
Angiogenesis Inhibitors and Immunomodulation in Renal Cell Cancers: The Past, Present, and Future

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Angiogenesis Is a Hallmark of Cancer Development
1.2. Immunomodulation and Angiogenesis: A Crucial Interface in Renal Cell Carcinomas
1.3. Drug Development of Advanced Clear-Cell Renal Cell Carcinoma
1.3.1. Cytokine Therapy
1.3.2. VEGF and Other Angiogenesis Inhibitor Monotherapies
1.3.3. Mammalian Target of Rapamycin (mTOR) Inhibitors
1.3.4. Immune Checkpoint Inhibitors
1.3.5. Combination VEGF Monoclonal Antibody and Immune Checkpoint Inhibitor
1.3.6. Combination Tyrosine Kinase Inhibitor and Immune Checkpoint Inhibitor
1.4. Non-Clear-Cell Renal Cell Carcinoma
1.5. Opportunities for Advancement in Renal Cell Carcinoma Drug Development
2. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides, J.V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib Plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Zitvogel, L.; Kroemer, G. Immunological Mechanisms Underneath the Efficacy of Cancer Therapy. Cancer Immunol. Res. 2016, 4, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Ramanujan, S.; Koenig, G.C.; Padera, T.P.; Stoll, B.R.; Jain, R.K. Local imbalance of proangiogenic and antiangiogenic factors: A potential mechanism of focal necrosis and dormancy in tumors. Cancer Res. 2000, 60, 1442–1448. [Google Scholar] [PubMed]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef]
- Ferrara, N.; Alitalo, K. Clinical applications of angiogenic growth factors and their inhibitors. Nat. Med. 1999, 5, 1359–1364. [Google Scholar] [CrossRef]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Bouck, N.; Stellmach, V.; Hsu, S.C. How Tumors Become Angiogenic. Adv. Cancer Res. 1996, 69, 135–174. [Google Scholar]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Bottaro, D.P.; Liotta, L.A. Out of air is not out of action. Nature 2003, 423, 593–595. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.H.; Shih, S.C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, M.; McSheehy, P.M.; Griffiths, J.R.; Bashford, C.L. Causes and consequences of tumour acidity and implications for treatment. Mol. Med. Today 2000, 6, 15–19. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K. Determinants of tumor blood flow: A review. Cancer Res. 1988, 48, 2641–2658. [Google Scholar]
- Griffioen, A.W. Anti-angiogenesis: Making the tumor vulnerable to the immune system. Cancer Immunol. Immunother. 2008, 57, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Comito, G.; Calvani, M.; Giannoni, E.; Bianchini, F.; Calorini, L.; Torre, E.; Migliore, C.; Giordano, S.; Chiarugi, P. HIF-1α Stabilization by Mitochondrial ROS Promotes Met-Dependent Invasive Growth and Vasculogenic Mimicry in Melanoma Cells. Free Radic. Biol. Med. 2011, 51, 893–904. [Google Scholar] [CrossRef]
- Eberhard, A.; Kahlert, S.; Goede, V.; Hemmerlein, B.; Plate, K.H.; Augustin, H.G. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for antiangiogenic tumor therapies. Cancer Res. 2000, 60, 1388–1393. [Google Scholar]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesely, M.D.; Schreiber, R.D. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finicle, B.T.; Jayashankar, V.; Edinger, A.L. Nutrient scavenging in cancer. Nat. Rev. Cancer 2018, 18, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. 2011, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- DeClerck, Y.A. Desmoplasia: A Response or a Niche? Cancer Discov. 2012, 2, 772–774. [Google Scholar] [CrossRef] [Green Version]
