
Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
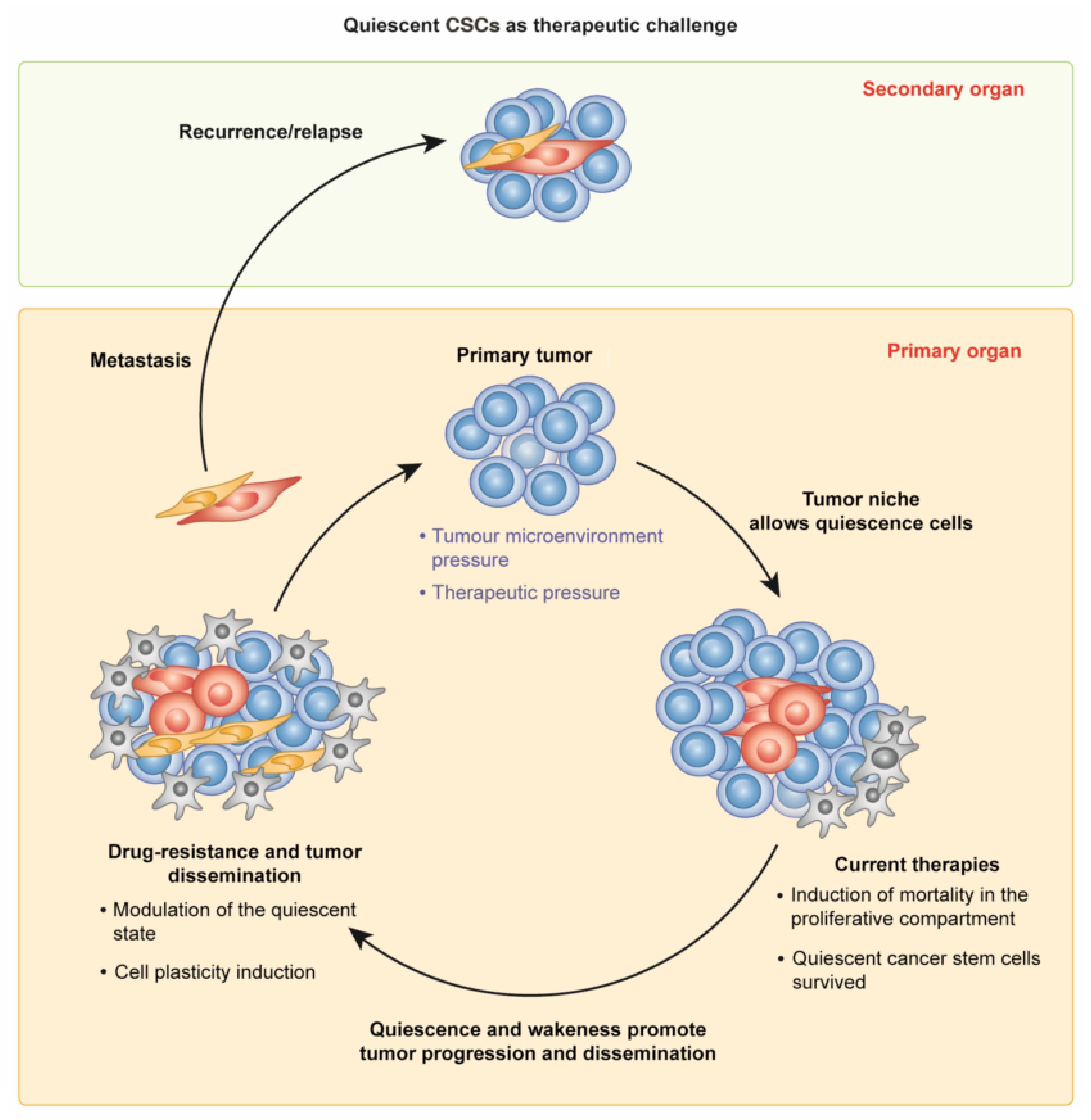
2. CSCs in Tumor Growth and Dissemination: The Quiescent and the Active State
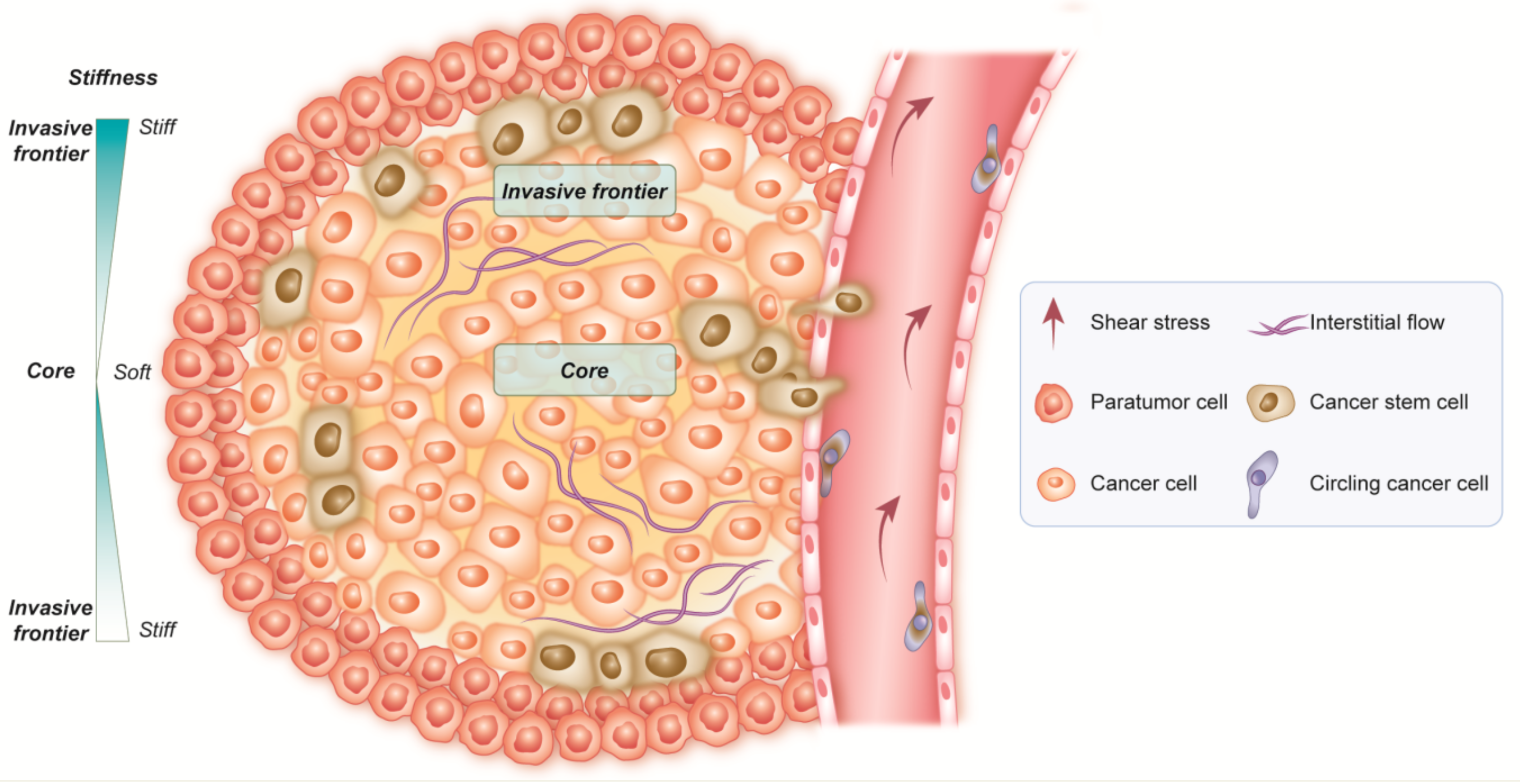
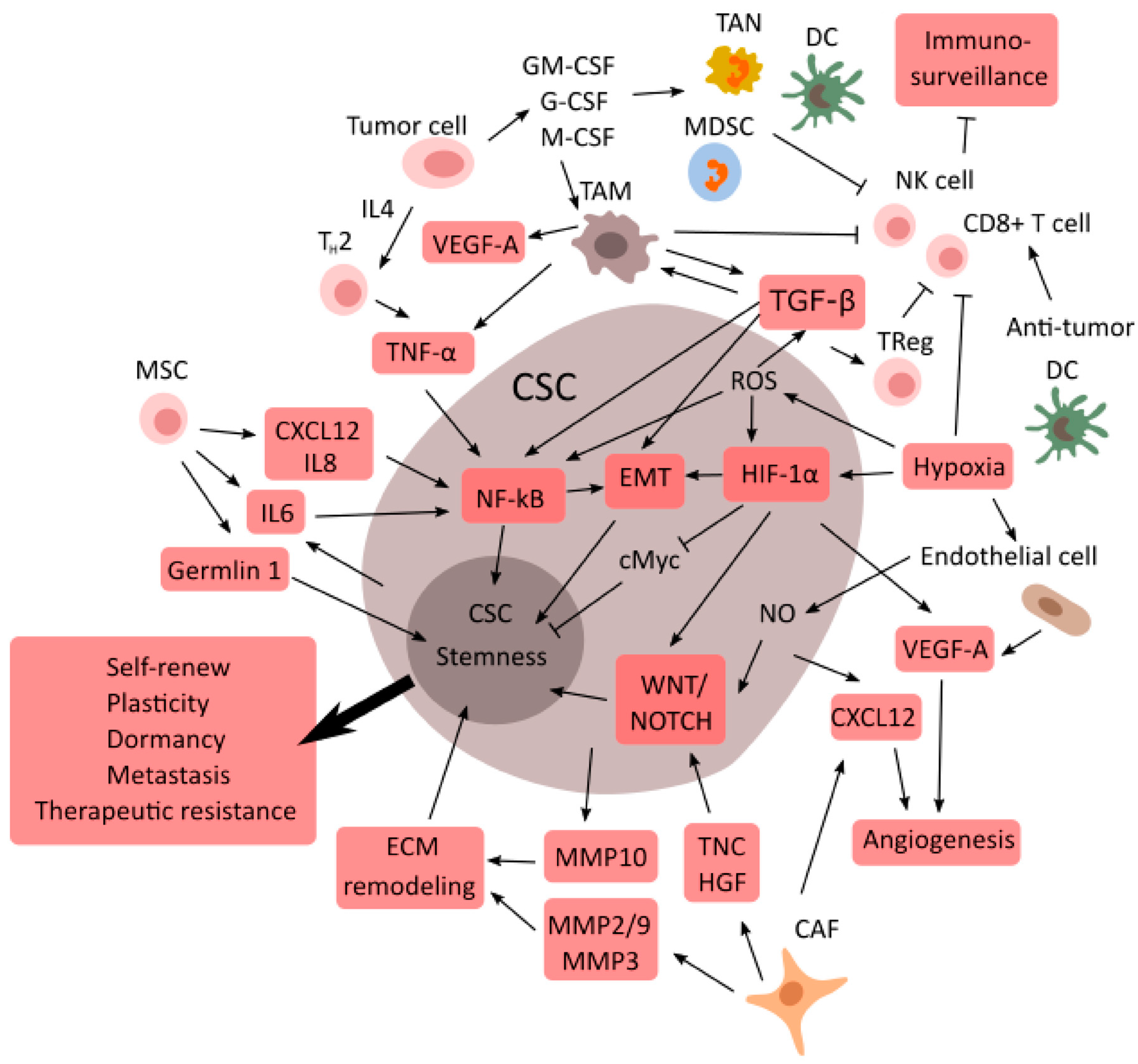
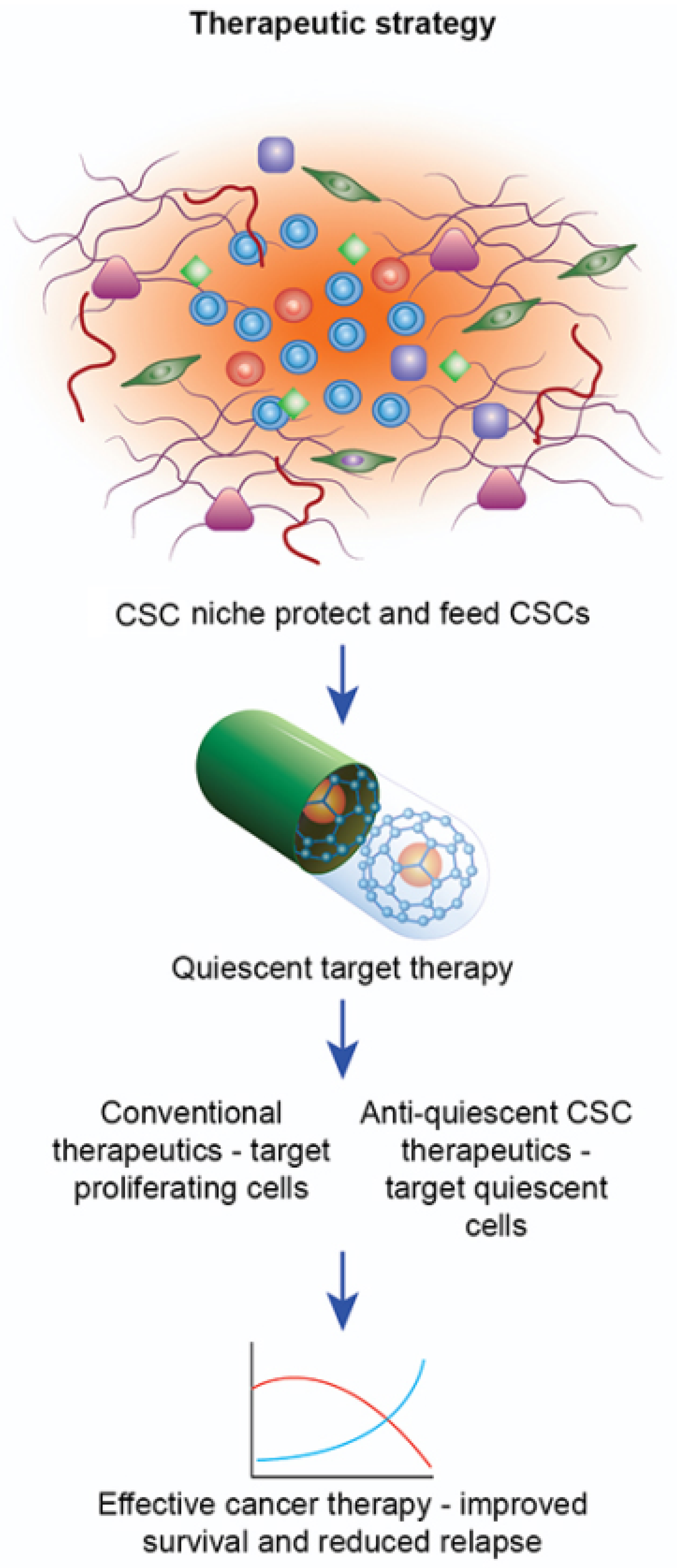
3. Cancer Resistance: CSCs and the Tumor Microenvironment
TME Supports CSCs
4. The Molecular Mechanisms Switching on CSCs and Metastasis
4.1. Horizontal Gene Transfers between Cells
4.2. Genetic Instability
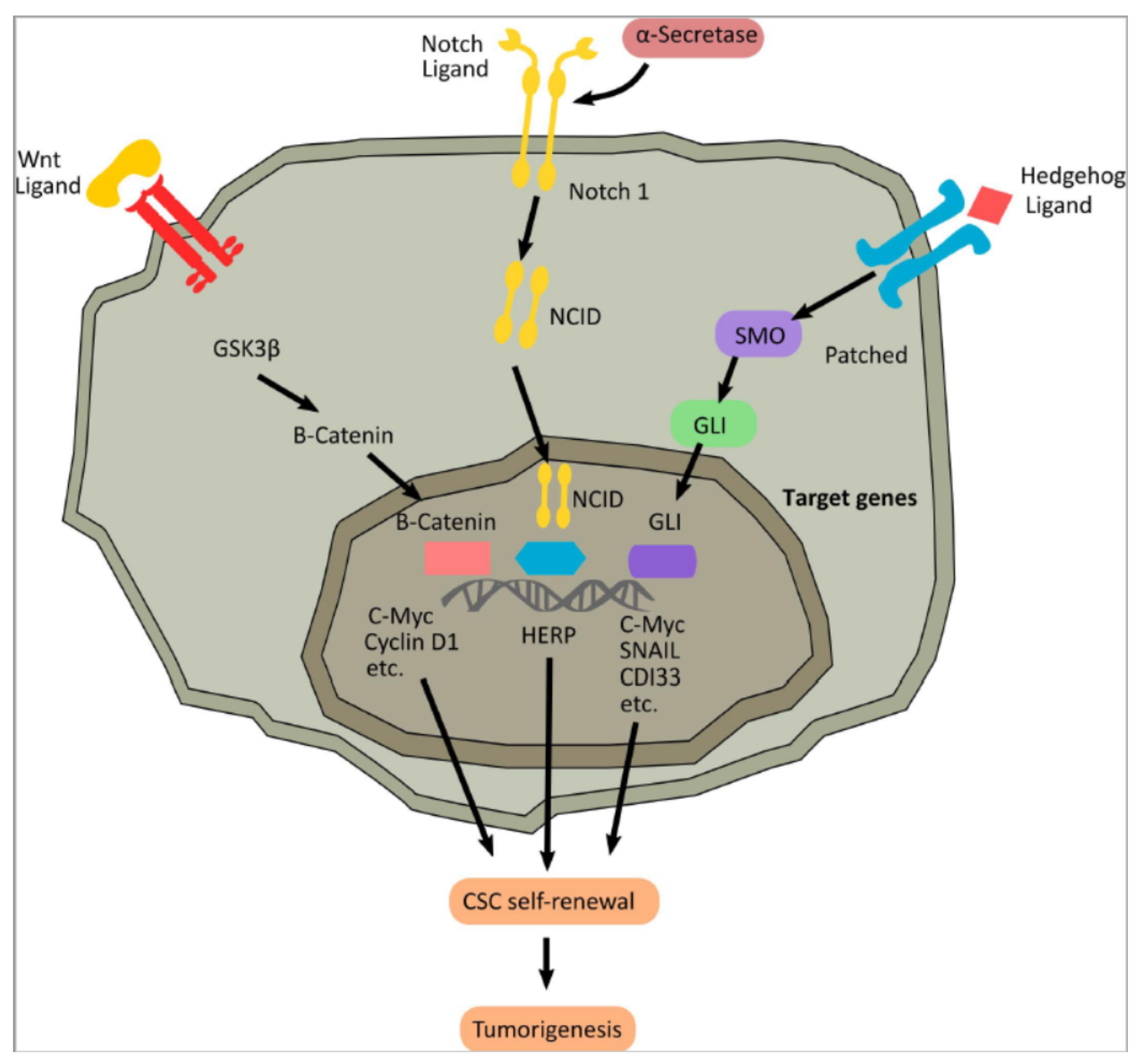
4.3. Molecular Pathways in Cancer Stem Cells
4.3.1. Wnt Signaling
4.3.2. The Hedgehog Signaling Pathway
4.3.3. The Notch Signaling Pathway
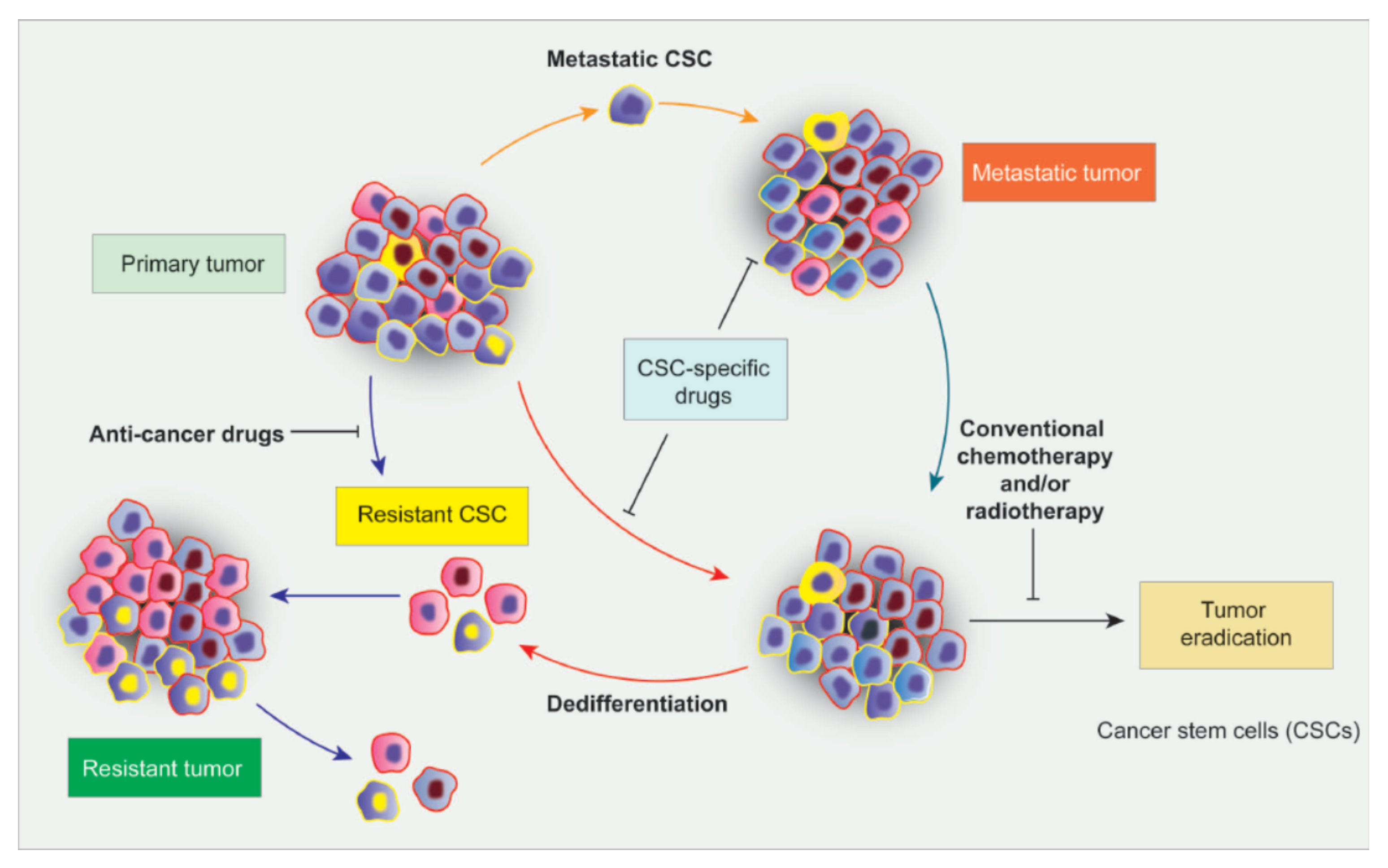
5. CSCs as Novel Targets for Cancer Therapy: New Perspectives to Control Tumorigenesis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Falzone, L.; Salomone, S.; Libra, M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Tanaka, F. Recurrence after surgery in patients with NSCLC. Transl. Lung Cancer Res. 2014, 3, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, R.; Tiselius, E.; Roudi, R.; Teghararian, O.; Suo, C.; Song, H. Immunotherapy (excluding checkpoint inhibitors) for stage I to III non-small cell lung cancer treated with surgery or radiotherapy with curative intent. Cochrane Database Syst. Rev. 2017, 12, CD011300. [Google Scholar] [CrossRef]
- Karuppasamy, R.; Veerappapillai, S.; Maiti, S.; Shin, W.H.; Kihara, D. Current progress and future perspectives of polypharmacology: From the view of non-small cell lung cancer. Semin. Cancer Biol. 2021, 68, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Antoni, D.; Mornex, F. Chemoradiotherapy of locally advanced nonsmall cell lung cancer: State of the art and perspectives. Curr. Opin. Oncol. 2016, 28, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, H.H.; Christensen, M.R.; Lassen, U.N. A systematic review of targeted agents for non-small cell lung cancer. Acta Oncol. 