
Hyperactivation of MAPK Induces Tamoxifen Resistance in SPRED2-Deficient ERα-Positive Breast Cancer




Abstract
Simple Summary
Abstract
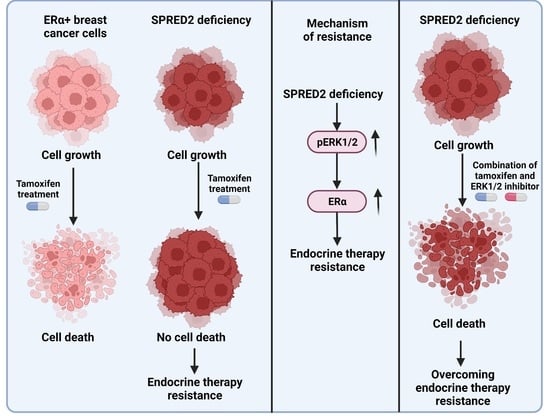
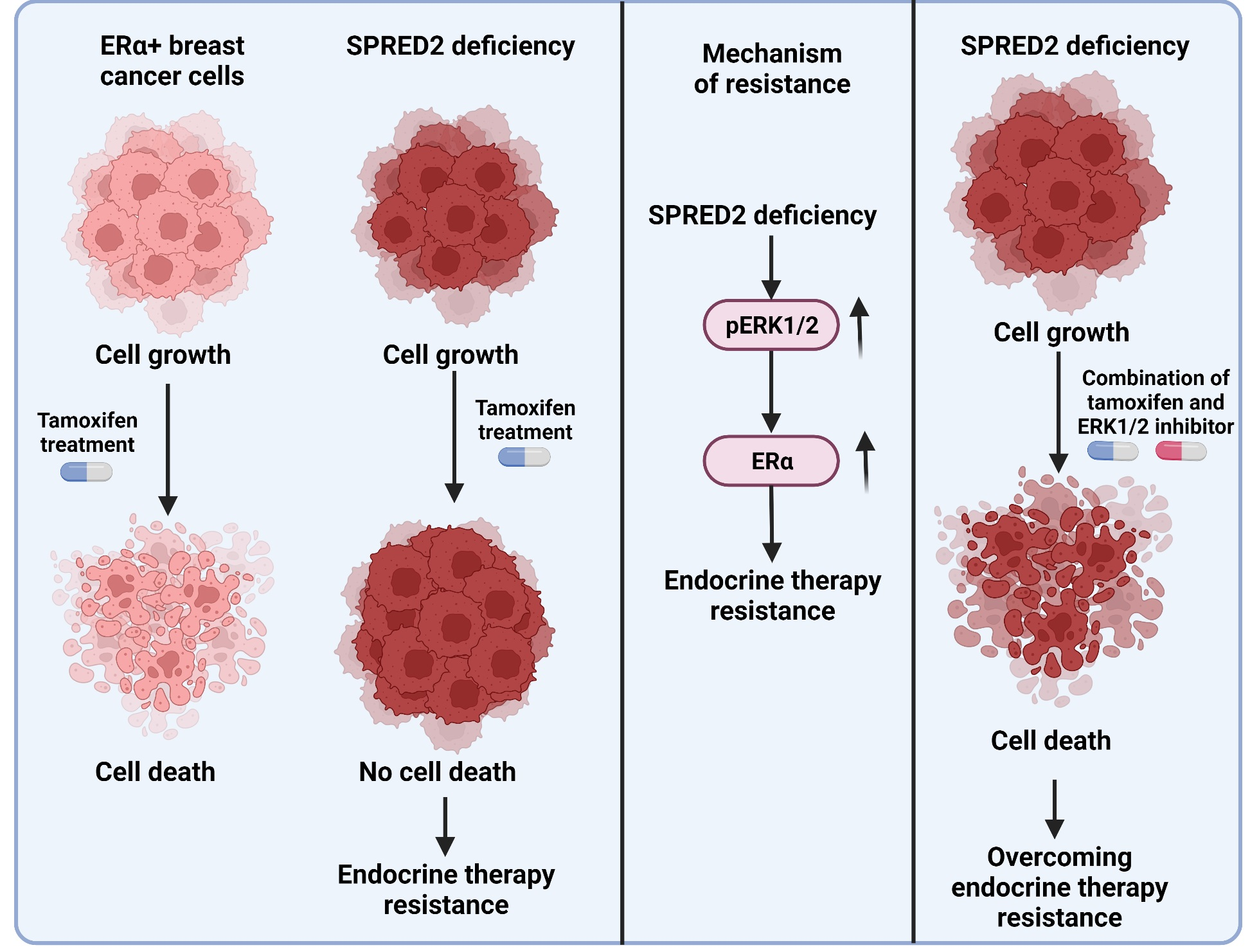
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Cell Treatments and Proliferation Assays
2.4. Lentiviral Transduction for sgRNA Gene Knockdowns and Generation of Single sgRNA Knockdown Clones
2.5. Lentiviral Transduction for shRNA-Mediated Knockdowns
2.6. Protein Extraction and Immunoblotting
2.7. Antibodies
2.8. Luciferase Reporter Assays
2.9. Wound Healing Assay
2.10. RNA Extraction and Real-Time RT-qPCR
2.11. Crystal Violet Cell Proliferation Assay
2.12. In Silico Data Analyses
2.13. Statistical Analyses
3. Results
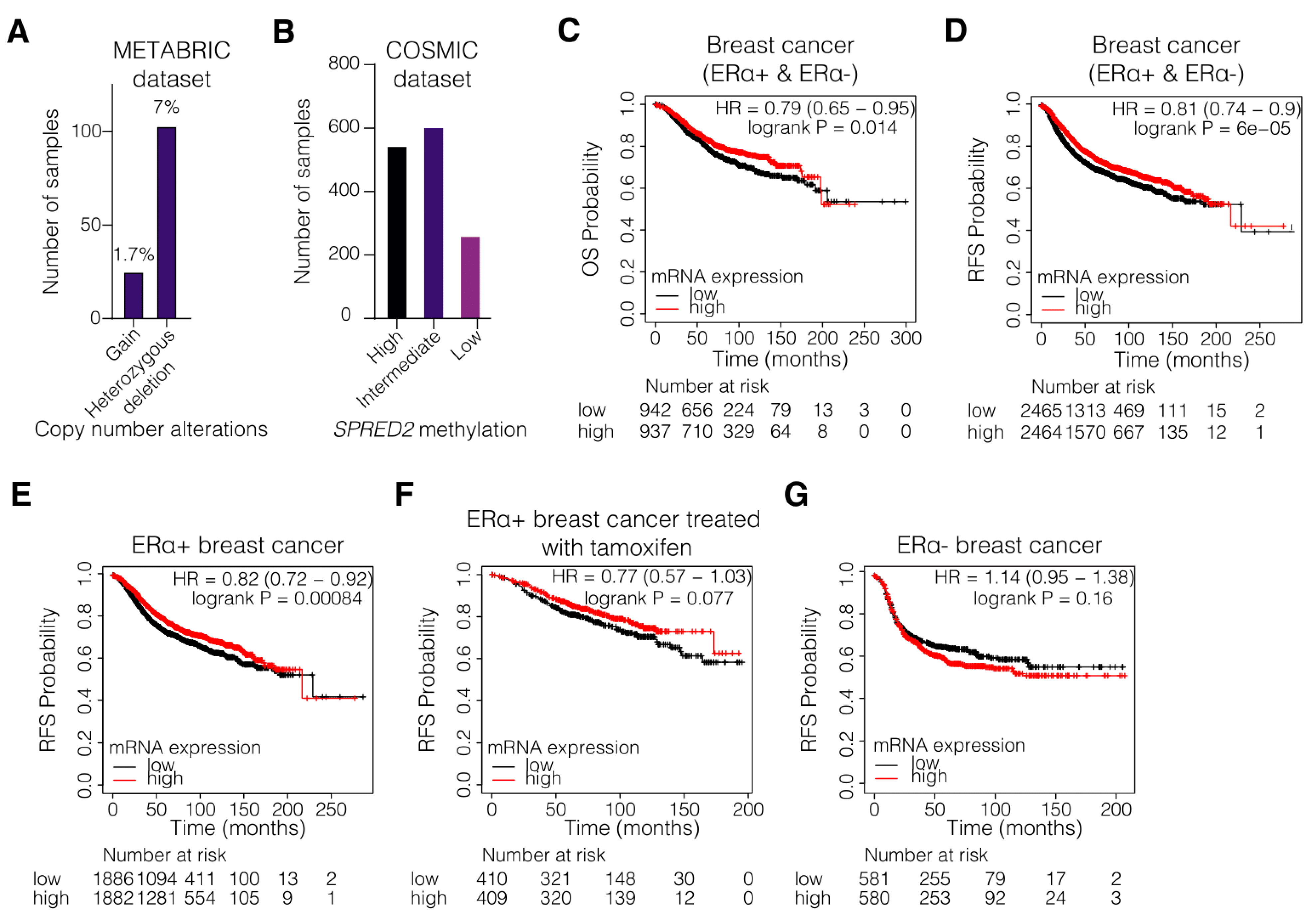
3.1. Hypermethylation and Decreased Expression of SPRED2 Are Associated with Poor Clinical Outcome of ERα+ BC
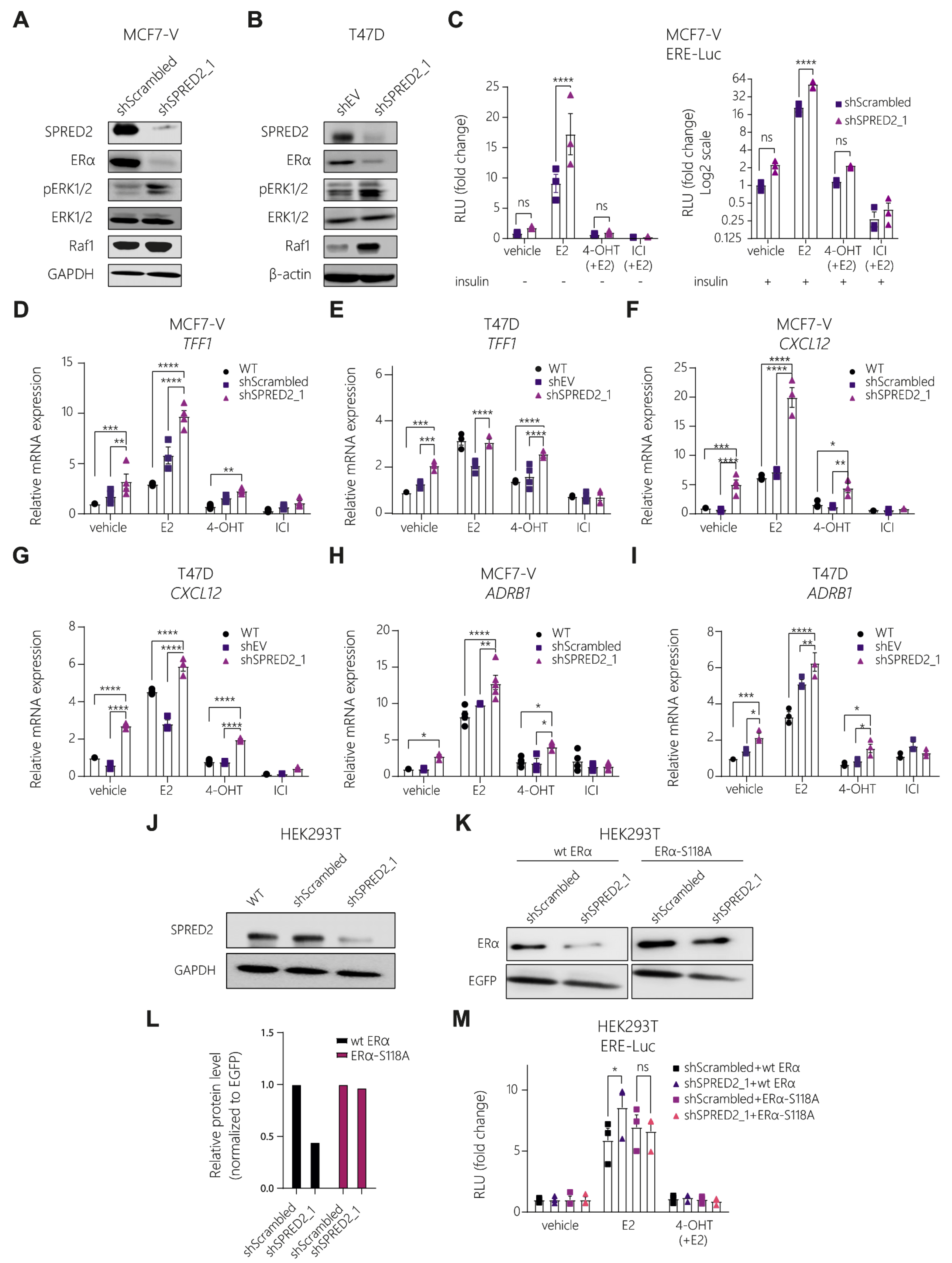
3.2. SPRED2 Depletion Promotes the Proliferation of BC Cell Lines and Their Resistance to Tamoxifen
3.3. Knockdown of SPRED2 Stimulates ERα Activity by Activation of ERK1/2
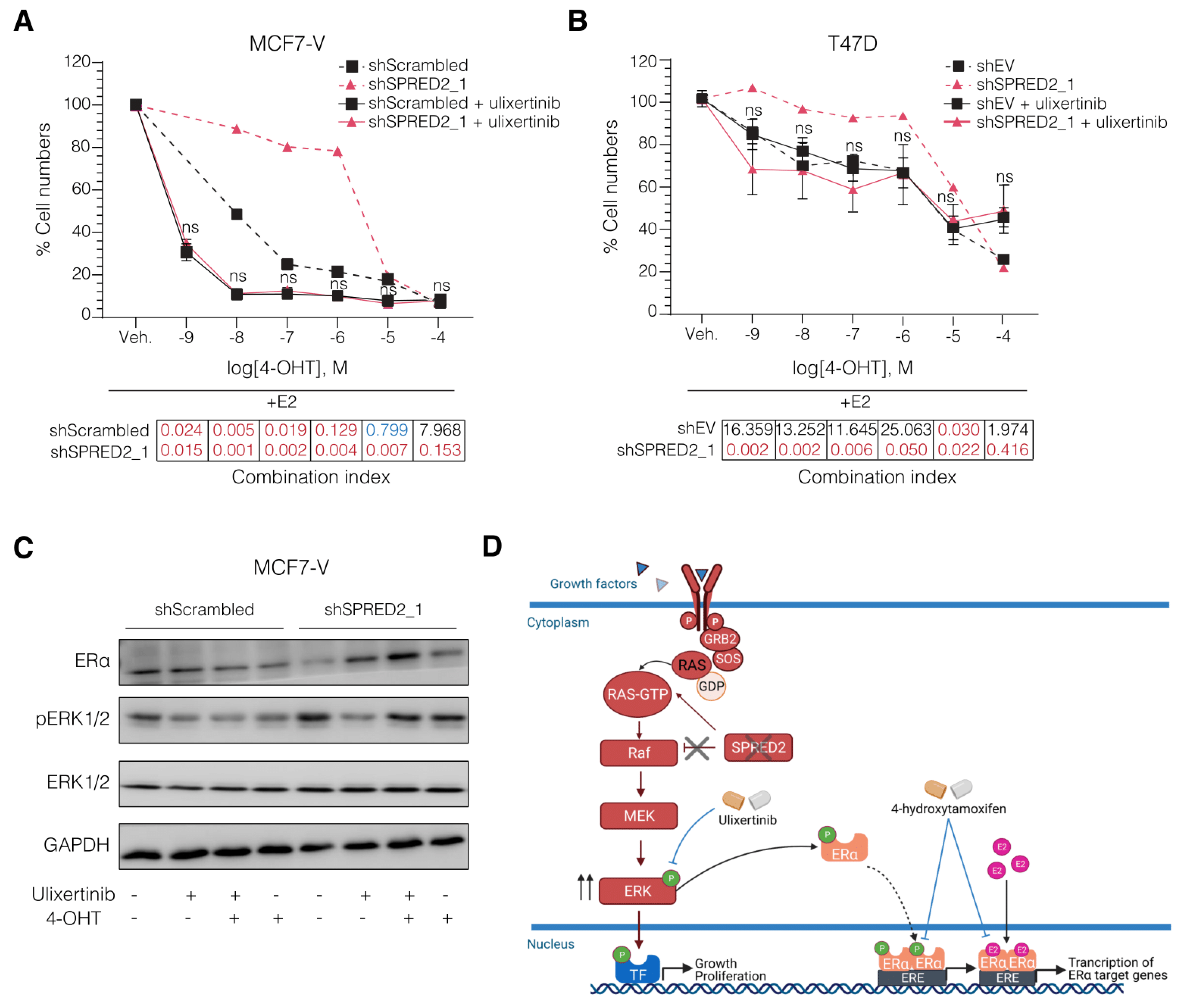
3.4. The ERK1/2 Inhibitor Ulixertinib Resensitizes SPRED2-Deficient BC Cells to 4-OHT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Azamjah, N.; Soltan-Zadeh, Y.; Zayeri, F. Global Trend of Breast Cancer Mortality Rate: A 25-Year Study. Asian Pac. J. Cancer Prev. 2019, 20, 2015–2020. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and nuclear estrogen receptor alpha actions: From tissue specificity to medical implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Farcas, A.M.; Nagarajan, S.; Cosulich, S.; Carroll, J.S. Genome-wide estrogen receptor activity in breast cancer. Endocrinology 2021, 162, 1–15. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Korach, K.S. Estrogen Receptors: New Directions in the New Millennium. Endocr. Rev. 2018, 39, 664–675. [Google Scholar] [CrossRef]
- Siersbaek, R.; Kumar, S.; Carroll, J.S. Signaling pathways and steroid receptors modulating estrogen receptor α function in breast cancer. Genes Dev. 2018, 32, 1141–1154. [Google Scholar] [CrossRef]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef]