- Whatcott, C.; Posner, R.; Hoff, D.V. Desmoplasia and Chemoresistance in Pancreatic Cancer. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P., Munshi, H., Eds.; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- Calorini, L.; Bianchini, F. Environmental control of invasiveness and metastatic dissemination of tumor cells: The role of tumor cell-host cell interactions. Cell Commun. Signal. 2010, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, A.L.; Munn, D.H. Creating immune privilege: Active local suppression that benefits friends, but protects foes. Nat. Rev. Immunol. 2008, 8, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Fricke, I.; Gabrilovich, D.I. Dendritic cells and tumor microenvironment: A dangerous liaison. Immunol. Investig. 2006, 35, 459–483. [Google Scholar] [CrossRef]
- Pruneri, G.; Vingiani, A.; Denkert, C. Tumor infiltrating lymphocytes in early breast cancer. Breast 2018, 37, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Pagès, F.; Galon, J.; Dieu-Nosjean, M.C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Kos, Z.; Roblin, E.; Kim, R.S.; Michiels, S.; Gallas, B.D.; Chen, W.; van de Vijver, K.K.; Goel, S.; Adams, S.; Demaria, S.; et al. Pitfalls in assessing stromal tumor infiltrating lymphocytes (sTILs) in breast cancer. NPJ Breast Cancer 2020, 6, 17. [Google Scholar] [CrossRef]
- Leffers, N.; Gooden, M.J.; de Jong, R.A.; Hoogeboom, B.N.; ten Hoor, K.A.; Hollema, H.; Boezen, H.M.; van der Zee, A.G.; Daemen, T.; Nijman, H.W.; et al. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol. Immunother. 2009, 58, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Şenbabaoğlu, Y.; Gejman, R.S.; Winer, A.G.; Liu, M.; Van Allen, E.M.; de Velasco, G.; Miao, D.; Ostrovnaya, I.; Drill, E.; Luna, A.; et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016, 17, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mennitto, A.; Huber, V.; Ratta, R.; Sepe, P.; de Braud, F.; Procopio, G.; Guadalupi, V.; Claps, M.; Stellato, M.; Daveri, E.; et al. Angiogenesis and Immunity in Renal Carcinoma: Can We Turn an Unhappy Relationship into a Happy Marriage? J. Clin. Med. 2020, 9, 930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall Survival and Updated Results for Sunitinib Compared with Interferon Alfa in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Hawkins, R.E.; Wagstaff, J.; Salman, P.; Mardiak, J.; Barrios, C.H.; Zarba, J.J.; Gladkov, O.A.; Lee, E.; Szczylik, C.; et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: Final overall survival results and safety update. Eur. J. Cancer 2013, 49, 1287–1296. [Google Scholar] [CrossRef]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A Randomized Trial of Bevacizumab, an Anti–Vascular Endothelial Growth Factor Antibody, for Metastatic Renal Cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Yagoda, A.; Petrylak, D.; Thompson, S. Cytotoxic chemotherapy for advanced renal cell carcinoma. Urol. Clin. N. Am. 1993, 20, 303–321. [Google Scholar] [CrossRef]
- Elson, P.J.; Kvols, L.K.; Vogl, S.E.; Glover, D.J.; Hahn, R.G.; Trump, D.L.; Carbone, P.P.; Earle, J.D.; Davis, T.E. Phase II trials of 5-day vinblastine infusion (NSC 49842), L-alanosine (NSC 153353), acivicin (NSC 163501), and aminothiadiazole (NSC 4728) in patients with recurrent or metastatic renal cell carcinoma. Investig. New Drugs 1988, 6, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Oliver, R.T.D.; Nethersell, A.B.W.; Bottomley, J.M. Unexplained Spontaneous Regression and Alpha-interferon as Treatment for Metastatic Renal Carcinoma. Br. J. Urol. 1989, 63, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, A.D.; Poletajew, S.; Wasiutyński, A. Reviews Spontaneous regression of renal cell carcinoma. Współczesna Onkol. 2013, 2, 123–127. [Google Scholar] [CrossRef]
