenvers, K.L. Getting to Know Ovarian Cancer Ascites: Opportunities for Targeted Therapy-Based Translational Research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef]
- Lane, D.; Matte, I.; Garde-Granger, P.; Laplante, C.; Carignan, A.; Rancourt, C.; Piché, A. Inflammation-Regulating Factors in Ascites as Predictive Biomarkers of Drug Resistance and Progression-Free Survival in Serous Epithelial Ovarian Cancers. BMC Cancer 2015, 15, 492. [Google Scholar] [CrossRef]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-Induced Exosomes Contribute to a More Aggressive and Chemoresistant Ovarian Cancer Phenotype: A Novel Mechanism Linking STAT3/Rab Proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour Cell Secretome in Chemoresistance and Tumour Recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef]
- Jafari, R.; Rahbarghazi, R.; Ahmadi, M.; Hassanpour, M.; Rezaie, J. Hypoxic Exosomes Orchestrate Tumorigenesis: Molecular Mechanisms and Therapeutic Implications. J. Transl. Med. 2020, 18, 474. [Google Scholar] [CrossRef]
- Venturella, M.; Criscuoli, M.; Carraro, F.; Naldini, A.; Zocco, D. Interplay between Hypoxia and Extracellular Vesicles in Cancer and Inflammation. Biology 2021, 10, 606. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.; Cristea, M.; DeRosa, M.; Eisenhauer, E.L.; et al. Ovarian Cancer, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 191–226. [Google Scholar] [CrossRef] [PubMed]
- McMullen, M.; Karakasis, K.; Madariaga, A.; Oza, A.M. Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers 2020, 12, 1607. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Khambhati, N.N.; Qi, M.X.; Patel, K.S.; Ravikumar, N.; Brandenburg, C.P.; Dawson, M.R. Alterations in Ovarian Cancer Cell Adhesion Drive Taxol Resistance by Increasing Microtubule Dynamics in a FAK-Dependent Manner. Sci. Rep. 2015, 5, 9529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Erkan, E.P.; Jamalzadeh, S.; Dai, J.; Andersson, N.; Kaipio, K.; Lamminen, T.; Mansuri, N.; Huhtinen, K.; Carpén, O.; et al. Longitudinal Single-Cell RNA-Seq Analysis Reveals Stress-Promoted Chemoresistance in Metastatic Ovarian Cancer. Sci. Adv. 2022, 8, eabm1831. [Google Scholar] [CrossRef]
- Kan, T.; Zhang, S.; Zhou, S.; Zhang, Y.; Zhao, Y.; Gao, Y.; Zhang, T.; Gao, F.; Wang, X.; Zhao, L.; et al. Single-Cell RNA-Seq Recognized the Initiator of Epithelial Ovarian Cancer Recurrence. Oncogene 2022, 41, 895–906. [Google Scholar] [CrossRef]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; DiSaia, P.; Gabra, H.; Glenn, P.; et al. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-Genome Characterization of Chemoresistant Ovarian Cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for Cancer Therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-α Prolyl Hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Sonveaux, P.; Copetti, T.; Saedeleer, C.J.D.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the Lactate Transporter MCT1 in Endothelial Cells Inhibits Lactate-Induced HIF-1 Activation and Tumor Angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-Inducible Factor 1 Activation by Aerobic Glycolysis Implicates the Warburg Effect in Carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-Y.; Song, H.S.; Park, S.-Y.; Chung, S.-H.; Kim, Y.-J. Pyruvate Promotes Tumor Angiogenesis through HIF-1-Dependent PAI-1 Expression. Int. J. Oncol. 2011, 38, 571–576. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pan, Y.; Mansfield, K.D.; Bertozzi, C.C.; Rudenko, V.; Chan, D.A.; Giaccia, A.J.; Simon, M.C. Multiple Factors Affecting Cellular Redox Status and Energy Metabolism Modulate Hypoxia-Inducible Factor Prolyl Hydroxylase Activity In Vivo and In Vitro. Mol. Cell. Biol. 2007, 27, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial Fission Causes Cisplatin Resistance under Hypoxic Conditions via ROS in Ovarian Cancer Cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef]
- McEvoy, L.M.; O’Toole, S.A.; Spillane, C.D.; Martin, C.M.; Gallagher, M.F.; Stordal, B.; Blackshields, G.; Sheils, O.; O’Leary, J.J. Identifying Novel Hypoxia-Associated Markers of Chemoresistance in Ovarian Cancer. BMC Cancer 2015, 15, 547. [Google Scholar] [CrossRef]
- Huang, L.; Ao, Q.; Zhang, Q.; Yang, X.; Xing, H.; Li, F.; Chen, G.; Zhou, J.; Wang, S.; Xu, G.; et al. Hypoxia Induced Paclitaxel Resistance in Human Ovarian Cancers via Hypoxia-Inducible Factor 1alpha. J. Cancer Res. Clin. Oncol. 2010, 136, 447–456. [Google Scholar] [CrossRef]
- Singh, S.K.; Mishra, M.K.; Singh, R. Hypoxia-Inducible Factor-1α Induces CX3CR1 Expression and Promotes the Epithelial to Mesenchymal Transition (EMT) in Ovarian Cancer Cells. J. Ovarian Res. 2019, 12, 42. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Guadamillas, M.C.; Cerezo, A.; Pozo, M.A. Overcoming Anoikis–Pathways to Anchorage-Independent Growth in Cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT Spectrum Defines an Anoikis-Resistant and Spheroidogenic Intermediate Mesenchymal State That Is Sensitive to e-Cadherin Restoration by a Src-Kinase Inhibitor, Saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K.; Balamurugan, K. Hypoxia, Partial EMT and Collective Migration: Emerging Culprits in Metastasis. Transl. Oncol. 2020, 13, 100845. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the Tumour Transition States Occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Montoya, C.A.; Donnenberg, V.S.; Sant, S. Interplay between Tumor Microenvironment and Partial EMT as the Driver of Tumor Progression. iScience 2021, 24, 102113. [Google Scholar] [CrossRef]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-Mapping Post-Hypoxic Tumor Cells Reveals a ROS-Resistant Phenotype That Promotes Metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef]