- Cole, M.P.; Jones, C.T.; Todd, I.D. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br. J. Cancer 1971, 25, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Jordan, V.C. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resist. 2019, 2, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; O’Malley, B.W. Coregulator function: A key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 2004, 25, 45–71. [Google Scholar] [CrossRef]
- Feng, Q.; O’Malley, B.W. Nuclear receptor modulation-role of coregulators in selective estrogen receptor modulator (SERM) actions. Steroids 2014, 90, 39–43. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef]
- McGlynn, L.M.; Kirkegaard, T.; Edwards, J.; Tovey, S.; Cameron, D.; Twelves, C.; Bartlett, J.M.; Cooke, T.G. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin. Cancer Res. 2009, 15, 1487–1495. [Google Scholar] [CrossRef]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000, 351, 289–305. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef]
- Bunone, G.; Briand, P.A.; Miksicek, R.J.; Picard, D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996, 15, 2174–2183. [Google Scholar] [CrossRef] [PubMed]
- Bennesch, M.A.; Segala, G.; Wider, D.; Picard, D. LSD1 engages a corepressor complex for the activation of the estrogen receptor α by estrogen and cAMP. Nucleic Acids Res. 2016, 44, 8655–8670. [Google Scholar] [CrossRef] [PubMed]
- Picard, D. Molecular mechanisms of cross-talk between growth factors and nuclear receptor signaling. Pure Appl. Chem. 2003, 75, 1743–1756. [Google Scholar] [CrossRef]
- Ignar-Trowbridge, D.M.; Nelson, K.G.; Bidwell, M.C.; Curtis, S.W.; Washburn, T.F.; McLachlan, J.A.; Korach, K.S. Coupling of dual signaling pathways: Epidermal growth factor action involves the estrogen receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4658–4662. [Google Scholar] [CrossRef] [PubMed]
- Ignar-Trowbridge, D.M.; Teng, C.T.; Ross, K.A.; Parker, M.G.; Korach, K.S.; McLachlan, J.A. Peptide growth factors elicit estrogen receptor-dependent transcriptional activation of an estrogen-responsive element. Mol. Endocrinol. 1993, 7, 992–998. [Google Scholar] [CrossRef][Green Version]