- Smith, K. Lowest dose interleukin-2 immunotherapy. Blood 1993, 81, 1414–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizza, P.; Moretti, F.; Belardelli, F. Recent advances on the immunomodulatory effects of IFN-α: Implications for cancer immunotherapy and autoimmunity. Autoimmunity 2010, 43, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, G.; Fisher, R.I.; Rosenberg, S.A.; Sznol, M.; Parkinson, D.R.; Louie, A.C. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 1995, 13, 688–696. [Google Scholar] [CrossRef]
- Medical Research Council Renal Cancer Collaborators. Interferon-alpha and survival in metastatic renal carcinoma: Early results of a randomised controlled trial. Lancet 1999, 353, 14–17. [Google Scholar] [CrossRef]
- Escudier, B.; Szczylik, C.; Hutson, T.E.; Demkow, T.; Staehler, M.; Rolland, F.; Negrier, S.; Laferriere, N.; Scheuring, U.J.; Cella, D.; et al. Randomized Phase II Trial of First-Line Treatment with Sorafenib Versus Interferon Alfa-2a in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2009, 27, 1280–1289. [Google Scholar] [CrossRef]
- Verzoni, E.; Escudier, B.; Hutson, T.E.; McDermott, D.F.; Pal, S.K.; Porta, C.; Rini, B.I.; Needle, M.N.; Atkins, M.B. TIVO-3: Durability of response and updated overall survival of tivozanib versus sorafenib in metastatic renal cell carcinoma (mRCC). J. Clin. Oncol. 2021, 39, 4546. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, M.D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S.; et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rollan, F.; et al. Sorafenib for Treatment of Renal Cell Carcinoma: Final Efficacy and Safety Results of the Phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; Powles, T.; Scheffold, C.; Choueiri, T.K. Long-term follow-up of overall survival for cabozantinib versus everolimus in advanced renal cell carcinoma. Br. J. Cancer 2018, 118, 1176–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; De Souza, P.; Merchan, J.R.; et al. Pazopanib versus Sunitinib in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Porta, C.; Bono, P.; Powles, T.; Eisen, T.; Sternberg, C.N.; Gschwend, J.E.; De Giorgi, U.; Parikh, O.; Hawkins, R.; et al. Randomized, Controlled, Double-Blind, Cross-Over Trial Assessing Treatment Preference for Pazopanib Versus Sunitinib in Patients with Metastatic Renal Cell Carcinoma: PISCES Study. J. Clin. Oncol. 2014, 32, 1412–1418. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib Versus Sunitinib as Initial Targeted Therapy for Patients with Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J. Clin. Oncol. 2017, 35, 591–597. [Google Scholar] [CrossRef]
- Bjarnason, G.A.; Knox, J.J.; Kollmannsberger, C.K.; Soulieres, D.; Ernst, D.S.; Zalewski, P.; Canil, C.M.; Winquist, E.; Hotte, S.J.; North, S.A.; et al. The efficacy and safety of sunitinib given on an individualised schedule as first-line therapy for metastatic renal cell carcinoma: A phase 2 clinical trial. Eur. J. Cancer 2019, 108, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Olsen, M.R.; Hudes, G.R.; Burke, J.M.; Edenfield, W.J.; Wilding, G.; Agarwal, N.; Thompson, J.A.; Cella, D.; et al. Randomized phase II trial of sunitinib on an intermittent versus continuous dosing schedule as first-line therapy for advanced renal cell carcinoma. J. Clin. Oncol. 2012, 30, 1371–1377. [Google Scholar] [CrossRef]
- Ornstein, M.C.; Wood, L.S.; Elson, P.; Allman, K.D.; Beach, J.; Martin, A.; Zanick, B.R.; Grivas, P.; Gilligan, T.; Garcia, J.A.; et al. A Phase II Study of Intermittent Sunitinib in Previously Untreated Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2017, 35, 1764–1769. [Google Scholar] [CrossRef]
- Thiery-Vuillemin, A.; Gravis, G.; Constans Schlurmann, F.; Bompas, E.; Rolland, F.; Gross-Goupil, M.; Vano, Y.A.; Guillot, A.; Barthélémy, P.; Joly, C.; et al. 720P Randomised phase II study to assess the efficacy and tolerability of sunitinib by dose administration regimen in anti-angiogenic naïve patients with metastatic renal cell carcinoma (mRCC): Interim analysis (IA) of SURF study. Ann. Oncol. 2020, 31, S565–S566. [Google Scholar] [CrossRef]