2018, 57, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Rath, B. Detection of circulating tumor cells in non-small cell lung cancer. J. Thorac. Dis. 2016, 8, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Mamdani, H.; Ahmed, S.; Armstrong, S.; Mok, T.; Jalal, S.I. Blood-based tumor biomarkers in lung cancer for detection and treatment. Transl. Lung Cancer Res. 2017, 6, 648–660. [Google Scholar] [CrossRef]
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: Implications for cancer therapy. Curr. Protoc. Pharmacol. 2013, 61, 14–25. [Google Scholar] [CrossRef]
- Lathia, J.D.; Liu, H. Overview of cancer stem cells and stemness for community oncologists. Target. Oncol. 2017, 12, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Matsui, W.H.; Chang, J.C. Cancer stem cells: A nuanced perspective. Medicine 2016, 95 (Suppl. S1), S26–S28. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Arya, R.K.; Maheshwari, S.; Singh, A.; Meena, S.; Pandey, P.; Dormond, O.; Datta, D. Tumor heterogeneity and cancer stem cell paradigm: Updates in concept, controversies and clinical relevance. Int. J. Cancer 2015, 136, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef]
- Schatton, T.; Frank, N.Y.; Frank, M.H. Identification and targeting of cancer stem cells. Bioessays 2009, 31, 1038–1049. [Google Scholar] [CrossRef]
- Beck, B.; Driessens, G.; Goossens, S.; Youssef, K.K.; Kuchnio, A.; Caauwe, A.; Sotiropoulou, P.A.; Loges, S.; Lapouge, G.; Candi, A.; et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 2011, 478, 399–403. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, J.D.; Barr, M.; Fennell, D.; Richard, D.; Reynolds, J.; O’Leary, J.; O’Byrne, K. The cancer stem-cell hypothesis: Its emerging role in lung cancer biology and its relevance for future therapy. J. Thorac. Oncol. 2012, 7, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Noren, H.; Jove, R.; Beljanski, V.; Grinnemo, K.H. Differences and similarities between cancer and somatic stem cells: Therapeutic implications. Stem. Cell Res. Ther. 2020, 11, 489. [Google Scholar] [CrossRef]