- Newton, C.J.; Buric, R.; Trapp, T.; Brockmeier, S.; Pagotto, U.; Stalla, G.K. The unliganded estrogen receptor (ER) transduces growth factor signals. J. Steroid Biochem. Mol. Biol. 1994, 48, 481–486. [Google Scholar] [CrossRef]
- Ma, Z.Q.; Santagati, S.; Patrone, C.; Pollio, G.; Vegeto, E.; Maggi, A. Insulin-like growth factors activate estrogen receptor to control the growth and differentiation of the human neuroblastoma cell line SK-ER3. Mol. Endocrinol. 1994, 8, 910–918. [Google Scholar] [CrossRef]
- Wakioka, T.; Sasaki, A.; Kato, R.; Shouda, T.; Matsumoto, A.; Miyoshi, K.; Tsuneoka, M.; Komiya, S.; Baron, R.; Yoshimura, A. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001, 412, 647–651. [Google Scholar] [CrossRef]
- Bundschu, K.; Walter, U.; Schuh, K. Getting a first clue about SPRED functions. Bioessays 2007, 29, 897–907. [Google Scholar] [CrossRef]
- Gong, J.; Yan, Z.; Liu, Q. Progress in experimental research on SPRED protein family. J. Int. Med. Res. 2020, 48, 300060520929170. [Google Scholar] [CrossRef]
- Kawazoe, T.; Taniguchi, K. The Sprouty/Spred family as tumor suppressors: Coming of age. Cancer Sci. 2019, 110, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Tora, L.; Mullick, A.; Metzger, D.; Ponglikitmongkol, M.; Park, I.; Chambon, P. The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J. 1989, 8, 1981–1986. [Google Scholar] [CrossRef]
- Ali, S.; Metzger, D.; Bornert, J.M.; Chambon, P. Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J. 1993, 12, 1153–1160. [Google Scholar] [CrossRef]
- Fankhauser, C.P.; Briand, P.A.; Picard, D. The hormone binding domain of the mineralocorticoid receptor can regulate heterologous activities in cis. Biochem. Biophys. Res. Commun. 1994, 200, 195–201. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Molloy, M.E.; Lewinska, M.; Williamson, A.K.; Nguyen, T.T.; Kuser-Abali, G.; Gong, L.; Yan, J.; Little, J.B.; Pandolfi, P.P.; Yuan, Z.M. ZBTB7A governs estrogen receptor alpha expression in breast cancer. J. Mol. Cell Biol. 2018, 10, 273–284. [Google Scholar] [CrossRef]