- Brugarolas, J. Renal-Cell Carcinoma—Molecular Pathways and Therapies. N. Engl. J. Med. 2007, 356, 185–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, M.H.; Molina, A.M.; Motzer, R.J. mTOR Inhibitors in Advanced Renal Cell Carcinoma. Hematol./Oncol. Clin. N. Am. 2011, 25, 835–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, Interferon Alfa, or Both for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannir, N.M.; Msaouel, P.; Ross, J.A.; Devine, C.E.; Chandramohan, A.; Gonzalez, G.M.N.; Wang, X.; Wang, J.; Corn, P.G.; Lim, Z.D.; et al. Temsirolimus versus Pazopanib (TemPa) in Patients with Advanced Clear-cell Renal Cell Carcinoma and Poor-risk Features: A Randomized Phase II Trial. Eur. Urol. Oncol. 2020, 3, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: Final results and analysis of prognostic factors. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef]
- Motzer, R.J.; Barrios, C.H.; Kim, T.M.; Falcon, S.; Cosgriff, T.; Harker, W.G.; Srimuninnimit, V.; Pittman, K.; Sabbatini, R.; Rha, S.Y.; et al. Phase II Randomized Trial Comparing Sequential First-Line Everolimus and Second-Line Sunitinib Versus First-Line Sunitinib and Second-Line Everolimus in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2014, 32, 2765–2772. [Google Scholar] [CrossRef] [Green Version]
- Knox, J.J.; Barrios, C.H.; Kim, T.M.; Cosgriff, T.; Srimuninnimit, V.; Pittman, K.; Sabbatini, R.; Rha, S.Y.; Flaig, T.W.; Page, R.D.; et al. Final overall survival analysis for the phase II RECORD-3 study of first-line everolimus followed by sunitinib versus first-line sunitinib followed by everolimus in metastatic RCC. Ann. Oncol. 2017, 28, 1339–1345. [Google Scholar] [CrossRef]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar]
- Bedke, J.; Gouttefangeas, C.; Singh-Jasuja, H.; Stevanović, S.; Behnes, C.-L.; Stenzl, A. Targeted therapy in renal cell carcinoma: Moving from molecular agents to specific immunotherapy. World J. Urol. 2014, 32, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.; Hughes, M.; Kammula, U.; Royal, R.; Sherry, R.M.; Topalian, S.L.; Suri, K.B.; Levy, C.; Allen, T.; Mavroukakis, S.; et al. Ipilimumab (Anti-CTLA4 Antibody) Causes Regression of Metastatic Renal Cell Cancer Associated with Enteritis and Hypophysitis. J. Immunother. 2007, 30, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F.; et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J. Clin. Oncol. 2015, 33, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Antonia, J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taulor, M.; Eder, J.P.; Jäger, D.; Pietanza, C.; Le Dung, T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Hammers, H.J.; Plimack, E.R.; Infante, J.R.; Rini, B.I.; McDermott, D.F.; Lewis, L.D.; Voss, M.H.; Sharma, P.; Pal, S.K.; Razak, A.R.A.; et al. Safety and Efficacy of Nivolumab in Combination with Ipilimumab in Metastatic Renal Cell Carcinoma: The CheckMate 016 Study. J. Clin. Oncol. 2017, 35, 3851–3858. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Burotto, M.; Choueiri, T.K.; Hammers, H.J.; Plimack, E.R.; Porta, C.G.; George, S.; Powles, T.B.; et al. 661P Conditional survival and 5-year follow-up in CheckMate 214: First-line nivolumab + ipilimumab (N+I) versus sunitinib (S) in advanced renal cell carcinoma (aRCC). Ann. Oncol. 2021, 32, S685–S687. [Google Scholar] [CrossRef]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef]
- Atkins, M.B.; Plimack, E.R.; Puzanov, I.; Fishman, M.N.; McDermott, D.F.; Cho, D.C.; Vaishampayan, U.; George, S.; Olencki, T.E.; Tarazi, J.C.; et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018, 19, 405–415. [Google Scholar] [CrossRef]