- Hodson, R. Precision medicine. Nature 2016, 537, S49. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.K.; Oluoha, O.; Patel, K.; VanderWalde, A. Precision medicine in oncology: A review of multi-tumor actionable molecular targets with an emphasis on non-small cell lung cancer. J. Pers. Med. 2021, 11, 518. [Google Scholar] [CrossRef] [PubMed]
- Koesling, D.; Bozzaro, C. Chronic pain patients’ need for recognition and their current struggle. Med. Health Care Philos. 2021, 24, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.R.; Wang, S.; Yan, S.C.; Zhang, Y.; Nelson, P.J.; Jia, H.L.; Qin, L.X.; Dong, Q.Z. Therapeutic strategies targeting cancer stem cells and their microenvironment. Front. Oncol. 2019, 9, 1104. [Google Scholar] [CrossRef]
- Vassalli, G. Aldehyde dehydrogenases: Not just markers, but functional regulators of stem cells. Stem. Cells Int. 2019, 2019, 3904645. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Visus, C.; Wang, Y.; Lozano-Leon, A.; Ferris, R.L.; Silver, S.; Szczepanski, M.J.; Brand, R.E.; Ferrone, C.R.; Whiteside, T.L.; Ferrone, S.; et al. Targeting ALDH (bright) human carcinoma-initiating cells with ALDH1A1-specific CD8? T cells. Clin. Cancer Res. 2011, 17, 6174–6184. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; Spinola, M.; Dodge, M.; Raso, M.G.; Behrens, C.; Gao, B.; Schuster, K.; Shao, C.; Larsen, J.E.; Sullivan, L.A.; et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010, 70, 9937–9948. [Google Scholar] [CrossRef]
- Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Sighinolfi, P.; Stefani, A.; Morandi, U.; Dominici, M.; Aramini, B. Isolation and identification of cancer stem-like cells in adenocarcinoma and squamous cell carcinoma of the lung: A pilot study. Front. Oncol. 2019, 9, 1394. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Guo, X.; Huang, Y. Correlation of cancer stem-cell markers OCT4, SOX2, and NANOG with clinicopathological features and prognosis in operative patients with rectal cancer. Yonsei Med. J. 2018, 59, 35–42. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in cancer: Stem cells, cancer stem cells, and their microenvironment. Stem. Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Sighinolfi, P.; Stefani, A.; Morandi, U.; Dominici, M.; Aramini, B. CD44+/EPCAM+ cells detect a subpopulation of ALDHhigh cells in human non-small cell lung cancer: A chance for targeting cancer stem cells? Oncotarget 2020, 11, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, C.; Yu, Y.; Li, W.; Ma, Z.; Wang, J.; Zhang, X.; Gao, H.; Liu, D. Photoactive silver nanoagents for backgroundless monitoring and precision killing of multidrug-resistant bacteria. Nanotheranostics 2021, 5, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer stem cells-origins and biomarkers: Perspectives for targeted personalized therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef]
- Franco, S.S.; Szczesna, K.; Iliou, M.S.; Al-Qahtani, M.; Mobasheri, A.; Kobolák, J.; Dinnyés, A. In vitro models of cancer stem cells and clinical applications. BMC Cancer 2016, 16 (Suppl. S2), 738. [Google Scholar] [CrossRef]
- Wang, K.; Wu, X.; Wang, J.; Huang, J. Cancer stem cell theory: Therapeutic implications for nanomedicine. Int. J. Nanomed. 2013, 8, 899–908. [Google Scholar]
- Tan, B.T.; Park, C.Y.; Ailles, L.E.; Weissman, I.L. The cancer stem cell hypothesis: A work in progress. Lab. Investig. 2006, 86, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Gómez-López, S.; Lerner, R.G.; Petritsch, C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell Mol. Life Sci. 2014, 71, 575–597. [Google Scholar] [CrossRef]
- Tassan, J.-P.; Kubiak, J.Z. Asymmetric Cell Division in Development, Differentiation and Cancer; Springer: Cham, Germany, 2017; Volume 61, pp. 351–373. Available online: http://link.springer.com/10.1007/978-3-319-53150-2 (accessed on 28 January 2022).