- Long, F.; Liu, W.; Jia, P.; Wang, H.; Jiang, G.; Wang, T. HIF-1α-Induced Autophagy Contributes to Cisplatin Resistance in Ovarian Cancer Cells. Pharmazie 2018, 73, 533–536. [Google Scholar] [CrossRef]
- Pagotto, A.; Pilotto, G.; Mazzoldi, E.L.; Nicoletto, M.O.; Frezzini, S.; Pastò, A.; Amadori, A. Autophagy Inhibition Reduces Chemoresistance and Tumorigenic Potential of Human Ovarian Cancer Stem Cells. Cell Death Dis. 2017, 8, e2943. [Google Scholar] [CrossRef]
- Qin, J.; Liu, Y.; Lu, Y.; Liu, M.; Li, M.; Li, J.; Wu, L. Hypoxia-Inducible Factor 1 Alpha Promotes Cancer Stem Cells-like Properties in Human Ovarian Cancer Cells by Upregulating SIRT1 Expression. Sci. Rep. 2017, 7, 10592. [Google Scholar] [CrossRef]
- Seo, E.J.; Kim, D.K.; Jang, I.H.; Choi, E.J.; Shin, S.H.; Lee, S.I.; Kwon, S.-M.; Kim, K.-H.; Suh, D.-S.; Kim, J.H. Hypoxia-NOTCH1-SOX2 Signaling Is Important for Maintaining Cancer Stem Cells in Ovarian Cancer. Oncotarget 2016, 7, 55624–55638. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.A.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The Unfolded Protein Response Protects Human Tumor Cells during Hypoxia through Regulation of the Autophagy Genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; Hulver, M.W.; Schmelz, E.M. Ovarian Tumor-Initiating Cells Display a Flexible Metabolism. Exp. Cell Res. 2014, 328, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian Cancer Spheroid Cells with Stem Cell-like Properties Contribute to Tumor Generation, Metastasis and Chemotherapy Resistance through Hypoxia-Resistant Metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.-X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef]
- Liang, D.; Ma, Y.; Liu, J.; Trope, C.G.; Holm, R.; Nesland, J.M.; Suo, Z. The Hypoxic Microenvironment Upgrades Stem-like Properties of Ovarian Cancer Cells. BMC Cancer 2012, 12, 201. [Google Scholar] [CrossRef]
- Duan, L.; Tao, J.; Yang, X.; Ye, L.; Wu, Y.; He, Q.; Duan, Y.; Chen, L.; Zhu, J. HVEM/HIF-1α Promoted Proliferation and Inhibited Apoptosis of Ovarian Cancer Cells under Hypoxic Microenvironment Conditions. J. Ovarian Res. 2020, 13, 40. [Google Scholar] [CrossRef]
- Rosen, D.G.; Mercado-Uribe, I.; Yang, G.; Bast, R.C.; Amin, H.M.; Lai, R.; Liu, J. The Role of Constitutively Active Signal Transducer and Activator of Transcription 3 in Ovarian Tumorigenesis and Prognosis. Cancer 2006, 107, 2730–2740. [Google Scholar] [CrossRef]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early Responses of the STAT3 Pathway to Platinum Drugs Are Associated with Cisplatin Resistance in Epithelial Ovarian Cancer. Braz. J. Med. Biol. Res. 2013, 46, 650–658. [Google Scholar] [CrossRef]
- Abubaker, K.; Luwor, R.B.; Escalona, R.; McNally, O.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Targeted Disruption of the JAK2/STAT3 Pathway in Combination with Systemic Administration of Paclitaxel Inhibits the Priming of Ovarian Cancer Stem Cells Leading to a Reduced Tumor Burden. Front. Oncol. 2014, 4, 75. [Google Scholar] [CrossRef]
- Selvendiran, K.; Bratasz, A.; Kuppusamy, M.L.; Tazi, M.F.; Rivera, B.K.; Kuppusamy, P. Hypoxia Induces Chemoresistance in Ovarian Cancer Cells by Activation of Signal Transducer and Activator of Transcription 3. Int. J. Cancer 2009, 125, 2198–2204. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Feng, J.; Hong, Z.; Chen, L.; Li, W.; Liao, S.; Wang, X.; Ji, T.; Wang, S.; Ma, D.; et al. Silencing of the STAT3 Signaling Pathway Reverses the Inherent and Induced Chemoresistance of Human Ovarian Cancer Cells. Biochem. Biophys. Res. Commun. 2013, 435, 188–194. [Google Scholar] [CrossRef]
- da Cunha, B.R.; Domingos, C.; Stefanini, A.C.B.; Henrique, T.; Polachini, G.M.; Castelo-Branco, P.; Tajara, E.H. Cellular Interactions in the Tumor Microenvironment: The Role of Secretome. J. Cancer 2019, 10, 4574–4587. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.-N.; Wang, G.; Hood, B.L.; Conrads, K.A.; Hamilton, C.A.; Maxwell, G.L.; Darcy, K.M.; Conrads, T.P. Identification of Candidate Circulating Cisplatin-Resistant Biomarkers from Epithelial Ovarian Carcinoma Cell Secretomes. Br. J. Cancer 2014, 110, 123–132. [Google Scholar] [CrossRef] [PubMed]
- López de Andrés, J.; Griñán-Lisón, C.; Jiménez, G.; Marchal, J.A. Cancer Stem Cell Secretome in the Tumor Microenvironment: A Key Point for an Effective Personalized Cancer Treatment. J. Hematol. Oncol. 2020, 13, 136. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef]
- Browning, L.; Patel, M.R.; Horvath, E.B.; Tawara, K.; Jorcyk, C.L. IL-6 and Ovarian Cancer: Inflammatory Cytokines in Promotion of Metastasis. Cancer Manag. Res. 2018, 10, 6685–6693. [Google Scholar] [CrossRef]
- Matte, I.; Lane, D.; Laplante, C.; Rancourt, C.; Piché, A. Profiling of Cytokines in Human Epithelial Ovarian Cancer Ascites. Am. J. Cancer Res. 2012, 2, 566–580. [Google Scholar]
- Granados, M.L.; Hudson, L.G.; Samudio-Ruiz, S.L. Contributions of the Epidermal Growth Factor Receptor to Acquisition of Platinum Resistance in Ovarian Cancer Cells. PLoS ONE 2015, 10, 0136893. [Google Scholar] [CrossRef]
- Lee, A.H.; Ghosh, D.; Quach, N.; Schroeder, D.; Dawson, M.R. Ovarian Cancer Exosomes Trigger Differential Biophysical Response in Tumor-Derived Fibroblasts. Sci. Rep. 2020, 10, 8686. [Google Scholar] [CrossRef]