- Wright, P.K.; May, F.E.; Darby, S.; Saif, R.; Lennard, T.W.; Westley, B.R. Estrogen regulates vesicle trafficking gene expression in EFF-3, EFM-19 and MCF-7 breast cancer cells. Int. J. Clin. Exp. Pathol. 2009, 2, 463–475. [Google Scholar] [PubMed]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Fan, J.; Ding, Z.; Li, R.; Lin, K.; Fu, J.; Huang, Q.; Zeng, Y.; Liu, J. Prognostic and Predictive Value of Transcription Factors Panel for Digestive System Carcinoma. Front. Oncol. 2021, 11, 670129. [Google Scholar] [CrossRef] [PubMed]
- Rueda, O.M.; Sammut, S.J.; Seoane, J.A.; Chin, S.F.; Caswell-Jin, J.L.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; Ali, H.R.; et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 2019, 567, 399–404. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, T.; Martin, N. CompuSyn for Drug Combinations: PC Software and User’s Guide: A Computer Program for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Values; ComboSyn Inc.: Paramus, NJ, USA, 2005. [Google Scholar]
- Newell-Price, J.; Clark, A.J.; King, P. DNA methylation and silencing of gene expression. Trends Endocrinol. Metab. 2000, 11, 142–148. [Google Scholar] [CrossRef]
- Gyorffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Ringner, M.; Fredlund, E.; Hakkinen, J.; Borg, A.; Staaf, J. GOBO: Gene expression-based outcome for breast cancer online. PLoS ONE 2011, 6, e17911. [Google Scholar] [CrossRef]
- Merot, Y.; Metivier, R.; Penot, G.; Manu, D.; Saligaut, C.; Gannon, F.; Pakdel, F.; Kah, O.; Flouriot, G. The relative contribution exerted by AF-1 and AF-2 transactivation functions in estrogen receptor α transcriptional activity depends upon the differentiation stage of the cell. J. Biol. Chem. 2004, 279, 26184–26191. [Google Scholar] [CrossRef] [PubMed]
- Wijayaratne, A.L.; McDonnell, D.P. The human estrogen receptor-α is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J. Biol. Chem. 2001, 276, 35684–35692. [Google Scholar] [CrossRef] [PubMed]
- Bennesch, M.A.; Picard, D. Minireview: Tipping the balance: Ligand-independent activation of steroid receptors. Mol. Endocrinol. 2015, 29, 349–363. [Google Scholar] [CrossRef] [PubMed]