- Vinogradova, T.V.; Chernov, I.P.; Monastyrskaya, G.S.; Kondratyeva, L.G.; Sverdlov, E.D. Cancer stem cells: Plasticity works against therapy. Acta Naturae 2015, 7, 46–55. [Google Scholar] [CrossRef]
- Saga, R.; Matsuya, Y.; Takahashi, R.; Hasegawa, K.; Date, H.; Hosokawa, Y. Analysis of the high-dose-range radioresistance of prostate cancer cells, including cancer stem cells, based on a stochastic model. J. Radiat. Res. 2019, 60, 298–307. [Google Scholar] [CrossRef]
- Dzobo, K.; Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Mwapagha, L.M.; Al-Awwad, N.; Dandara, C.; Parker, M.I. Cancer stem cell hypothesis for therapeutic innovation in clinical oncology? Taking the root out, not chopping the leaf. OMICS J. Integr. Biol. 2016, 20, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Boue, S.; Izpisua Belmonte, J.C. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J. Cell reversal from a differentiated to a stem-like state at cancer initiation. Front. Oncol. 2020, 10, 541. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Impact of the tumor microenvironment on tumor heterogeneity and consequences for cancer cell plasticity and stemness. Cancers 2020, 12, 3716. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Lehnert, H.; Ungefroren, H.; Hass, R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol. Cancer 2017, 16, 28. [Google Scholar] [CrossRef]
- Zheng, X.; Yu, C.; Xu, M. Linking tumor microenvironment to plasticity of cancer stem cells: Mechanisms and application in cancer therapy. Front. Oncol. 2021, 11, 678333. [Google Scholar] [CrossRef]
- Olmeda, F.; Ben Amar, M. Clonal pattern dynamics in tumor: The concept of cancer stem cells. Sci. Rep. 2019, 9, 15607. [Google Scholar] [CrossRef]
- Coller, H.A. Regulation of cell cycle entry and exit: A single cell perspective. Compr. Physiol. 2019, 10, 317–344. [Google Scholar] [CrossRef]
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019, 21, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Gasch, C.; Ffrench, B.; O’Leary, J.J.; Gallagher, M.F. Catching moving targets: Cancer stem cell hierarchies, therapy-resistance & considerations for clinical intervention. Mol. Cancer 2017, 16, 43. [Google Scholar]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Zeniou, M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; Niu, G.; Huber, P.; Carter, W.B. Tumor-induced upregulation of twist, snail, and slug represses the activity of the human VE-cadherin promoter. Arch. Biochem. Biophys. 2009, 482, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Merrell, A.J.; Stanger, B.Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef]
- Luo, M.; Li, J.F.; Yang, Q.; Zhang, K.; Wang, Z.W.; Zheng, S.; Zhou, J.J. Stem cell quiescence and its clinical relevance. World J. Stem. Cells. 2020, 12, 1307–1326. [Google Scholar] [CrossRef]
- Sun, L.Y.; Pang, C.Y.; Li, D.K.; Liao, C.H.; Huang, W.C.; Wu, C.C.; Chou, Y.Y.; Li, W.W.; Chen, S.Y.; Liu, H.W.; et al. Antioxidants cause rapid expansion of human adipose-derived mesenchymal stem cells via CDK and CDK inhibitor regulation. J. Biomed. Sci. 2013, 20, 53. [Google Scholar] [CrossRef]
- Li, L.; Bhatia, R. Stem cell quiescence. Clin. Cancer Res. 2011, 17, 4936–4941. [Google Scholar] [CrossRef] [PubMed]
- Jandial, R.; Waters, D.J.; Chen, M.Y. Cancer stem cells can arise from differentiated neoplastic cells. Neurosurgery 2011, 69, N22. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Wade Harper, J.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Sage, J. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 2012, 26, 1409–1420. [Google Scholar] [CrossRef]
- Kim, E.; Cheng, Y.; Bolton-Gillespie, E.; Cai, X.; Ma, C.; Tarangelo, A.; Le, L.; Jambhekar, M.; Raman, P.; Hayer, K.E.; et al. Rb family proteins enforce the homeostasis of quiescent hematopoietic stem cells by repressing Socs3 expression. J. Exp. Med. 2017, 214, 1901–1912. [Google Scholar] [CrossRef]
- Imayoshi, I.; Sakamoto, M.; Yamaguchi, M.; Mori, K.; Kageyama, R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 2010, 30, 3489–3498. [Google Scholar] [CrossRef]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol. Proced. Online 2014, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Serviss, J.T.; Johnsson, P.; Grandér, D. An emerging role for long non-coding RNAs in cancer metastasis. Front. Genet 2014, 5, 234. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Ruth, J.R.; Pant, D.K.; Pan, T.C.; Seidel, H.E.; Baksh, S.C.; Keister, B.A.; Singh, R.; Sterner, C.J.; Bakewell, S.J.; Moody, S.E.; et al. Cellular dormancy in minimal residual disease following targeted therapy. Breast Cancer Res. BCR 2021, 23, 63. [Google Scholar] [CrossRef] [PubMed]
- Summers, M.A.; McDonald, M.M.; Croucher, P.I. Cancer cell dormancy in metastasis. Cold Spring Harb. Perspect. Med. 2020, 10, a037556. [Google Scholar] [CrossRef]
- Davies, A.E.; Albeck, J.G. Microenvironmental signals and biochemical information processing: Cooperative determinants of intratumoral plasticity and heterogeneity. Front. Cell Dev. Biol. 2018, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Ullrich, J.E. Thrombospondin 1 and Its diverse roles as a regulator of extracellular matrix in fibrotic disease. J. Histochem. Cytochem. 2019, 67, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massague, J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The metabolic flexibility of quiescent CSC: Implications for chemotherapy resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Shen, S.; Clairambault, J. Cell plasticity in cancer cell populations. F1000Research 2020, 9, 635. [Google Scholar] [CrossRef]
- Saxena, S.; Singh, R.K. Chemokines orchestrate tumor cells and the microenvironment to achieve metastatic heterogeneity. Cancer Metastasis Rev. 2021, 40, 447–476. [Google Scholar] [CrossRef] [PubMed]
- Kleffel, S.; Schatton, T. Tumor dormancy and cancer stem cells: Two sides of the same coin? Adv. Exp. Med. Biol. 2013, 734, 145–179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Xin, L.; Liang, A.; Fu, Y. Cancer stem cell hypothesis: A brief summary and two proposals. Cytotechnology 2013, 65, 505–512. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Qiao, B.; Smith, R.A.; Gopalan, V.; Lam, A.K. Cancer stem cell: Fundamental experimental pathological concepts and updates. Exp. Mol. Pathol. 2015, 98, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug resistance driven by cancer stem cells and their niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, L.; Dong, L.; Wang, X. Emerging role of exosome signalling in maintaining cancer stem cell dynamic equilibrium. J. Cell Mol. Med. 2018, 22, 3719–3728. [Google Scholar] [CrossRef]
- Picco, N.; Gatenby, R.A.; Anderson, A.R.A. Stem cell plasticity and niche dynamics in cancer progression. IEEE Trans. Biomed. Eng. 2017, 64, 528–537. [Google Scholar] [CrossRef]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Tu, V.Y.; Ayari, A.; O’Connor, R.S. Beyond the lactate paradox: How lactate and acidity impact T cell therapies against cancer. Antibodies 2021, 10, 25. [Google Scholar] [CrossRef]
- Marquardt, S.; Solanki, M.; Spitschak, A.; Vera, J.; Pützer, B.M. Emerging functional markers for cancer stem cell-based therapies: Understanding signaling networks for targeting metastasis. Semin. Cancer Biol. 2018, 53, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Rodriguez-Aznar, E.; Ferreira, M.S.V.; Frappart, P.O.; Dittrich, T.; Tiwary, K.; Meessen, S.; Lerma, L.; Daiss, N.; Schulte, L.A.; et al. Telomerase and pluripotency factors jointly regulate stemness in pancreatic cancer stem cells. Cancers 2021, 13, 3145. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef]
- Tian, B.R.; Lin, W.F.; Zhang, Y. Effects of biomechanical forces on the biological behavior of cancer stem cells. J. Cancer 2021, 12, 5895–5902. [Google Scholar] [CrossRef]
- Chim, L.K.; Mikos, A.G. Biomechanical forces in tissue engineered tumor models. Curr. Opin. Biomed. Eng. 2018, 6, 42–50. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix stiffness-mediated effects on stemness characteristics occurring in HCC cells. Oncotarget 2016, 7, 32221–32231. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Johansson, S. Beta1 integrins restrict the growth of foci and spheroids. Histochem. Cell Biol. 2012, 138, 881–894. [Google Scholar] [CrossRef]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef]
- Jing, N.; Gao, W.Q.; Fang, Y.X. Regulation of formation, stemness and therapeutic resistance of cancer stem cells. Front. Cell Dev. Biol. 2021, 9, 641498. [Google Scholar] [CrossRef]