- Alharbi, M.; Lai, A.; Sharma, S.; Kalita-de Croft, P.; Godbole, N.; Campos, A.; Guanzon, D.; Salas-Burgos, A.; Carrion, F.; Zuñiga, F.A.; et al. Extracellular Vesicle Transmission of Chemoresistance to Ovarian Cancer Cells Is Associated with Hypoxia-Induced Expression of Glycolytic Pathway Proteins, and Prediction of Epithelial Ovarian Cancer Disease Recurrence. Cancers 2021, 13, 3388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, X.L.; Qu, Y.; Wu, J.; Zhu, Y.Q.; Sun, W.J.; Li, L.Z. Autocrine Production of Interleukin-6 Confers Cisplatin and Paclitaxel Resistance in Ovarian Cancer Cells. Cancer Lett. 2010, 295, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zong, X.; Mitra, S.; Mitra, A.K.; Matei, D.; Nephew, K.P. IL-6 Mediates Platinum-Induced Enrichment of Ovarian Cancer Stem Cells. JCI Insight 2018, 3, 122360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gu, X.; Xia, L.; Zhou, Y.; Bouamar, H.; Yang, J.; Ding, X.; Zwieb, C.; Zhang, J.; Hinck, A.P.; et al. A Novel TGFβ Trap Blocks Chemotherapeutics-Induced TGFβ1 Signaling and Enhances Their Anticancer Activity in Gynecologic Cancers. Clin Cancer Res. 2018, 24, 2780–2793. [Google Scholar] [CrossRef]
- Lane, D.; Matte, I.; Rancourt, C.; Piché, A. Prognostic Significance of IL-6 and IL-8 Ascites Levels in Ovarian Cancer Patients. BMC Cancer 2011, 11, 210. [Google Scholar] [CrossRef]
- Li, Z.; Yan-Qing, W.; Xiao, Y.; Shi-Yi, L.; Meng-Qin, Y.; Shu, X.; Dong-Yong, Y.; Ya-Jing, Z.; Yan-Xiang, C. Exosomes Secreted by Chemoresistant Ovarian Cancer Cells Promote Angiogenesis. J. Ovarian Res. 2021, 14, 7. [Google Scholar] [CrossRef]
- Hergueta-Redondo, M.; Peinado, H. The Influence of Secreted Factors and Extracellular Vesicles in Ovarian Cancer Metastasis. Eur. J. Cancer Suppl. 2020, 15, 38–48. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Ernst, M.; Putoczki, T.L. Molecular Pathways: IL11 as a Tumor-Promoting Cytokine—Translational Implications for Cancers. Clin. Cancer Res. 2014, 20, 5579–5588. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, B.; Zhang, K.; Song, Y.; Zhang, L.; Xi, M. IL-27 Suppresses SKOV3 Cells Proliferation by Enhancing STAT3 and Inhibiting the Akt Signal Pathway. Mol. Immunol. 2016, 78, 155–163. [Google Scholar] [CrossRef]
- Liu, C.; Wang, Y.; Song, H.; Li, Q.; Zhang, Y.; Chen, P.; Song, Y.; Su, M.; Huang, Q.; Wang, M.; et al. Genetic Association of Interleukin-31 Gene Polymorphisms with Epithelial Ovarian Cancer in Chinese Population. Dis. Markers 2018, 2018, e3503858. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 Produced by Tumor Microenvironment Promotes Self-Renewal of CD133+ Cancer Stem-like Cells in Ovarian Cancer. Oncogene 2015, 34, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The Tumor Microenvironment and Its Role in Promoting Tumor Growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Macciò, A.; Madeddu, C. Inflammation and Ovarian Cancer. Cytokine 2012, 58, 133–147. [Google Scholar] [CrossRef]
- Thibault, B.; Castells, M.; Delord, J.-P.; Couderc, B. Ovarian Cancer Microenvironment: Implications for Cancer Dissemination and Chemoresistance Acquisition. Cancer Metastasis Rev. 2014, 33, 17–39. [Google Scholar] [CrossRef]
- Xu, L.; Xie, K.; Mukaida, N.; Matsushima, K.; Fidler, I.J. Hypoxia-Induced Elevation in Interleukin-8 Expression by Human Ovarian Carcinoma Cells. Cancer Res. 1999, 59, 5822–5829. [Google Scholar]
- Xu, L.; Fidler, I.J. Acidic PH-Induced Elevation in Interleukin 8 Expression by Human Ovarian Carcinoma Cells 1. Cancer Res. 2000, 60, 4610–4616. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Regulation of NF-ΚB by TNF Family Cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-KappaB Activation Pathways, Emerging Molecular Targets for Cancer Prevention and Therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.G.; Zeineldin, R.; Silberberg, M.; Stack, M.S. Activated Epidermal Growth Factor Receptor in Ovarian Cancer. Cancer Treat. Res. 2009, 149, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zhang, H.; Jia, Z.; Cui, M.; Tian, J. Chemoresistance and Targeting of Growth Factors/Cytokines Signalling Pathways: Towards the Development of Effective Therapeutic Strategy for Endometrial Cancer. Am. J. Cancer Res. 2018, 8, 1317–1331. [Google Scholar] [PubMed]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular Adaptation to Hypoxia through Hypoxia Inducible Factors and Beyond. Nat. Re-Views Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Cheng, J.C.; Klausen, C.; Leung, P.C.K. Hypoxia-Inducible Factor 1 Alpha Mediates Epidermal Growth Factor-Induced down-Regulation of E-Cadherin Expression and Cell Invasion in Human Ovarian Cancer Cells. Cancer Lett. 2013, 329, 197–206. [Google Scholar] [CrossRef]
- Wong, C.; Wellman, T.L.; Lounsbury, K.M. VEGF and HIF-1α Expression Are Increased in Advanced Stages of Epithelial Ovarian Cancer. Gynecol. Oncol. 2003, 91, 513–517. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF Targets the Tumour Cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Blanc, L.; Vidal, M. New Insights into the Function of Rab GTPases in the Context of Exosomal Secretion. Small GTPases 2018, 9, 95–106. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, J.; Miao, Y.; Zhang, Q. The Effect of Extracellular Vesicles on the Regulation of Mitochondria under Hypoxia. Cell Death Dis. 2021, 12, 358. [Google Scholar] [CrossRef]