- Amin, A.; Plimack, E.R.; Ernstoff, M.S.; Lewis, L.D.; Bauer, T.M.; McDermott, D.F.; Carducci, M.; Kollmannsberger, C.; Rini, B.I.; Heng, D.Y.C.; et al. Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: The CheckMate 016 study. J. Immunother. Cancer 2018, 6, 109. [Google Scholar] [CrossRef]
- Chowdhury, S.; Infante, J.R.; Hawkins, R.; Voss, M.H.; Perini, R.; Arkenau, T.; Voskoboynik, M.; Aimone, P.; Naeije, I.; Reising, A.; et al. A Phase I/II Study to Assess the Safety and Efficacy of Pazopanib and Pembrolizumab Combination Therapy in Patients with Advanced Renal Cell Carcinoma. Clin. Genitourin. Cancer 2021, 19, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Bellmunt, J.; Négrier, S.; Bajetta, E.; Melichar, B.; Bracarda, S.; Ravaud, A.; Golding, S.; Jethwa, S.; Sneller, V.; et al. Phase III Trial of Bevacizumab Plus Interferon Alfa-2a in Patients with Metastatic Renal Cell Carcinoma (AVOREN): Final Analysis of Overall Survival. J. Clin. Oncol. 2010, 28, 2144–2150. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Halabi, S.; Rosenberg, J.E.; Stadler, W.M.; Vaena, D.A.; Archer, L.; Atkins, J.N.; Picus, J.; Czaykowski, P.; Dutcher, J.; et al. Phase III Trial of Bevacizumab Plus Interferon Alfa versus Interferon Alfa Monotherapy in Patients with Metastatic Renal Cell Carcinoma: Final Results of CALGB 90206. J. Clin. Oncol. 2010, 28, 2137–2143. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Waddell, T.; Gafanov, R.; Pouliot, F.; Nosov, D.; Melichar, B.; Soulieres, D.; Borchiellini, D.; et al. Pembrolizumab (pembro) plus axitinib (axi) versus sunitinib as first-line therapy for advanced clear cell renal cell carcinoma (ccRCC): Results from 42-month follow-up of Keynote-426. J. Clin. Oncol. 2021, 39, 4500. [Google Scholar] [CrossRef]
- Ahrens, M.; Scheich, S.; Hartmann, A.; Bergmann, L. Non-Clear Cell Renal Cell Carcinoma—Pathology and Treatment Options. Oncol. Res. Treat. 2019, 42, 128–135. [Google Scholar] [CrossRef]
- McGregor, B.A.; McKay, R.R.; Braun, D.A.; Werner, L.; Gray, K.; Flaifel, A.; Signoretti, S.; Hirsch, M.S.; Steinharter, J.A.; Bakouny, Z.; et al. Results of a Multicenter Phase II Study of Atezolizumab and Bevacizumab for Patients with Metastatic Renal Cell Carcinoma with Variant Histology and/or Sarcomatoid Features. J. Clin. Oncol. 2020, 38, 63–70. [Google Scholar] [CrossRef]
- Lee, J.-L.; Ziobro, M.; Gafanov, R.; Matveev, V.B.; Suarez, C.; Donskov, F.; Pouliot, F.; Alekseev, B.Y.; Wiechno, P.J.; Tomczak, P. KEYNOTE-427 cohort B: First-line pembrolizumab (pembro) monotherapy for advanced non-clear cell renal cell carcinoma (NCC-RCC). J. Clin. Oncol. 2019, 37, 4569. [Google Scholar] [CrossRef]
- Bellmunt, J.; Dutcher, J. Targeted therapies and the treatment of non-clear cell renal cell carcinoma. Ann. Oncol. 2013, 24, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- De Velasco, G.; McKay, R.R.; Lin, X.; Moreira, R.B.; Simantov, R.; Choueiri, T.K. Comprehensive Analysis of Survival Outcomes in Non-Clear Cell Renal Cell Carcinoma Patients Treated in Clinical Trials. Clin. Genitourin. Cancer 2017, 15, 652–660.e1. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Jonasch, E.; Albiges, L.; Altinmakas, E.; Ng, C.S.; Matin, S.F.; Wang, X.; Qiao, W.; Dubauskas Lim, Z.; Tamboli, P.; et al. Everolimus Versus Sunitinib Prospective Evaluation in Metastatic Non–Clear Cell Renal Cell Carcinoma (ESPN): A Randomized Multicenter Phase 2 Trial. Eur. Urol. 2016, 69, 866–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gedye, C.; Pook, D.W.; Krieger, L.E.M.; Harris, C.A.; Goh, J.C.; Kichenadasse, G.; Gurney, H.; Underhill, C.; Parnis, F.; Joshua, A.M.; et al. UNISON-nivolumab then ipilimumab + nivolumab in advanced non-clear cell renal cell carcinoma (ANZUP 1602): Part 1—Nivolumab monotherapy. J. Clin. Oncol. 2021, 39, 325. [Google Scholar] [CrossRef]