- Khalaf, K.; Hana, D.; Chou, J.T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the tumor microenvironment involved in immune resistance and drug resistance. Front. Immunol. 2021, 12, 656364. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Seo, B.R.; Fischbach, C.; Gourdon, D. Fibronectin mechanobiology regulates tumorigenesis. Cell Mol. Bioeng. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, C.; Shi, Y.; Wu, Q.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.F.; et al. Targeting glioma stem cell-derived pericytes disrupts the blood-tumor barrier and improves chemotherapeutic efficacy. Cell Stem Cell 2017, 21, 591–603.e4. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]
- Keyvani-Ghamsari, S.; Khorsandi, K.; Rasul, A.; Zaman, M.K. Current understanding of epigenetics mechanism as a novel target in reducing cancer stem cells resistance. Clin. Epigenetics 2021, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Lutz, C.; van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011, 469, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Sun, M.; Li, G.H.; Wu, Y.Z.; Wang, Y.; Jin, F.; Zhang, Y.Y.; Yang, L.; Wang, D.L. Activation of the phosphorylation of ATM contributes to radioresistance of glioma stem cells. Oncol. Rep. 2013, 30, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892, Erratum in N. Engl. J. Med. 2012, 367, 976. [Google Scholar] [CrossRef]
- Jin, L.; Vu, T.; Yuan, G.; Datta, P.K. STRAP Promotes Stemness of Human Colorectal Cancer via Epigenetic Regulation of the NOTCH pathway. Cancer Res. 2017, 77, 5464–5478. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Triaca, V.; Carito, V.; Fico, E.; Rosso, P.; Fiore, M.; Ralli, M.; Lambiase, A.; Greco, A.; Tirassa, P. Cancer stem cells-driven tumor growth and immune escape: The Janus face of neurotrophins. Aging 2019, 11, 11770–11792. [Google Scholar] [CrossRef]
- Ilkhanizadeh, S.; Weiss, W.A. Starvation favors glioma stem cells. Nat. Neurosci. 2013, 16, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Shimono, Y.; Funakoshi, Y.; Imamura, Y.; Toyoda, M.; Kiyota, N.; Kono, S.; Takao, S.; Mukohara, T.; Minami, H. Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene 2019, 38, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Sighinolfi, P.; Pinelli, M.; Lovati, E.; Stefani, A.; Morandi, U.; et al. Correlating tumor-infiltrating lymphocytes and lung cancer stem cells: A cross-sectional study. Ann. Transl. Med. 2019, 7, 619. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell 2017, 20, 450–461.e4. [Google Scholar] [CrossRef] [PubMed]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef]
- Tallerico, R.; Todaro, M.; Di Franco, S.; Maccalli, C.; Garofalo, C.; Sottile, R.; Palmieri, C.; Tirinato, L.; Pangigadde, P.N.; La Rocca, R.; et al. Human NK cells selective targeting of colon cancer-initiating cells: A role for natural cytotoxicity receptors and MHC class I molecules. J. Immunol. 2013, 190, 2381–2390. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Nör, J.E. Head and neck cancer stem cells. J. Dent. Res. 2012, 91, 334–340. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2017, 36, 1707–1720. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Morandi, U.; Dominici, M.; Aramini, B. Cancer stem-neuroendocrine cells in an atypical carcinoid case report. Transl. Lung Cancer Res. 2019, 8, 1157–1162. [Google Scholar] [CrossRef]
- Masciale, V.; Banchelli, F.; Grisendi, G.; D’Amico, R.; Maiorana, A.; Stefani, A.; Morandi, U.; Dominici, M.; Aramini, B. New Perspectives in different gene expression profiles for early and locally advanced non-small cell lung cancer stem cells. Front. Oncol. 2021, 11, 613198. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.X.; Yu, Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol. Sin. 2015, 36, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell plasticity and heterogeneity in cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Bloom, A.B.; Zaman, M.H. Influence of the microenvironment on cell fate determination and migration. Physiol. Genom. 2014, 46, 309–314. [Google Scholar] [CrossRef]
- Pastò, A.; Consonni, F.M.; Sica, A. Influence of innate immunity on cancer cell stemness. Int. J. Mol. Sci. 2020, 21, 3352. [Google Scholar] [CrossRef] [PubMed]
- Kaveh, K.; Kohandel, M.; Sivaloganathan, S. Replicator dynamics of cancer stem cell: Selection in the presence of differentiation and plasticity. Math. Biosci. 2016, 272, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer stem cells and radioresistance: DNA repair and beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef]
- Olivares-Urbano, M.A.; Griñán-Lisón, C.; Marchal, J.A.; Núñez, M.I. CSC Radioresistance: A Therapeutic challenge to improve radiotherapy effectiveness in cancer. Cells 2020, 9, 1651. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer stem cells: The architects of the tumor ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.; Ho, N.P.; Lee, T.K. Cancer stem cells and their microenvironment: Biology and therapeutic implications. Stem Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef] [PubMed]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23 Pt B, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Application of highly immunocompromised mice for the establishment of patient-derived xenograft (PDX) models. Cells 2019, 8, 889. [Google Scholar] [CrossRef]
- Horowitz, N.B.; Mohammad, I.; Moreno-Nieves, U.Y.; Koliesnik, I.; Tran, Q.; Sunwoo, J.B. Humanized mouse models for the advancement of innate lymphoid cell-based cancer immunotherapies. Front. Immunol. 2021, 12, 648580. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Bayik, D.; Lathia, J.D. Cancer stem cell-immune cell crosstalk in tumour progression. Nat. Rev. Cancer 2021, 21, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Rowley, D.A.; Schreiber, H. Targeting stroma to treat cancers. Semin. Cancer Biol. 2012, 22, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Aspizua, S.; González-Masa, A.; Conti, C.J.; García, M.; Chacón-Solano, E.; Larcher, F.; Del Río, M. Humanization of tumor stroma by tissue engineering as a tool to improve squamous cell carcinoma xenograft. Int. J. Mol. Sci. 2020, 21, 1951. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Guo, M.; Cai, C.; Zhao, G.; Qiu, X.; Zhao, H.; Ma, Q.; Tian, L.; Li, X.; Hu, Y.; Liao, B.; et al. Hypoxia promotes migration and induces CXCR4 expression via HIF-1? Activation in human osteosarcoma. PLoS ONE 2014, 9, e90518. [Google Scholar] [CrossRef]
- Nguyen, V.H.L.; Hough, R.; Bernaudo, S.; Peng, C. Wnt/?-catenin signalling in ovarian cancer: Insights into its hyperactivation and function in tumorigenesis. J. Ovarian Res. 2019, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Teeuwssen, M.; Fodde, R. Wnt signaling in ovarian cancer stemness, EMT and Therapy Resistance. J. Clin. Med. 2019, 8, 1658. [Google Scholar] [CrossRef]
- Tanabe, A.; Sahara, H. The metabolic heterogeneity and flexibility of cancer stem cells. Cancers 2020, 12, 2780. [Google Scholar] [CrossRef]
- Morrison, A.J. Cancer cell metabolism connects epigenetic modifications to transcriptional regulation. FEBS J. 2021. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic adaptations in cancer stem cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef] [PubMed]
- Penkert, J.; Ripperger, T.; Schieck, M.; Schlegelberger, B.; Steinemann, D.; Illig, T. On metabolic reprogramming and tumor biology: A comprehensive survey of metabolism in breast cancer. Oncotarget 2016, 7, 67626–67649. [Google Scholar] [CrossRef]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic reprogramming of cancer associated fibroblasts: The slavery of stromal fibroblasts. Biomed Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Pantel, K. Tumor cell dissemination: Emerging biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2021, 124, 13–26. [Google Scholar] [CrossRef]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2017, 11, 62–78. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Haase, G.; Ben-Ze’ev, A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Research 2016, 5, 699. [Google Scholar] [CrossRef]
- de Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Massagué, J. Extracellular matrix players in metastatic niches. EMBO J. 2012, 31, 254–256. [Google Scholar] [CrossRef]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef]
- Cho, C.; Kelsh-Lasher, R.; Ambesi, A.; McKeown-Longo, P.J. Cryptic activity within the Type III1 domain of fibronectin regulates tissue inflammation and angiogenesis. Curr. Top Pept. Protein Res. 2015, 16, 37–47. [Google Scholar]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer stem cell plasticity—A deadly deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Sánchez, A.; Suárez-Martínez, E.; Sánchez-Díaz, L.; Carnero, A. Therapeutic targeting of signaling pathways related to cancer stemness. Front. Oncol. 2020, 10, 1533. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning cancer fate: Tumor microenvironment’s role in cancer stem cell quiescence and reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Kyriakou, T.C.; Papageorgis, P. Mechanisms of metastatic tumor dormancy and implications for cancer therapy. Int. J. Mol. Sci. 2019, 20, 6158. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Nam, J.S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef]