- Milman, N.; Ginini, L.; Gil, Z. Exosomes and Their Role in Tumorigenesis and Anticancer Drug Resistance. Drug Resist. Updates 2019, 45, 1–12. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Role of the RAB7 Protein in Tumor Progression and Cisplatin Chemoresistance. Cancers 2019, 11, 1096. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Lin, Y.-S.; Pan, Y.-C.; Tsai, P.-H.; Wu, C.-Y.; Kuo, P.-L. Hypoxic Lung Cancer-Secreted Exosomal MiR-23a Increased Angiogenesis and Vascular Permeability by Targeting Prolyl Hydroxylase and Tight Junction Protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M. Emerging Roles of Extracellular Vesicles in Cellular Senescence and Aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef]
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-Inducible Factors and RAB22A Mediate Formation of Microvesicles That Stimulate Breast Cancer Invasion and Metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242. [Google Scholar] [CrossRef]
- Ohta, T.; Takahashi, T.; Shibuya, T.; Amita, M.; Henmi, N.; Takahashi, K.; Kurachi, H. Inhibition of the Rho/ROCK Pathway Enhances the Efficacy of Cisplatin through the Blockage of Hypoxia-Inducible Factor-1α in Human Ovarian Cancer Cells. Cancer Biol. Ther. 2012, 13, 25–33. [Google Scholar] [CrossRef]
- Shao, C.; Yang, F.; Miao, S.; Liu, W.; Wang, C.; Shu, Y.; Shen, H. Role of Hypoxia-Induced Exosomes in Tumor Biology. Mol. Cancer 2018, 17, 120. [Google Scholar] [CrossRef]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-Term Single Treatment of Chemotherapy Results in the Enrichment of Ovarian Cancer Stem Cell-like Cells Leading to an Increased Tumor Burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of Taxane Resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.; van Deemter, L.; Smit, J.J.; van der Valk, M.A.; Voordouw, A.C.; Spits, H.; van Tellingen, O.; et al. Normal Viability and Altered Pharmacokinetics in Mice Lacking Mdr1-Type (Drug-Transporting) P-Glycoproteins. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef]
- Du, F.; Wu, X.; Liu, Y.; Wang, T.; Qi, X.; Mao, Y.; Jiang, L.; Zhu, Y.; Chen, Y.; Zhu, R.; et al. Acquisition of Paclitaxel Resistance via PI3K-dependent Epithelial-mesenchymal Transition in A2780 Human Ovarian Cancer Cells. Oncol. Rep. 2013, 30, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Hu, Y.; Fu, Y.; Zhou, T.; You, J.; Du, J.; Zheng, L.; Cao, J.; Ying, M.; Dai, X.; et al. Targeting Slug-Mediated Non-Canonical Activation of c-Met to Overcome Chemo-Resistance in Metastatic Ovarian Cancer Cells. Acta Pharm. Sin. B 2019, 9, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to Paclitaxel Induces Epithelial-Mesenchymal Transition and Enhances Metastatic Potential for Epithelial Ovarian Carcinoma Cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Cianfrocca, R.; Spinella, F.; Di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of Chemoresistance and EMT Phenotype Is Linked with Activation of the Endothelin A Receptor Pathway in Ovarian Carcinoma Cells. Clin. Cancer Res. 2011, 17, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.D.; Keller, A.; Zhou, B.; Tian, R.; Bruce, J.E. Cellular Interactome Dynamics during Paclitaxel Treatment. Cell Rep. 2019, 29, 2371–2383.e5. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-Associated Reprogramming Promotes Cancer Stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many Roads Lead to Oncogene-Induced Senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Pellegrini, M.; Pallini, R.; Levi, A.; Falchetti, M.L. Cancer Response to Therapy-Induced Senescence: A Matter of Dose and Timing. Cancers 2021, 13, 484. [Google Scholar] [CrossRef]
- Dalton, W.B.; Nandan, M.O.; Moore, R.T.; Yang, V.W. Human Cancer Cells Commonly Acquire DNA Damage during Mitotic Arrest. Cancer Res. 2007, 67, 11487–11492. [Google Scholar] [CrossRef]
- Xuan, B.; Ghosh, D.; Cheney, E.M.; Clifton, E.M.; Dawson, M.R. Dysregulation in Actin Cytoskeletal Organization Drives Increased Stiffness and Migratory Persistence in Polyploidal Giant Cancer Cells. Sci. Rep. 2018, 8, 11935. [Google Scholar] [CrossRef] [PubMed]
- Hain, K.O.; Colin, D.J.; Rastogi, S.; Allan, L.A.; Clarke, P.R. Prolonged Mitotic Arrest Induces a Caspase-Dependent DNA Damage Response at Telomeres That Determines Cell Survival. Sci. Rep. 2016, 6, 26766. [Google Scholar] [CrossRef] [PubMed]
- Sonego, M.; Pellizzari, I.; Dall’Acqua, A.; Pivetta, E.; Lorenzon, I.; Benevol, S.; Bomben, R.; Spessotto, P.; Sorio, R.; Gattei, V.; et al. Common Biological Phenotypes Characterize the Acquisition of Platinum-Resistance in Epithelial Ovarian Cancer Cells. Sci. Rep. 2017, 7, 7104. [Google Scholar] [CrossRef]
- Yoon, M.-J.; Cha, H.; Ahn, J.; Lee, D.; Jeong, H.-S.; Koo, H.S.; Kang, Y.-J. Dysfunctional Activity of Classical DNA End-Joining Renders Acquired Resistance to Carboplatin in Human Ovarian Cancer Cells. Cancer Lett. 2021, 520, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.M.; Rizzo, S.; van der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA Methylation Drivers of Acquired Cisplatin Resistance in Ovarian Cancer Identified by Methylome and Expression Profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S. Three Steps to the Immortality of Cancer Cells: Senescence, Polyploidy and Self-Renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Faheem, M.M.; Seligson, N.D.; Ahmad, S.M.; Rasool, R.U.; Gandhi, S.G.; Bhagat, M.; Goswami, A. Convergence of Therapy-Induced Senescence (TIS) and EMT in Multistep Carcinogenesis: Current Opinions and Emerging Perspectives. Cell Death Discov. 2020, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Nam, J.-S. The Force Awakens: Metastatic Dormant Cancer Cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The Dynamic Nature of Senescence in Cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Fitsiou, E.; Soto-Gamez, A.; Demaria, M. Biological Functions of Therapy-Induced Senescence in Cancer. Semin. Cancer Biol. 2021, S1044-579X(21)00071-7. [Google Scholar] [CrossRef]