- Lee, C.-H.; Voss, M.H.; Carlo, M.I.; Chen, Y.-B.; Reznik, E.; Knezevic, A.; Lefkowitz, R.A.; Shapnik, N.; Tassone, D.; Dadoun, C.; et al. Nivolumab plus cabozantinib in patients with non-clear cell renal cell carcinoma: Results of a phase 2 trial. J. Clin. Oncol. 2021, 39, 4509. [Google Scholar] [CrossRef]
- Pal, S.K.; Tangen, C.; Thompson, I.M., Jr.; Balzer-Haas, N.; George, D.J.; Heng, D.Y.C.; Shuch, B.; Stein, M.; Tretiakova, M.; Humphrey, P.; et al. A comparison of sunitinib with cabozantinib, crizotinib, and savolitinib for treatment of advanced papillary renal cell carcinoma: A randomised, open-label, phase 2 trial. Lancet 2021, 397, 695–703. [Google Scholar] [CrossRef]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Epaillard, N.; Simonaggio, A.; Elaidi, R.; Azzouz, F.; Braychenko, E.; Thibault, C.; Sun, C.M.; Moreira, M.; Oudard, S.; Vano, Y.A. BIONIKK: A phase 2 biomarker driven trial with nivolumab and ipilimumab or VEGFR tyrosine kinase inhibitor (TKI) in naïve metastatic kidney cancer. Bull. Cancer 2020, 107, eS22–eS27. [Google Scholar] [CrossRef]
- Vano, Y.; Elaidi, R.T.; Bennamoun, M.; Chevreau, C.M.; Borchiellini, D.; Pannier, D.; Maillet, D.; Gross-Goupil, M.; Tournigand, C.; Laguerre, B.; et al. LBA25 Results from the phase II biomarker driven trial with nivolumab (N) and ipilimumab or VEGFR tyrosine kinase inhibitor (TKI) in naive metastatic kidney cancer (m-ccRCC) patients (pts): The BIONIKK trial. Ann. Oncol. 2020, 31, S1157. [Google Scholar] [CrossRef]
- Derosa, L.; Routy, B.; Fidelle, M.; Iebba, V.; Alla, L.; Pasolli, E.; Segata, N.; Desnoyer, A.; Pietrantonio, F.; Ferrere, G.; et al. Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients. Eur. Urol. 2020, 78, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Semrad, T.J.; Groshen, S.; Luo, C.; Pal, S.; Vaishampayan, U.; Joshi, M.; Quinn, D.I.; Mack, P.C.; Gandara, D.R.; Lara, P.N.; et al. Randomized Phase 2 Study of Trebananib (AMG 386) with or without Continued Anti-Vascular Endothelial Growth Factor Therapy in Patients with Renal Cell Carcinoma Who Have Progressed on Bevacizumab, Pazopanib, Sorafenib, or Sunitinib—Results of NCI/CTEP Protocol 9048. Kidney Cancer 2019, 3, 51–61. [Google Scholar] [PubMed] [Green Version]
- Choueiri, T.K.; Bauer, T.M.; McDermott, D.F.; Arrowsmith, E.; Roy, A.; Perini, R.F.; Vickery, D.; Tykodi, S.S. Phase 2 study of the oral hypoxia-inducible factor 2 alfa inhibitor MK-6482 in combination with cabozantinib in patients with advanced clear cell renal cell carcinoma. In Proceedings of the 2021 Genitourinary Cancers Symposium, Virtual Meeting, 11–13 February 2021. [Google Scholar]
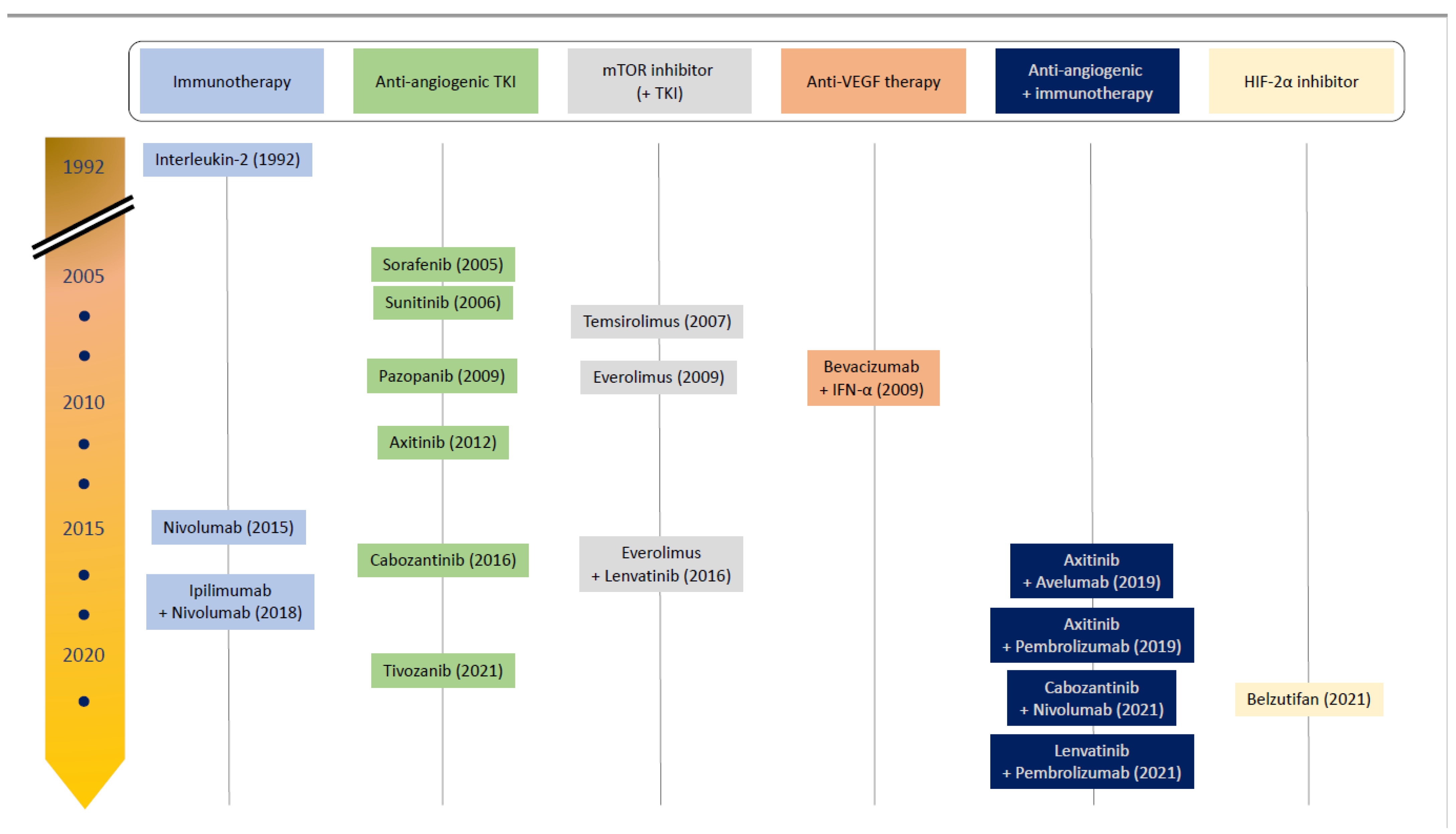
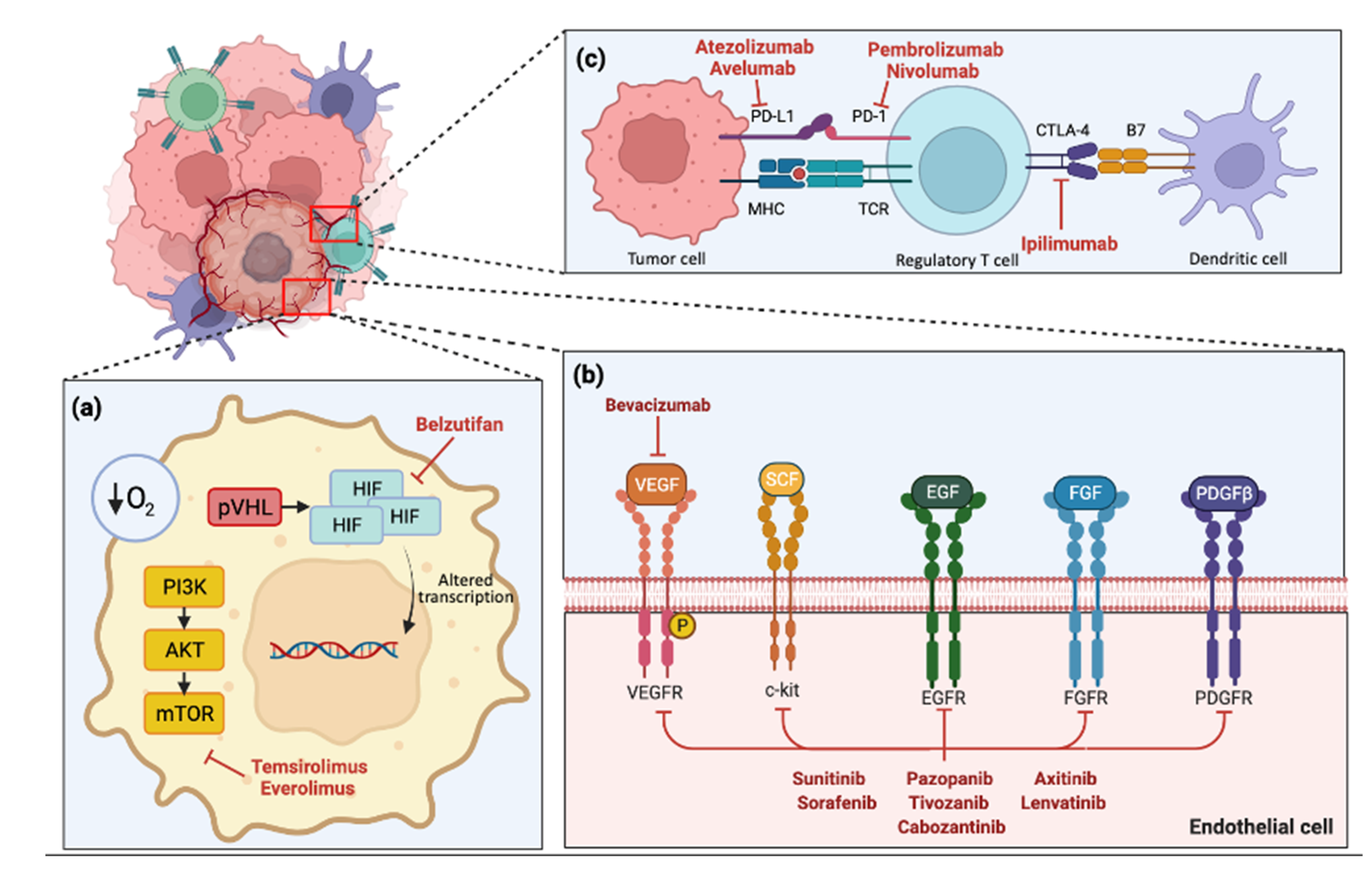

{kind=link}
{kind=link}
| Trial | Study Population | Drugs | Enrollment | Primary Outcome Measures | Key Outcomes |
|---|---|---|---|---|---|
| IMmotion151 [107] NCT02420821 | Previously untreated, advanced RCC with clear-cell or sarcomatoid histology Any IMDC risk group | Arm 1: Atezolizumab 1200 mg plus bevacizumab 15 mg/kg IV Q3W Arm 2: Sunitinib 50 mg PO daily, 4 weeks on treatment followed by 2 weeks off treatment | 915 | PFS in the PD-L1-positive population and OS in ITT population | mPFS in PDL1 + ve patients: atezolizumab + bevacizumab vs. sunitinib: 11.2 mo vs. 7.7 mo. HR 0.74; 95%CI 0.57–0.96; p = 0.0217 OS in ITT (interim): atezolizumab + bevacizumab vs. sunitinib: 43% vs. 42%. HR 0.93; 95%CI 0.76–1.14; p = 0.4751 |