- Dayton, J.B.; Piccolo, S.R. Classifying cancer genome aberrations by their mutually exclusive effects on transcription. BMC Med. Genom. 2017, 10 (Suppl. S4), 66. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zhou, X.; Yang, J.; Shi, H.; Li, H.; Zhao, X.; Ma, X. The role of tumor-stroma interactions in drug resistance within tumor microenvironment. Front. Cell Dev. Biol. 2021, 9, 637675. [Google Scholar] [CrossRef]
- He, J.; Xiong, L.; Li, Q.; Lin, L.; Miao, X.; Yan, S.; Hong, Z.; Yang, L.; Wen, Y.; Deng, X. 3D modeling of cancer stem cell niche. Oncotarget 2017, 9, 1326–1345. [Google Scholar] [CrossRef]
- Baram, T.; Rubinstein-Achiasaf, L.; Ben-Yaakov, H.; Ben-Baruch, A. Inflammation-driven breast tumor cell plasticity: Stemness/EMT therapy resistance and dormancy. Front. Oncol. 2021, 10, 614468. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Morandi, U.; Dominici, M.; Haider, K.H. Cancer stem cells and macrophages: Molecular connections and future perspectives against cancer. Oncotarget 2021, 12, 230–250. [Google Scholar] [CrossRef]
- Pawelek, J.M. Tumour cell hybridization and metastasis revisited. Melanoma Res. 2000, 10, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Prinz, F.; Bauer, M.; Spiegel, M.; Neubert, W.J.; Gregor, M.; Schulze-Osthoff, K.; Lauer, U. Sendai virus infection induces apoptosis through activation of caspase-8 (FLICE) and caspase-3 (CPP32). J. Virol. 1999, 73, 702–708. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Hochedlinger, K. Induced pluripotency: History, mechanisms, and applications. Genes Dev. 2010, 24, 2239–2263. [Google Scholar] [CrossRef]
- Delespaul, L.; Gélabert, C.; Lesluyes, T.; Le Guellec, S.; Pérot, G.; Leroy, L.; Baud, J.; Merle, C.; Lartigue, L.; Chibon, F. Cell-cell fusion of mesenchymal cells with distinct differentiations triggers genomic and transcriptomic remodelling toward tumour aggressiveness. Sci. Rep. 2020, 10, 21634. [Google Scholar] [CrossRef]
- Gauck, D.; Keil, S.; Niggemann, B.; Zänker, K.S.; Dittmar, T. Hybrid clone cells derived from human breast epithelial cells and human breast cancer cells exhibit properties of cancer stem/initiating cells. BMC Cancer 2017, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.H.; Gao, X.; Luo, D.; Zhou, X.D.; Xiong, W.; Liu, G.X. EMT and acquisition of stem cell-like properties are involved in spontaneous formation of tumorigenic hybrids between lung cancer and bone marrow-derived mesenchymal stem cells. PLoS ONE 2014, 9, e87893. [Google Scholar] [CrossRef] [PubMed]
- Merle, C.; Lagarde, P.; Lartigue, L.; Chibon, F. Acquisition of cancer stem cell capacities after spontaneous cell fusion. BMC Cancer 2021, 21, 241. [Google Scholar] [CrossRef]
- Jiang, E.; Yan, T.; Xu, Z.; Shang, Z. Tumor microenvironment and cell fusion. Biomed Res. Int. 2019, 2019, 5013592. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; Hernadez de la Cruz, O.N.; Lopez-Gonzalez, J.S. Contribution of angiogenesis to inflammation and cancer. Front. Oncol. 2019, 9, 1399. [Google Scholar] [CrossRef] [PubMed]
- Ram, Y.; Hadany, L. Evolution of stress-induced mutagenesis in the presence of horizontal gene transfer. Am. Nat. 2019, 194, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Emamalipour, M.; Seidi, K.; Zununi Vahed, S.; Jahanban-Esfahlan, A.; Jaymand, M.; Majdi, H.; Amoozgar, Z.; Chitkushev, L.T.; Javaheri, T.; Jahanban-Esfahlan, R.; et al. Horizontal gene transfer: From evolutionary flexibility to disease progression. Front. Cell Dev. Biol. 2020, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auer, M.; Ulz, P.; Geigl, J.B.; Speicher, M.R. Circulating tumor cells and DNA as liquid biopsies. Genome Med. 2013, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.T.; Cheng, J.; Feng, X.; He, S.J.; Wang, Y.W.; Huang, Q. The viable circulating tumor cells with cancer stem cells feature, where is the way out? J. Exp. Clin. Cancer Res. 2018, 37, 38. [Google Scholar] [CrossRef]
- Rycaj, K.; Tang, D.G. Cell-of-origin of cancer versus cancer stem cells: Assays and interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and cancer stem cells: Unleashing, hijacking, and restricting cellular plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Fagnocchi, L.; Zippo, A. Tumorigenic cell reprogramming and cancer plasticity: Interplay between signaling, microenvironment, and epigenetics. Stem Cells Int. 2018, 2018, 4598195. [Google Scholar] [CrossRef] [PubMed]
- Lagasse, E. Cancer stem cells with genetic instability: The best vehicle with the best engine for cancer. Gene Ther. 2008, 15, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.P.; Hawkins, J.R.; Boyle, J.; Bridger, J.M. The genomic health of human pluripotent stem cells: Genomic instability and the consequences on nuclear organization. Front. Genet. 2019, 9, 623. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Jasper, H.; Rudolph, K.L. Aging-induced stem cell mutations as drivers for disease and cancer. Cell Stem Cell 2015, 16, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.E.; Shung, C.Y.; Saylor, K.W.; Müllendorff, K.A.; Weiss, J.B.; Wong, M.H. Lessons from development: A role for asymmetric stem cell division in cancer. Stem Cell Res. 2010, 4, 3–9, Erratum in Stem Cell Res. 2010, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Venkei, Z.G.; Yamashita, Y.M. Emerging mechanisms of asymmetric stem cell division. J. Cell Biol. 2018, 217, 3785–3795. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular chaperones in cancer stem cells: Determinants of stemness and potential targets for antitumor therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting notch, hedgehog, and wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Červenka, I.; Čajánek, L. The connections of Wnt pathway components with cell cycle and centrosome: Side effects or a hidden logic? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 614–637. [Google Scholar] [CrossRef] [PubMed]
- Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Screpanti, I.; Bellavia, D. Wnt, notch, and TGF-? Pathways impinge on hedgehog signaling complexity: An open window on cancer. Front. Genet. 2019, 10, 711. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xiao, G.; Hu, J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci. 2014, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell. 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/?-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT signaling in tumors: The way to evade drugs and immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef] [PubMed]
- Tai, D.; Wells, K.; Arcaroli, J.; Vanderbilt, C.; Aisner, D.L.; Messersmith, W.A.; Lieu, C.H. Targeting the WNT signaling pathway in cancer therapeutics. Oncologist 2015, 20, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic. Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Gooding, A.J.; Schiemann, W.P. Epithelial-mesenchymal transition programs and cancer stem cell phenotypes: Mediators of breast cancer therapy resistance. Mol. Cancer Res. 2020, 18, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma stem-like cells, metabolic strategy to kill a challenging target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog signaling in the maintenance of cancer stem cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Fields, A.P. Molecular pathways: Novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin. Cancer Res. 2015, 21, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.M. Notch signaling and Notch signaling modifiers. Int. J. Biochem. Cell Biol. 2011, 43, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- van Tetering, G.; Vooijs, M. Proteolytic cleavage of Notch: “HIT and RUN”. Curr. Mol. Med. 2011, 11, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Grogan, S.P.; Olee, T.; Hiraoka, K.; Lotz, M.K. Repression of chondrogenesis through binding of notch signaling proteins HES-1 and HEY-1 to N-box domains in the COL2A1 enhancer site. Arthritis Rheum. 2008, 58, 2754–2763. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Chung, G.; Hamamori, Y.; Kedes, L. HERP1 is a cell type-specific primary target of Notch. J. Biol. Chem. 2002, 277, 6598–6607. [Google Scholar] [CrossRef] [PubMed]
- Xiu, M.X.; Liu, Y.M. The role of oncogenic Notch2 signaling in cancer: A novel therapeutic target. Am. J. Cancer Res. 2019, 9, 837–854. [Google Scholar]