- de Launoit, Y.; Baert, J.L.; Chotteau-Lelievre, A.; Monte, D.; Coutte, L.; Mauen, S.; Firlej, V.; Degerny, C.; Verreman, K. The Ets transcription factors of the PEA3 group: Transcriptional regulators in metastasis. Biochim. Biophys. Acta 2006, 1766, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996, 74, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Thrane, S.; Lykkesfeldt, A.E.; Larsen, M.S.; Sorensen, B.S.; Yde, C.W. Estrogen receptor α is the major driving factor for growth in tamoxifen-resistant breast cancer and supported by HER/ERK signaling. Breast Cancer Res. Treat. 2013, 139, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Heightman, T.D.; Berdini, V.; Braithwaite, H.; Buck, I.M.; Cassidy, M.; Castro, J.; Courtin, A.; Day, J.E.H.; East, C.; Fazal, L.; et al. Fragment-Based Discovery of a Potent, Orally Bioavailable Inhibitor That Modulates the Phosphorylation and Catalytic Activity of ERK1/2. J. Med. Chem. 2018, 61, 4978–4992. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Blair, S.L.; Burstein, H.J.; Dang, C.; Elias, A.D.; et al. NCCN Guidelines(R) Insights: Breast Cancer, Version 4.2021. J. Natl. Compr. Canc. Netw. 2021, 19, 484–493. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Cakir, M.; Grossman, A.B. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert. Opin. Ther. Targets 2009, 13, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Khavari, T.A.; Rinn, J. Ras/Erk MAPK signaling in epidermal homeostasis and neoplasia. Cell Cycle 2007, 6, 2928–2931. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Yamamoto, T.; Tsuchiya, Y.; Nishida, E. ERK MAP kinase in G cell cycle progression and cancer. Cancer Sci. 2006, 97, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, N.; Valencia, T.; Warren, A.Y.; Gnanapragasam, V.J. Evidence for downregulation of the negative regulator SPRED2 in clinical prostate cancer. Br. J. Cancer 2013, 108, 597–601. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Kong, F.; Xiao, F.; Li, Y.; Wang, H.; Zhang, S.; Huang, D.; Wang, L.; Yang, Y. Spred2 inhibits epithelial-mesenchymal transition of colorectal cancer cells by impairing ERK signaling. Oncol. Rep. 2020, 44, 174–184. [Google Scholar] [CrossRef]
- Villar, V.; Kocic, J.; Santibanez, J.F. Spred2 inhibits TGF-β1-induced urokinase type plasminogen activator expression, cell motility and epithelial mesenchymal transition. Int. J. Cancer 2010, 127, 77–85. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, C.; Shen, Q.; Munoz-Medellin, D.; Kim, H.; Brown, P.H. AP-1 blockade in breast cancer cells causes cell cycle arrest by suppressing G1 cyclin expression and reducing cyclin-dependent kinase activity. Oncogene 2004, 23, 8238–8246. [Google Scholar] [CrossRef][Green Version]