- Pawlik, W.; Pawlik, J.; Kozłowski, M.; Łuczkowska, K.; Kwiatkowski, S.; Kwiatkowska, E.; Machaliński, B.; Cymbaluk-Płoska, A. The Clinical Importance of IL-6, IL-8, and TNF-α in Patients with Ovarian Carcinoma and Benign Cystic Lesions. Diagnostics 2021, 11, 1625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, W.; Wang, X.; Wang, X.; Sun, H. Prognostic Value of Serum IL-8 and IL-10 in Patients with Ovarian Cancer Undergoing Chemotherapy. Oncol. Lett. 2019, 17, 2365–2369. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, B.; Goode, E.L.; Kalli, K.R.; Knutson, K.L.; DeRycke, M.S. The Immune System in the Pathogenesis of Ovarian Cancer. Crit. Rev. Immunol. 2013, 33, 137–164. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Thompson, R.; Wilson, J.L.; Robinson, S.; Hagemann, T.; Fatah, R.; Gould, D.; Ayhan, A.; Balkwill, F. The Inflammatory Cytokine Tumor Necrosis Factor-α Generates an Autocrine Tumor-Promoting Network in Epithelial Ovarian Cancer Cells. Cancer Res. 2007, 67, 585–592. [Google Scholar] [CrossRef]
- Coward, J.; Kulbe, H.; Chakravarty, P.; Leader, D.; Vassileva, V.; Leinster, D.A.; Thompson, R.; Schioppa, T.; Nemeth, J.; Vermeulen, J.; et al. Interleukin-6 as a Therapeutic Target in Human Ovarian Cancer. Clin. Cancer Res. 2011, 17, 6083–6096. [Google Scholar] [CrossRef]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-Dependent EGFR Activation Induces the Co-Expression of IL-6 and PAI-1 via the NFkB Pathway in Advanced-Stage Epithelial Ovarian Cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-ΚB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Wu, C.-J.; Sundararajan, V.; Sheu, B.-C.; Huang, R.Y.-J.; Wei, L.-H. Activation of STAT3 and STAT5 Signaling in Epithelial Ovarian Cancer Progression: Mechanism and Therapeutic Opportunity. Cancers 2019, 12, 24. [Google Scholar] [CrossRef]
- Gui, T.; Shen, K. The Epidermal Growth Factor Receptor as a Therapeutic Target in Epithelial Ovarian Cancer. Cancer Epidemiol. 2012, 36, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Zhang, X.; Paladino, D.; Sengupta, B.; Ahmad, S.; Holloway, R.W.; Ingersoll, S.B.; Turkson, J. Hyperactive EGF Receptor, Jaks and Stat3 Signaling Promote Enhanced Colony-Forming Ability, Motility and Migration of Cisplatin-Resistant Ovarian Cancer Cells. Oncogene 2012, 31, 2309–2322. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.C.; Haisley, C.; Hurteau, J.; Moser, T.L.; Whitaker, R.; Bast, R.C.; Stack, M.S. Regulation of Invasion of Epithelial Ovarian Cancer by Transforming Growth Factor-Beta. Gynecol. Oncol. 2001, 80, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-β Induces Global Changes in DNA Methylation during the Epithelial-to-Mesenchymal Transition in Ovarian Cancer Cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Merlot, B.; Lucot, J.-P.; Collinet, P.; Vinatier, D.; Fournier, I.; Salzet, M. Epithelial-Mesenchymal Transition in Ovarian Cancer. Cancer Lett. 2010, 291, 59–66. [Google Scholar] [CrossRef]
- Chan, M.W.; Huang, Y.-W.; Hartman-Frey, C.; Kuo, C.-T.; Deatherage, D.; Qin, H.; Cheng, A.S.; Yan, P.S.; Davuluri, R.V.; Huang, T.H.-M.; et al. Aberrant Transforming Growth Factor Beta1 Signaling and SMAD4 Nuclear Translocation Confer Epigenetic Repression of ADAM19 in Ovarian Cancer. Neoplasia 2008, 10, 908–919. [Google Scholar] [CrossRef]
- Yeh, K.-T.; Chen, T.-H.; Yang, H.-W.; Chou, J.-L.; Chen, L.-Y.; Yeh, C.-M.; Chen, Y.-H.; Lin, R.-I.; Su, H.-Y.; Chen, G.C.W.; et al. Aberrant TGFβ/SMAD4 Signaling Contributes to Epigenetic Silencing of a Putative Tumor Suppressor, RunX1T1 in Ovarian Cancer. Epigenetics 2011, 6, 727–739. [Google Scholar] [CrossRef]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal Lysosomal Trafficking and Enhanced Exosomal Export of Cisplatin in Drug-Resistant Human Ovarian Carcinoma Cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef]
- Namee, N.M.; O’Driscoll, L. Extracellular vesicles and anti-cancer drug resistance. Biochim. Biophys. Acta (BBA) Rev. Cancer 2018, 1870, 123–136. [Google Scholar] [CrossRef]
- Samuel, P.; Mulcahy, L.A.; Furlong, F.; McCarthy, H.O.; Brooks, S.A.; Fabbri, M.; Pink, R.C.; Carter, D.R.F. Cisplatin Induces the Release of Extracellular Vesicles from Ovarian Cancer Cells That Can Induce Invasiveness and Drug Resistance in Bystander Cells. Philos. Trans. R Soc. Lond. B Biol. Sci. 2018, 373, 20170065. [Google Scholar] [CrossRef]
- Crow, J.; Atay, S.; Banskota, S.; Artale, B.; Schmitt, S.; Godwin, A.K. Exosomes as Mediators of Platinum Resistance in Ovarian Cancer. Oncotarget 2017, 8, 11917–11936. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- van Schooneveld, E.; Wildiers, H.; Vergote, I.; Vermeulen, P.B.; Dirix, L.Y.; Van Laere, S.J. Dysregulation of MicroRNAs in Breast Cancer and Their Potential Role as Prognostic and Predictive Biomarkers in Patient Management. Breast Cancer Res. 2015, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Stevic, I.; Müller, V.; Ni, Q.; Oliveira-Ferrer, L.; Pantel, K.; Schwarzenbach, H. Exosomal MicroRNAs as Tumor Markers in Epithelial Ovarian Cancer. Mol. Oncol. 2018, 12, 1935–1948. [Google Scholar] [CrossRef]