| KEYNOTE-426 [4] NCT02853331 | Previously untreated, advanced RCC with clear-cell component with or without sarcomatoid features Any IMDC risk group | Arm 1: Pembrolizumab 200 mg IV Q3W and axitinib 5 mg PO twice daily Arm 2: Sunitinib 50 mg PO daily, 4 weeks on treatment followed by 2 weeks off treatment | 861 | PFS | mPFS: pembrolizumab + axitinib vs. sunitinib: 15.1 mo vs. 11.1 mo. HR 0.69; 95%CI 0.57–0.84; p < 0.001 |
| JAVELIN Renal 101 [108] NCT02684006 | Previously untreated, advanced RCC with a clear-cell component | Arm 1: Avelumab 10 mg/kg IV Q2W plus axitinib 5 mg PO BD Arm 2: Sunitinib 50 mg PO daily, 4 weeks on treatment followed by 2 weeks off treatment | 886 | PFS and OS in patients with PD-L1-positive tumors | 63.2% had PD-L1-positive tumors. mPFS in PD-L1-positive tumors: avelumab + axitinib vs. sunitinib: 13.8 mo vs. 7.2 mo. HR 0.61; 95%CI 0.47–0.79; p < 0.001. OS in PD-L1-positive tumors: avelumab + axitinib vs. sunitinib: HR 0.82; 95%CI 0.53–1.28; p = 0.38 |
| CLEAR [3] NCT02811861 | Previously untreated, advanced RCC with a clear-cell component Any IMDC risk group | Arm 1: Lenvatinib 18 mg PO daily plus everolimus 5 mg PO daily Arm 2: Lenvatinib 20 mg PO daily plus pembrolizumab 200 mg IV Q3W Arm 3: Sunitinib 50 mg PO daily, 4 weeks on treatment followed by 2 weeks off treatment | 1069 | PFS | Lenvatinib + pembrolizumab vs. sunitinib: 23.9 mo vs. 9.2 mo; HR 0.39; 95%CI 0.32–0.49; p < 0.001) Lenvatinib plus everolimus vs. sunitinib: 14.7 mo vs. 9.2 mo; HR 0.65; 95%CI 0.53–0.80; p < 0.001) |
| CheckMate 9ER [2] NCT03141177 | Previously untreated, advanced RCC with a clear-cell component Any IMDC risk group | Arm 1: Nivolumab 240 mg IV Q2W plus cabozantinib 40 mg PO daily Arm 2: Sunitinib 50 mg PO daily, 4 weeks on treatment followed by 2 weeks off treatment | 651 | PFS | Nivolumab + cabozantinib vs. sunitinib: 16.6 mo vs. 8.3 mo; HR 0.51; 95%CI 0.41–0.64; p < 0.001) |
| Trial | Phase | Study Population | Drugs | Estimated Enrollment | Primary Outcome Measures | Estimated Primary Study Completion |
|---|---|---|---|---|---|---|
| NCT02724878 | II | Unresectable/metastatic nccRCC Untreated or previously treated | Atezolizumab 1200 mg IV Q3W + bevacizumab 15 mg/kg IV Q3W | 60 | ORR | April 2021 * |
| LENKYN NCT04267120 | II | Unresectable/metastatic nccRCC Previously untreated | Pembrolizumab 200 mg IV Q3W + lenvatinib 20 mg daily PO | 34 | ORR | July 2024 |
| KEYNOTE-B61 NCT04704219 | II | Unresectable/metastatic nccRCC Previously untreated | Pembrolizumab 400 mg IV Q6W + lenvatinib 20 mg daily PO | 152 | ORR | August 2024 |
| CA209-9KU NCT03635892 | II | Unresectable/metastatic nccRCC 0–1 previous lines of treatment | Cabozantinib 40 mg daily PO + nivolumab 240 mg Q2W | 97 | ORR | August 2022 |
| NCT04413123 | II | Unresectable/metastatic nccRCC Untreated or previously treated | Cabozantinib PO daily + nivolumab IV Q3W + ipilimumab IV Q3W 4 cycles, then maintenance cabozantinib and nivolumab | 40 | ORR | December 2021 |
| NCT04385654 | II | Metastatic nccRCC T2–4N0 or TxN1+ or nuclear grade > 3 | Toripalimab 240 mg Q3W IV + axitinib 5 mg BD PO for 6 weeks followed by resection of primary tumor | 40 | MPR, pCR, pNR | December 2021 |
| ICONIC NCT03866382 | II | Rare GU tumors (nccRCC cohorts include sarcomatoid, RMC, CDC, papillary, chromophobe, tRCC) | Nivolumab Q3W IV + ipilimumab Q3W IV + cabozantinib daily PO for 4 cycles, followed by maintenance nivolumab + cabozantinib | 224 | ORR | February 2023 |
| CONTACT-03 NCT04338269 | III | Unresectable/metastatic RCC, incl. nccRCC cohort (papillary, chromophobe, unclassified, sarcomatoid) | Arm 1: Atezolizumab 1200 mg IV Q3W + cabozantinib 60 mg daily PO Arm 2: Cabozantinib 60 mg daily PO | 500 | PFS, OS | December 2022 |
| NCT03595124 | II | Unresectable/metastatic tRCC | Arm 1: Axitinib BD PO + nivolumab Q2W IV Arm 2: Axitinib BD PO Arm 3: Nivolumab Q2W IV | 70 | PFS | Suspended (poor accrual) |