- Lu, C.; Shen, Q.; DuPre, E.; Kim, H.; Hilsenbeck, S.; Brown, P.H. cFos is critical for MCF-7 breast cancer cell growth. Oncogene 2005, 24, 6516–6524. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast 2003, 12, 362–367. [Google Scholar] [CrossRef]
- Kurokawa, H.; Lenferink, A.E.; Simpson, J.F.; Pisacane, P.I.; Sliwkowski, M.X.; Forbes, J.T.; Arteaga, C.L. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000, 60, 5887–5894. [Google Scholar] [PubMed]
- Chung, Y.L.; Sheu, M.L.; Yang, S.C.; Lin, C.H.; Yen, S.H. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int. J. Cancer 2002, 97, 306–312. [Google Scholar] [CrossRef]
- Johnston, S.R.; Lu, B.; Scott, G.K.; Kushner, P.J.; Smith, I.E.; Dowsett, M.; Benz, C.C. Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin. Cancer Res. 1999, 5, 251–256. [Google Scholar]
- Smith, L.M.; Wise, S.C.; Hendricks, D.T.; Sabichi, A.L.; Bos, T.; Reddy, P.; Brown, P.H.; Birrer, M.J. cJun overexpression in MCF-7 breast cancer cells produces a tumorigenic, invasive and hormone resistant phenotype. Oncogene 1999, 18, 6063–6070. [Google Scholar] [CrossRef]
- Zhou, Y.; Yau, C.; Gray, J.W.; Chew, K.; Dairkee, S.H.; Moore, D.H.; Eppenberger, U.; Eppenberger-Castori, S.; Benz, C.C. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer 2007, 7, 59. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Hutcheson, I.R.; Britton, D.; Knowlden, J.M.; Jones, H.E.; Harper, M.E.; Hiscox, S.E.; Barrow, D.; Gee, J.M. Growth factor signalling networks in breast cancer and resistance to endocrine agents: New therapeutic strategies. J. Steroid Biochem. Mol. Biol. 2005, 93, 257–262. [Google Scholar] [CrossRef]
- Lewis, M.; Prouzet-Mauleon, V.; Lichou, F.; Richard, E.; Iggo, R.; Turcq, B.; Mahon, F.X. A genome-scale CRISPR knock-out screen in chronic myeloid leukemia identifies novel drug resistance mechanisms along with intrinsic apoptosis and MAPK signaling. Cancer Med. 2020, 9, 6739–6751. [Google Scholar] [CrossRef]