- Dorayappan, K.D.P.; Wallbillich, J.J.; Cohn, D.E.; Selvendiran, K. The Biological Significance and Clinical Applications of Exosomes in Ovarian Cancer. Gynecol. Oncol. 2016, 142, 199–205. [Google Scholar] [CrossRef]
- Falcone, G.; Felsani, A.; D’Agnano, I. Signaling by Exosomal MicroRNAs in Cancer. J. Exp. Clin. Cancer Res. 2015, 34, 1–10. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes Derived from Hypoxic Epithelial Ovarian Cancer Cells Deliver MicroRNAs to Macrophages and Elicit a Tumor-Promoted Phenotype. Cancer Lett. 2018, 435, 80–91. [Google Scholar] [CrossRef]
- Kan, C.W. Elevated Levels of Circulating MicroRNA-200 Family Members Correlate with Serous Epithelial Ovarian Cancer. BMC Cancer 2012, 12, 627. [Google Scholar] [CrossRef]
- Xiao, M. Let-7e Sensitizes Epithelial Ovarian Cancer to Cisplatin through Repressing DNA Double Strand Break Repair. J. Ovarian Res. 2017, 10, 24. [Google Scholar] [CrossRef]
- Chen, X. Exosomes Derived from Hypoxic Epithelial Ovarian Cancer Deliver MicroRNA-940 to Induce Macrophage M2 Polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef]
- Kinose, Y.; Sawada, K.; Nakamura, K.; Sawada, I.; Toda, A.; Nakatsuka, E.; Hashimoto, K.; Mabuchi, S.; Takahashi, K.; Kurachi, H.; et al. The Hypoxia-Related MicroRNA MiR-199a-3p Displays Tumor Suppressor Functions in Ovarian Carcinoma. Oncotarget 2015, 6, 11342–11356. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, L.-Z.; Qian, X.; Chen, Q.; Jiang, Y.; Li, D.; Lai, L.; Jiang, B.-H. MiR-145 Directly Targets P70S6K1 in Cancer Cells to Inhibit Tumor Growth and Angiogenesis. Nucleic Acids Res. 2012, 40, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Tian, J.; Zhang, L.; Chen, Y.; Hao, Q. Involvement of MicroRNA-93, a New Regulator of PTEN/Akt Signaling Pathway, in Regulation of Chemotherapeutic Drug Cisplatin Chemosensitivity in Ovarian Cancer Cells. FEBS Lett. 2012, 586, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hu, S.; Wang, J.; Cai, J.; Xiao, L.; Yu, L.; Wang, Z. MiR-27a Modulates MDR1/P-Glycoprotein Expression by Targeting HIPK2 in Human Ovarian Cancer Cells. Gynecol. Oncol. 2010, 119, 125–130. [Google Scholar] [CrossRef]
- Li, N.; Yang, L.; Wang, H.; Yi, T.; Jia, X.; Chen, C.; Xu, P. MiR-130a and MiR-374a Function as Novel Regulators of Cisplatin Resistance in Human Ovarian Cancer A2780 Cells. PLoS ONE 2015, 10, e0128886. [Google Scholar] [CrossRef]
- Li, X.; Chen, W.; Jin, Y.; Xue, R.; Su, J.; Mu, Z.; Li, J.; Jiang, S. MiR-142-5p Enhances Cisplatin-Induced Apoptosis in Ovarian Cancer Cells by Targeting Multiple Anti-Apoptotic Genes. Biochem. Pharmacol. 2019, 161, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal MiRNA Confers Chemo Resistance via Targeting Cav1/p-Gp/M2-Type Macrophage Axis in Ovarian Cancer. EBioMedicine 2018, 38, 100–112. [Google Scholar] [CrossRef]
- Amini-Farsani, Z.; Sangtarash, M.H.; Shamsara, M.; Teimori, H. MiR-221/222 Promote Chemoresistance to Cisplatin in Ovarian Cancer Cells by Targeting PTEN/PI3K/AKT Signaling Pathway. Cytotechnology 2018, 70, 203–213. [Google Scholar] [CrossRef]
- Weiner-Gorzel, K.; Dempsey, E.; Milewska, M.; McGoldrick, A.; Toh, V.; Walsh, A.; Lindsay, S.; Gubbins, L.; Cannon, A.; Sharpe, D.; et al. Overexpression of the MicroRNA MiR-433 Promotes Resistance to Paclitaxel through the Induction of Cellular Senescence in Ovarian Cancer Cells. Cancer Med. 2015, 4, 745–758. [Google Scholar] [CrossRef]
- Alharbi, M. MiRNa Signature in Small Extracellular Vesicles and Their Association with Platinum Resistance and Cancer Recurrence in Ovarian Cancer. Nanomedicine 2020, 28, 102207. [Google Scholar] [CrossRef]
- Huh, J.H.; Kim, T.H.; Kim, K.; Song, J.-A.; Jung, Y.J.; Jeong, J.-Y.; Lee, M.J.; Kim, Y.K.; Lee, D.H.; An, H.J. Dysregulation of MiR-106a and MiR-591 Confers Paclitaxel Resistance to Ovarian Cancer. Br. J. Cancer 2013, 109, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kong, W.; He, L.; Zhao, J.-J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V.; et al. MicroRNA Expression Profiling in Human Ovarian Cancer: MiR-214 Induces Cell Survival and Cisplatin Resistance by Targeting PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Pei, M.L.; Zhao, Z.X.; Shuang, T. Dysregulation of Lnc-SNHG1 and MiR-216b-5p Correlate with Chemoresistance and Indicate Poor Prognosis of Serous Epithelial Ovarian Cancer. J. Ovarian Res. 2020, 13, 144. [Google Scholar] [CrossRef]
- Xiao, S.; Li, Y.; Pan, Q.; Ye, M.; He, S.; Tian, Q.; Xue, M. MiR-34c/SOX9 Axis Regulates the Chemoresistance of Ovarian Cancer Cell to Cisplatin-Based Chemotherapy. J. Cell Biochem. 2019, 120, 2940–2953. [Google Scholar] [CrossRef]
- Jiang, J.; Xie, C.; Liu, Y.; Shi, Q.; Chen, Y. Up-Regulation of MiR-383-5p Suppresses Proliferation and Enhances Chemosensitivity in Ovarian Cancer Cells by Targeting TRIM27. Biomed. Pharmacother. 2019, 109, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Biamonte, F.; Santamaria, G.; Sacco, A.; Perrone, F.M.; Di Cello, A.; Battaglia, A.M.; Salatino, A.; Di Vito, A.; Aversa, I.; Venturella, R.; et al. MicroRNA Let-7g Acts as Tumor Suppressor and Predictive Biomarker for Chemoresistance in Human Epithelial Ovarian Cancer. Sci. Rep. 2019, 9, 5668. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.-N.; Yan, M.-D.; Lai, H.-C.; Huang, R.-L.; Chou, Y.-C.; Lin, W.-C.; Yeh, L.-T.; Lin, Y.-W. Downregulation of MiR-29 Contributes to Cisplatin Resistance of Ovarian Cancer Cells. Int. J. Cancer 2014, 134, 542–551. [Google Scholar] [CrossRef]