| Trial | Phase | Population | Treatment | Estimated Enrollment | Primary Endpoints | Estimated Primary Study Completion |
|---|---|---|---|---|---|---|
| COSMIC 313 NCT03937219 | III | Previously untreated, unresectable/metastatic renal cell carcinoma with a clear-cell component IMDC intermediate or poor risk | Arm 1: Cabozantinib + nivolumab + ipilimumab (4 doses) followed by cabozantinib + nivolumab Arm 2: Cabozantinib-matched placebo + nivolumab + ipilimumab (4 doses) followed by cabozantinib-matched placebo + nivolumab | 840 | PFS | November 2021 |
| CONTACT-03 NCT04338269 | III | Advanced, untreated RCC ccRCC and nccRCC cohorts | Arm 1: Atezolizumab 1200 mg IV Q3W + cabozantinib 60 mg PO daily Arm 2: Cabozantinib 60 mg PO daily | 500 | PFS, OS | December 2022 |
| PEDIGREE NCT03793166 | III | Unresectable/metastatic RCC with clear-cell component, including patients with sarcomatoid features IMDC intermediate and poor risk | Induction ipilimumab + nivolumab. In patients with non-CR/non-PD: Arm 1: maintenance nivolumab alone Arm 2: maintenance nivolumab plus cabozantinib | 1046 | OS | September 2022 |
| TiNivo-2 NCT04987203 | III | Advanced RCC with a clear component, progressed during or following at least 6 weeks of ICI treatment in the first- or second- line setting | Arm 1: Nivolumab IV Q4W plus tivozanib 1.34 mg PO daily, 3 weeks on treatment followed by 1 week off treatment Arm 2: Tivozanib 1.34 mg PO daily, 3 weeks on treatment followed by 1 week off treatment | 326 | PFS | July 2024 |
| TIDE-A NCT04698213 | II | Metastatic RCC with predominately clear-cell subtype with primary tumor resected | Axitinib 5 mg PO BD + avelumab 10 mg/kg IV Q2W | 75 | ORR | September 2023 |
| NCT03172754 | I/II | Untreated, advanced RCC with predominately clear-cell subtype | Axitinib + nivolumab | 98 | TRAE, ORR | April 2023 |
| NCT03149822 | I/II | Advanced or metastatic RCC | Cabozantinib + pembrolizumab | 45 | ORR | December 2020 |
| MK-3475-03A NCT04626479 | I/II | Untreated, locally advanced or metastatic clear-cell RCC | Pembrolizumab + favezelimab Pembrolizumab + belzutifan Pembrolizumab + MK-4830 Pembrolizumab + lenvatinib Lenvatinib + belzutifan Pembrolizumab + quavonlimab | 390 | TRAE | June 2025 |
| MK-3475-03B NCT04626518 | I/II | Locally advanced or metastatic ccRCC | Pembrolizumab + favezelimab Pembrolizumab + belzutifan Pembrolizumab + MK-4830 Pembrolizumab + lenvatinib Lenvatinib + belzutifan Pembrolizumab + quavonlimab | 370 | TRAE | May 2025 |
| NCT03634540 | II | Locally advanced or metastatic ccRCC Cohort 1: previously treated Cohort 2: immunotherapy-naïve | Belzutifan 120 mg PO daily + cabozantinib 60 mg PO daily | 118 | ORR | August 2025 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasherman, L.; Siu, D.H.W.; Woodford, R.; Harris, C.A. Angiogenesis Inhibitors and Immunomodulation in Renal Cell Cancers: The Past, Present, and Future. Cancers 2022, 14, 1406. https://doi.org/10.3390/cancers14061406
Kasherman L, Siu DHW, Woodford R, Harris CA. Angiogenesis Inhibitors and Immunomodulation in Renal Cell Cancers: The Past, Present, and Future. Cancers. 2022; 14(6):1406. https://doi.org/10.3390/cancers14061406
Chicago/Turabian StyleKasherman, Lawrence, Derrick Ho Wai Siu, Rachel Woodford, and Carole A. Harris. 2022. "Angiogenesis Inhibitors and Immunomodulation in Renal Cell Cancers: The Past, Present, and Future" Cancers 14, no. 6: 1406. https://doi.org/10.3390/cancers14061406
APA StyleKasherman, L., Siu, D. H. W., Woodford, R., & Harris, C. A. (2022). Angiogenesis Inhibitors and Immunomodulation in Renal Cell Cancers: The Past, Present, and Future. Cancers, 14(6), 1406. https://doi.org/10.3390/cancers14061406