- Kumar, V.; Vashishta, M.; Kong, L.; Wu, X.; Lu, J.J.; Guha, C.; Dwarakanath, B.S. The role of notch, hedgehog, and wnt signaling pathways in the resistance of tumors to anticancer therapies. Front. Cell Dev. Biol. 2021, 9, 650772. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.F.; Zhang, Y.C.; Peng, X.Q.; Long, Z.; Ming, Y.Z.; He, L.Y. Silencing Notch-1 induces apoptosis and increases the chemosensitivity of prostate cancer cells to docetaxel through Bcl-2 and Bax. Oncol. Lett. 2012, 3, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Das, T.; Damodaran, C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br. J. Cancer 2013, 109, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, H.; Zhang, J.; Chen, Y.; Wang, M.; Ma, J.; Hong, L.; Liu, N.; Fan, Q.; Lu, X.; et al. Increased notch signaling enhances radioresistance of malignant stromal cells induced by glioma stem/progenitor cells. PLoS ONE 2015, 10, e0142594. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, R.; Bentivegna, A. Role of notch signaling pathway in glioblastoma pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Núñez, F.J.; Haase, S.; McClellan, B.L.; Faisal, S.M.; Carney, S.V.; Yu, J.; Alghamri, M.S.; Asad, A.S.; Candia, A.J.N.; et al. Current approaches for glioma gene therapy and virotherapy. Front. Mol. Neurosci. 2021, 14, 621831. [Google Scholar] [CrossRef] [PubMed]
- Kenda Suster, N.; Virant-Klun, I. Presence and role of stem cells in ovarian cancer. World J. Stem. Cells 2019, 11, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.; Cowden Dahl, K.D. Can stemness and chemoresistance be therapeutically targeted via signaling pathways in ovarian cancer? Cancers 2018, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Meisel, C.T.; Porcheri, C.; Mitsiadis, T.A. Cancer stem cells, quo vadis? The notch signaling pathway in tumor initiation and progression. Cells 2020, 9, 1879. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Zhao, G.; Ilagan, M.X. An overview of notch signaling in adult tissue renewal and maintenance. Curr. Alzheimer. Res. 2012, 9, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, Z.Y.; Guo, X.T.; Xiao, X.H.; Xiong, L.X. Notch and breast cancer metastasis: Current knowledge, new sights and targeted therapy. Oncol. Lett. 2019, 18, 2743–2755. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, W.; Huang, K.; Wang, Y.; Li, J.; Yang, X. MicroRNA-34a inhibits cells proliferation and invasion by downregulating Notch1 in endometrial cancer. Oncotarget 2017, 8, 111258–111270. [Google Scholar] [CrossRef] [PubMed]
- Kontomanolis, E.N.; Kalagasidou, S.; Pouliliou, S.; Anthoulaki, X.; Georgiou, N.; Papamanolis, V.; Fasoulakis, Z.N. The notch pathway in breast cancer progression. Sci. World J. 2018, 2018, 2415489. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Nic An tSaoir, C.B.; Blayney, J.K.; Oram, L.C.; Crawford, N.T.; D’Costa, Z.C.; Quinn, J.E.; Kennedy, R.D.; Harkin, D.P.; Mullan, P.B. BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res. 2013, 41, 8601–8614. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhang, J.; Yarden, Y.; Fu, L. The key roles of cancer stem cell-derived extracellular vesicles. Signal Transduct. Target Ther. 2021, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Rajbhandari, N.; Wagner, K.U. Cancer cell dormancy in novel mouse models for reversible pancreatic cancer: A lingering challenge in the development of targeted therapies. Cancer Res. 2014, 74, 2138–2143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, L.; Li, J.C.; Yang, H.; Zhang, X.; Liu, L.L.; Li, Y.; Zeng, T.T.; Zhu, Y.H.; Li, X.D.; Li, Y.; et al. Expansion of cancer stem cell pool initiates lung cancer recurrence before angiogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E8948–E8957. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Dai, Y.; Grant, S.; Dent, P. Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol. Ther. 2012, 13, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Muriithi, W.; Macharia, L.W.; Heming, C.P.; Echevarria, J.L.; Nyachieo, A.; Filho, P.N.; Neto, V.M. ABC transporters and the hallmarks of cancer: Roles in cancer aggressiveness beyond multidrug resistance. Cancer Biol. Med. 2020, 17, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm. Sin. B 2021, 11, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Fang, X.; Luo, Q.; Ouyang, G. Cancer stem cells: A potential target for cancer therapy. Cell Mol. Life Sci. 2015, 72, 3411–3424. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Sullivan, J.P.; Girard, L.; Augustyn, A.; Yenerall, P.; Rodriguez-Canales, J.; Liu, H.; Behrens, C.; Shay, J.W.; Wistuba, I.I.; et al. Essential role of aldehyde dehydrogenase 1A3 for the maintenance of non-small cell lung cancer stem cells is associated with the STAT3 pathway. Clin. Cancer Res. 2014, 20, 4154–4166. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Aramini, B.; Masciale, V.; Bianchi, D.; Manfredini, B.; Banchelli, F.; D’Amico, R.; Bertolini, F.; Dominici, M.; Morandi, U.; Maiorana, A. ALDH expression in angiosarcoma of the lung: A potential marker of aggressiveness? Front. Med. 2020, 7, 544158. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Akhter, S.; Greig, N.H.; Kamal, M.A.; Midoux, P.; Pichon, C. Engineered nanoparticles against MDR in cancer: The state of the art and its prospective. Curr. Pharm. Des. 2016, 22, 4360–4373. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, Y.; Zhao, M. Exosome-based cancer therapy: Implication for targeting cancer stem cells. Front. Pharmacol. 2017, 7, 533. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Huang, X.; Mo, J.; Zhao, W. Drug delivery using nanoparticles for cancer stem-like cell targeting. Front. Pharmacol. 2016, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Golinelli, G.; Mastrolia, I.; Aramini, B.; Masciale, V.; Pinelli, M.; Pacchioni, L.; Casari, G.; Dall’Ora, M.; Soares, M.B.P.; Damasceno, P.K.F.; et al. Arming mesenchymal stromal/stem cells against cancer: Has the time come? Front. Pharmacol. 2020, 11, 529921. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target Ther. 2020, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Haider, K.H.; Aramini, B. Mircrining the injured heart with stem cell-derived exosomes: An emerging strategy of cell-free therapy. Stem Cell Res. Ther. 2020, 11, 23. [Google Scholar] [CrossRef]
- Diao, D.; Zhai, J.; Yang, J.; Wu, H.; Jiang, J.; Dong, X.; Passaro, A.; Aramini, B.; Rao, S.; Cai, K. Delivery of gefitinib with an immunostimulatory nanocarrier improves therapeutic efficacy in lung cancer. Transl. Lung Cancer Res. 2021, 10, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Aramini, B.; Masciale, V.; Haider, K.H. Defining lung cancer stem cells exosomal payload of miRNAs in clinical perspective. World J. Stem. Cells 2020, 12, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in exosome isolation techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Aramini, B.; Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Dominici, M.; Haider Kh, H. Targeting cancer stem cells: New perspectives for a cure to cancer? In Handbook of Stm Cells; Haider Kh, H., Ed.; Springer Nature: Cham, Germany, 2022; Chapter 45. [Google Scholar]
- Gaur, A.B.; Holbeck, S.L.; Colburn, N.H.; Israel, M.A. Downregulation of Pdcd4 by mir-21 facilitates glioblastoma proliferation in vivo. Neuro. Oncol. 2011, 13, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Aramini, B.; Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Dominici, M.; Haider Kh, H. Future perspectives of exosomal payload of miRNAs in lung cancer. In Handbook of Stem Cells; Haider Kh, H., Ed.; Springer Nature: Cham, Germany, 2022; Chapter 47. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aramini, B.; Masciale, V.; Grisendi, G.; Bertolini, F.; Maur, M.; Guaitoli, G.; Chrystel, I.; Morandi, U.; Stella, F.; Dominici, M.; et al. Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence. Cancers 2022, 14, 976. https://doi.org/10.3390/cancers14040976
Aramini B, Masciale V, Grisendi G, Bertolini F, Maur M, Guaitoli G, Chrystel I, Morandi U, Stella F, Dominici M, et al. Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence. Cancers. 2022; 14(4):976. https://doi.org/10.3390/cancers14040976
Chicago/Turabian StyleAramini, Beatrice, Valentina Masciale, Giulia Grisendi, Federica Bertolini, Michela Maur, Giorgia Guaitoli, Isca Chrystel, Uliano Morandi, Franco Stella, Massimo Dominici, and et al. 2022. "Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence" Cancers 14, no. 4: 976. https://doi.org/10.3390/cancers14040976
APA StyleAramini, B., Masciale, V., Grisendi, G., Bertolini, F., Maur, M., Guaitoli, G., Chrystel, I., Morandi, U., Stella, F., Dominici, M., & Haider, K. H. (2022). Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence. Cancers, 14(4), 976. https://doi.org/10.3390/cancers14040976