- Duan, L.; Yan, Y.; Wang, G.; Xing, Y.L.; Sun, J.; Wang, L.L. ΜiR-182-5p Functions as a Tumor Suppressor to Sensitize Human Ovarian Cancer Cells to Cisplatin through Direct Targeting the Cyclin Dependent Kinase 6 (CDK6). J. BUON 2020, 25, 2279–2286. [Google Scholar]
- Shuang, T.; Wang, M.; Shi, C.; Zhou, Y.; Wang, D. Down-Regulated Expression of MiR-134 Contributes to Paclitaxel Resistance in Human Ovarian Cancer Cells. FEBS Lett. 2015, 589, 3154–3164. [Google Scholar] [CrossRef]
- Kanlikilicer, P.; Rashed, M.H.; Bayraktar, R.; Mitra, R.; Ivan, C.; Aslan, B.; Zhang, X.; Filant, J.; Silva, A.M.; Rodriguez-Aguayo, C.; et al. Ubiquitous Release of Exosomal Tumor Suppressor MiR-6126 from Ovarian Cancer Cells. Cancer Res. 2016, 76, 7194–7207. [Google Scholar] [CrossRef]
- Au Yeung, C.L.; Co, N.-N.; Tsuruga, T.; Yeung, T.-L.; Kwan, S.-Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.-K.; et al. Exosomal Transfer of Stroma-Derived MiR21 Confers Paclitaxel Resistance in Ovarian Cancer Cells through Targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, H.; Yin, X.; Yang, M.; Wei, H.; Chen, Q.; Feng, F.; Liu, Y.; Xu, W.; Li, Y. Macrophages Derived Exosomes Deliver MiR-223 to Epithelial Ovarian Cancer Cells to Elicit a Chemoresistant Phenotype. J. Exp. Clin. Cancer Res. 2019, 38, 81. [Google Scholar] [CrossRef] [PubMed]
- Shender, V.O.; Pavlyukov, M.S.; Ziganshin, R.H.; Arapidi, G.P.; Kovalchuk, S.I.; Anikanov, N.A.; Altukhov, I.A.; Alexeev, D.G.; Butenko, I.O.; Shavarda, A.L.; et al. Proteome-Metabolome Profiling of Ovarian Cancer Ascites Reveals Novel Components Involved in Intercellular Communication. Mol. Cell Proteom. 2014, 13, 3558–3571. [Google Scholar] [CrossRef] [PubMed]
- Lund, R.J.; Huhtinen, K.; Salmi, J.; Rantala, J.; Nguyen, E.V.; Moulder, R.; Goodlett, D.R.; Lahesmaa, R.; Carpén, O. DNA Methylation and Transcriptome Changes Associated with Cisplatin Resistance in Ovarian Cancer. Sci. Rep. 2017, 7, 1469. [Google Scholar] [CrossRef]
- Kato, H.; Arakawa, A.; Suzumori, K.; Kataoka, N.; Young, S.R. FISH Analysis of BRCA1 Copy Number in Paraffin-Embedded Ovarian Cancer Tissue Samples. Exp. Mol. Pathol. 2004, 76, 138–142. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; deFazio, A.; Beroukhim, R.; Mermel, C.; George, J.; Getz, G.; Tothill, R.; Okamoto, A.; Raeder, M.B.; Harnett, P.; et al. Integrated Genome-Wide DNA Copy Number and Expression Analysis Identifies Distinct Mechanisms of Primary Chemoresistance in Ovarian Carcinomas. Clin. Cancer Res. 2009, 15, 1417–1427. [Google Scholar] [CrossRef]
- Moufarrij, S.; Dandapani, M.; Arthofer, E.; Gomez, S.; Srivastava, A.; Lopez-Acevedo, M.; Villagra, A.; Chiappinelli, K.B. Epigenetic Therapy for Ovarian Cancer: Promise and Progress. Clin. Epigenetics 2019, 11, 7. [Google Scholar] [CrossRef]
- Makhija, S.; Sit, A.; Edwards, R.; Aufman, K.; Weiss, H.; Kanbour-Shakir, A.; Gooding, W.; D’Angelo, G.; Ferrell, R.; Raja, S.; et al. Identification of Genetic Alterations Related to Chemoresistance in Epithelial Ovarian Cancer. Gynecol. Oncol. 2003, 90, 3–9. [Google Scholar] [CrossRef]
- Baker, V.V.; Borst, M.P.; Dixon, D.; Hatch, K.D.; Shingleton, H.M.; Miller, D. C-Myc Amplification in Ovarian Cancer. Gynecol. Oncol. 1990, 38, 340–342. [Google Scholar] [CrossRef]
- Shih, I.-M.; Sheu, J.J.-C.; Santillan, A.; Nakayama, K.; Yen, M.J.; Bristow, R.E.; Vang, R.; Parmigiani, G.; Kurman, R.J.; Trope, C.G.; et al. Amplification of a Chromatin Remodeling Gene, Rsf-1/HBXAP, in Ovarian Carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 14004–14009. [Google Scholar] [CrossRef]
- Park, J.T.; Li, M.; Nakayama, K.; Mao, T.-L.; Davidson, B.; Zhang, Z.; Kurman, R.J.; Eberhart, C.G.; Shih, I.-M.; Wang, T.-L. Notch3 Gene Amplification in Ovarian Cancer. Cancer Res. 2006, 66, 6312–6318. [Google Scholar] [CrossRef] [PubMed]
- Laferrière, N.B.; Brown, D.L. Effects of Taxol on the Polymerization and Posttranslational Modification of Class III Beta-Tubulin in P19 Embryonal Carcinoma Cells. Biochem. Cell Biol. 1995, 73, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting Cancer Metabolism in the Era of Precision Oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| miRNAs | Function and Respective Targets |
|---|---|
| Hypoxia: miRNAs altered in hypoxia/hypoxic tissue and associated functional change | |
| miRNA-181d-5p [189] | Increased expression in hypoxia-induced EVs; this enhanced M2 macrophage polarization and HGSOC cell migration and invasion |
| miRNA-940 [192] | Increased expression in hypoxia, HGSOC patient ascites, and exosomes; HGSOC cell–macrophage exosome exchange enhanced M2 phenotype polarization |
| miRNA-199a-3p [193] | Decreased expression reduced c-Met and AKT activity; this decreased proliferation, adhesion, and invasiveness |
| miRNA-145 [194] | Suppressed HGSOC; downregulated HIF-1 and VEGF via p70S6K1 |
| Therapy-induced: miRNAs altered in chemoresistant HGSOC cells or chemoresistance and associated functional change | |
| miRNA-93 [195] | Increased expression in chemoresistant HGSOC cells; this altered cell survival mechanisms via PTEN |
| miRNA-27a [196] | Increased expression in chemoresistant HGSOC cells; this increased MDR and PGP protein expression; inhibiting expression increased cell apoptosis via HIPK2 regulation |
| miRNAs-130a/374 [197] | Increased expression reduced cisplatin sensitivity; miR-130a knockdown inhibited MDR1 expression and upregulated PTEN expression |