- Liu, X.Y.; Yang, Y.F.; Wu, C.T.; Xiao, F.J.; Zhang, Q.W.; Ma, X.N.; Li, Q.F.; Yan, J.; Wang, H.; Wang, L.S. Spred2 is involved in imatinib-induced cytotoxicity in chronic myeloid leukemia cells. Biochem. Biophys. Res. Commun. 2010, 393, 637–642. [Google Scholar] [CrossRef]
- Ma, X.N.; Liu, X.Y.; Yang, Y.F.; Xiao, F.J.; Li, Q.F.; Yan, J.; Zhang, Q.W.; Wang, L.S.; Li, X.Y.; Wang, H. Regulation of human hepatocellular carcinoma cells by Spred2 and correlative studies on its mechanism. Biochem. Biophys. Res. Commun. 2011, 410, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Aksamitiene, E.; Kholodenko, B.N.; Kolch, W.; Hoek, J.B.; Kiyatkin, A. PI3K/Akt-sensitive MEK-independent compensatory circuit of ERK activation in ER-positive PI3K-mutant T47D breast cancer cells. Cell. Signal. 2010, 22, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ge, C.C.; Wang, J.; Wu, X.X.; Li, X.M.; Li, W.; Wang, S.S.; Liu, T.; Hou, J.Z.; Sun, H.; et al. MEK inhibitor, PD98059, promotes breast cancer cell migration by inducing beta-catenin nuclear accumulation. Oncol. Rep. 2017, 38, 3055–3063. [Google Scholar] [CrossRef] [PubMed]
- Germann, U.; Furey, B.; Roix, J.; Markland, W.; Hoover, R.; Aronov, A.; Hale, M.; Chen, G.; Martinez-Botella, G.; Alargova, R. The selective ERK inhibitor BVD-523 is active in models of MAPK pathway-dependent cancers, including those with intrinsic and acquired drug resistance. In Proceedings of the 106th Annual Meeting of the American Association for Cancer Research, Philadelphia, PA, USA, 18–22 April 2015; pp. 18–22. [Google Scholar]
- Jin, F.; Gao, D.; Wu, Q.; Liu, F.; Chen, Y.; Tan, C.; Jiang, Y. Exploration of N-(2-aminoethyl)piperidine-4-carboxamide as a potential scaffold for development of VEGFR-2, ERK-2 and Abl-1 multikinase inhibitor. Bioorg. Med. Chem. 2013, 21, 5694–5706. [Google Scholar] [CrossRef]
- Ding, W.-J.; Tao, W.-P.; Zeng, T.; Deng, J.-J.; Lei, H.-B.; Ge, W. Selective ERK inhibitor ulixertinib inhibits proliferation and induces apoptosis in lymphoma cell lines. Int. J. Clin. Exp. Med. 2016, 9, 10955–10962. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| shRNA Name | Target Sequence 5′–3′ | TRC Clone ID 2 |
|---|---|---|
| shSPRED2_1 | CAACAGCTACAGACAGTTCTT | TRCN0000056832 |