| miRNA-142-5p [198] | Increased expression enhanced HGSOC cell platinum sensitivity via modulation of antiapoptotic proteins |
| miRNA-1246 [199] | Increased expression in paclitaxel-resistant HGSOC cells and in patients with severe prognosis; this inhibited CAV-1 expression via the PDGFB receptor and altered cell proliferation |
| miRNA-221/222 [200] | Increased expression conferred cisplatin resistance via the PTEN/PI3K/AKT signaling pathway |
| miRNA-433 [201] | Increased expression induced paclitaxel resistance, HGSOC, and poor survival; this modulated HGSOC cell senescence and CDK6 activation |
| miRNA-891-5p [202] | Increased expression in HGSOC patients and patients who exhibited carboplatin resistance; miRNA associated with DNA repair proteins and MYC regulator genes |
| miRNAs-200a-c [190] | Increased expression in chemoresistant HGSOC patients; can serve as another diagnostic tool in addition to serum biomarker CA125 |
| miRNA-106a/591 [203] | Increased miRNA-106a expression and decreased miRNA-591 expression in taxol-resistant cells; miRNA-106a targeted BCL-10 and caspase-7; miRNA-591 targeted ZEB1 |
| miRNA-214 [204] | Decreased expression in chemoresistant HGSOC cells; played a crucial role in developing cisplatin resistance via PTEN |
| miRNA-216b-5p [205] | Decreased expression in taxol-resistant HGSOC cells; overexpression of miRNA and knockdown of SNHG1 led to taxol sensitivity |
| miRNA-34c [206] | Decreased expression in chemoresistant cells; directly targets SOX9, B-catenin, and c-MYC |
| miRNA-383-5p [207] | Decreased expression reduced chemosensitivity via TRIM27; this modulated cell proliferation and HGSOC growth |
| Let-7g [208] | Decreased expression in chemoresistant HGSOC patients; this induced EMT and resistance to platinum therapy |
| Let-7i [51] | Decreased expression in cisplatin-resistant HGSOC; this activated BRCA1, RAD51, and DNA damage repair pathways |
| miRNA-29 [209] | Decreased expression in cisplatin-resistant cells; this targeted ECM proteins, such as COL1A1, and modulated ERK1/2 and GSK3B |
| miRNA-182-5p [210] | Decreased expression in cisplatin-resistant HGSOC cells; this miRNA targeted CDK6 |
| miRNA-134 [211] | Decreased expression in taxol-resistant HGSOC cells; this targeted KAP2 and modulated cell survival and apoptosis |
| miRNA-6126 [212] | Decreased expression correlated to poor prognosis; highly regulated in exosomes; overexpression reduced angiogenic phenotypes and migration; also acted as a tumor suppressor via integrin β1 |
| miRNA-30a-5p [195] | Decreased expression in cisplatin-resistant HGSOC cells; this elevated apoptosis; exosome miRNA exchange altered chemosensitivity via SOX9 |
| Overlapping miRNAs: miRNAs altered in both hypoxic and chemoresistant HGSOC cells | |
| miRNA-21-3p [213] | Increased expression suppressed HGSOC cell apoptosis via APAF1 binding |
| miRNA-223 [214] | Increased expression in hypoxia-induced exosomes; this promoted drug resistance in HGSOC cells via the PTEN–PI3K/AKT pathway |
| miRNA-125b [189] | Increased expression in hypoxia-induced exosomes; this enhanced M2 macrophage polarization and increased HGSOC cell migration and invasion |
| miRNA-210 [215] | Increased expression enhanced cancer cell viability and proliferation by targeting PTPN1 |
| Refractory | Resistant | Both/Not Distinguished |
|---|---|---|
| Genomic | ||
| CpG methylation [157,216] | BRCA1/2 mutation/amplification [59] | |
| BRCA1 deletion [217,218] | ||
| NF1 [218] | ||
| RB1 [218] | ||
| CDK12 [218] | ||
| CSMD3 [218] | ||
| FAT3 [218] | ||
| GABRA6 [218] | ||
| CCNE1 amplification [58,59,218] | ||
| TP53 mutation [219] | ||
| IGF2R deletion [220] | ||
| MYC amplification [217,221] | ||
| MDR1 [59] | ||
| Rsf-1/HBXAP [222] | ||
| NOTCH3 [223] | ||
| Transcriptional | ||
| JUN [56] | MDR1 [59] | FOXM1 [218] |
| FOS [56] | ꞵ-tubulin III [140] | NOTCH [218] |
| TNF [56] | p38a [182] | SNAIL [183] |
| CXCR4 [56] | GSTpi [104] | SLUG [183] |
| SNAI1 [56] | BCL-2 [172] | N-CAD [183] |
| VIM [56] | Survivin [172] | p53 [67] |
| GADD45B [56] | SMAD4 [183] | IL6 [56,216] |
| MCL1 [56] | ||
| HIFs [60] | ||
| Tetraspanins [137] | ||
| SNAREs [137] | ||
| Rabs [137] | ||
| Translational/post-translational | ||
| AP-1 [120] | ꞵ-tubulin III [224] | STAT3 [90] |
| NF-kB [120] | JNK [182] | Akt/mTOR [113] |
| Cytokines | ||
| IL-6 [110] | ||
| IL-8 [110] | ||
| IL-11 [110] | ||
| IL-17 [110] | ||
| IL-27 [110] | ||
| IL-31 [110] | ||
| Growth factors | ||
| TGF-β [104,105,106] | ||
| EGF [104,105,106] | ||
| VEGF [104,105,106] | ||
| TNF-α [118] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, A.H.; Mejia Peña, C.; Dawson, M.R. Comparing the Secretomes of Chemorefractory and Chemoresistant Ovarian Cancer Cell Populations. Cancers 2022, 14, 1418. https://doi.org/10.3390/cancers14061418
Lee AH, Mejia Peña C, Dawson MR. Comparing the Secretomes of Chemorefractory and Chemoresistant Ovarian Cancer Cell Populations. Cancers. 2022; 14(6):1418. https://doi.org/10.3390/cancers14061418
Chicago/Turabian StyleLee, Amy H., Carolina Mejia Peña, and Michelle R. Dawson. 2022. "Comparing the Secretomes of Chemorefractory and Chemoresistant Ovarian Cancer Cell Populations" Cancers 14, no. 6: 1418. https://doi.org/10.3390/cancers14061418
APA StyleLee, A. H., Mejia Peña, C., & Dawson, M. R. (2022). Comparing the Secretomes of Chemorefractory and Chemoresistant Ovarian Cancer Cell Populations. Cancers, 14(6), 1418. https://doi.org/10.3390/cancers14061418