| shSPRED2_2 | GCAATCGAAGACCTTATAGAA | TRCN0000056828 |
| shScrambled | CCTAAGGTTAAGTCGCCCTCG 1 |
| Antibody | Supplier | Working Dilutions |
|---|---|---|
| Anti-ERα | Bethyl Laboratories (cat. # A300-498A) | 1:1000 |
| Anti-SPRED2 | Sigma-Aldrich (cat. # S7320) | 1:1000 |
| Anti-GAPDH (6C5) | Abcam (cat. # ab8245) | 1:30,000 |
| Anti-ERK 2 (C-14) | Santa Cruz Biotechnology (cat. # sc-154) | 1:200 |
| Anti-p-ERK (E-4) | Santa Cruz Biotechnology (cat. # sc-733) | 1:1000 |
| Anti-RAF1 (C-12) | Santa Cruz Biotechnology (cat. # sc-133) | 1:500 |
| Anti-β-actin | Millipore (cat. # MABT825) | 1:5000 |
| Anti-EGFP | Roche (cat. # 11814460001) | 1:5000 |
| IgGs, rabbit and mouse | Sigma-Aldrich (cat. # I5006 and I5381) | 1: 10,000–1: 30,000 |
| Gene | Forward Primer 5′–3′ | Reverse Primer 5′–3′ |
|---|---|---|
| SPRED2 | GGGACAGGCGTCTAGGTGAAC | AAAGCCGCTTCGTCCATTGC |
| CXCL12 | CCCAGGTGCTACACCCTTTT | CAGGAATGGGGCTCCTTCAG |
| ADRB1 | CCGGGAACAGGAACACAC | GAAAGCAAAAGGAAATATGTC |
| TFF11 | CAATTCTGTCTTTCACGGGG | CACCATGGAGAACAAGGTGA |
| ESR12 | GCTCTTGGACAGGAACCAGG | AAGATCTCCACCATGCCCTCT |
| β-actin (normalization control) 3 | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT |
| ETV44 | AGGAACAGACGGACTTCGCCTA | CTGGGAATGGTCGCAGAGGTTT |
| ETV5 | GGGTTCTTTGGGGTTTGTTT | CGCAGGGGAAAGTATTTCAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vafeiadou, V.; Hany, D.; Picard, D. Hyperactivation of MAPK Induces Tamoxifen Resistance in SPRED2-Deficient ERα-Positive Breast Cancer. Cancers 2022, 14, 954. https://doi.org/10.3390/cancers14040954
Vafeiadou V, Hany D, Picard D. Hyperactivation of MAPK Induces Tamoxifen Resistance in SPRED2-Deficient ERα-Positive Breast Cancer. Cancers. 2022; 14(4):954. https://doi.org/10.3390/cancers14040954
Chicago/Turabian StyleVafeiadou, Vasiliki, Dina Hany, and Didier Picard. 2022. "Hyperactivation of MAPK Induces Tamoxifen Resistance in SPRED2-Deficient ERα-Positive Breast Cancer" Cancers 14, no. 4: 954. https://doi.org/10.3390/cancers14040954
APA StyleVafeiadou, V., Hany, D., & Picard, D. (2022). Hyperactivation of MAPK Induces Tamoxifen Resistance in SPRED2-Deficient ERα-Positive Breast Cancer. Cancers, 14(4), 954. https://doi.org/10.3390/cancers14040954